 Asuman Gedikbasi1,2
Asuman Gedikbasi1,2 Guven Toksoy3Meryem Karaca2
Guven Toksoy3Meryem Karaca2 Cagri Gulec3Mehmet Cihan Balci2
Cagri Gulec3Mehmet Cihan Balci2 Dilek Gunes2Seda Gunes2Ayca Dilruba Aslanger3Gokcen Unverengil4
Dilek Gunes2Seda Gunes2Ayca Dilruba Aslanger3Gokcen Unverengil4 Birsen Karaman1,3Seher Basaran3Mubeccel Demirkol2Gulden Fatma Gokcay2
Birsen Karaman1,3Seher Basaran3Mubeccel Demirkol2Gulden Fatma Gokcay2 Zehra Oya Uyguner3*
Zehra Oya Uyguner3*- 1Department of Pediatric Basic Sciences, Institute of Child Health Istanbul University, Istanbul, Türkiye
- 2Division of Pediatric Nutrition and Metabolism, Department of Pediatrics, Istanbul Faculty of Medicine, Istanbul University, Istanbul, Türkiye
- 3Department of Medical Genetics, Istanbul Faculty of Medicine, Istanbul University, Istanbul, Türkiye
- 4Department of Pathology, Istanbul Faculty of Medicine, Istanbul University, Istanbul, Türkiye
Background: Mitochondrial diseases are the most common group of inherited metabolic disorders, causing difficulties in definite diagnosis due to clinical and genetic heterogeneity. Clinical components are predominantly associated with pathogenic variants shown in nuclear or mitochondrial genomes that affect vital respiratory chain function. The development of high-throughput sequencing technologies has accelerated the elucidation of the genetic etiology of many genetic diseases that previously remained undiagnosed.
Methods: Thirty affected patients from 24 unrelated families with clinical, radiological, biochemical, and histopathological evaluations considered for mitochondrial diseases were investigated. DNA isolated from the peripheral blood samples of probands was sequenced for nuclear exome and mitochondrial DNA (mtDNA) analyses. MtDNA sequencing was also performed from the muscle biopsy material in one patient. For segregation, Sanger sequencing is performed for pathogenic alterations in five other affected family members and healthy parents.
Results: Exome sequencing revealed 14 different pathogenic variants in nine genes encoding mitochondrial function peptides (AARS2, EARS2, ECHS1, FBXL4, MICOS13, NDUFAF6, OXCT1, POLG, and TK2) in 12 patients from nine families and four variants in genes encoding important for muscle structure (CAPN3, DYSF, and TCAP) in six patients from four families. Three probands carried pathogenic mtDNA variations in two genes (MT-ATP6 and MT-TL1). Nine variants in five genes are reported for the first time with disease association: (AARS2: c.277C>T/p.(R93*), c.845C>G/p.(S282C); EARS2: c.319C>T/p.(R107C), c.1283delC/p.(P428Lfs*); ECHS1: c.161G>A/p.(R54His); c.202G>A/p.(E68Lys); NDUFAF6: c.479delA/p.(N162Ifs*27); and OXCT1: c.1370C>T/p.(T457I), c.1173-139G>T/p.(?).
Conclusion: Bi-genomic DNA sequencing clarified genetic etiology in 67% (16/24) of the families. Diagnostic utility by mtDNA sequencing in 13% (3/24) and exome sequencing in 54% (13/24) of the families prioritized searching for nuclear genome pathologies for the first-tier test. Weakness and muscle wasting observed in 17% (4/24) of the families underlined that limb-girdle muscular dystrophy, similar to mitochondrial myopathy, is an essential point for differential diagnosis. The correct diagnosis is crucial for comprehensive genetic counseling of families. Also, it contributes to making treatment-helpful referrals, such as ensuring early access to medication for patients with mutations in the TK2 gene.
Introduction
Mitochondrial diseases (MDs), caused by mutations in either nuclear or mitochondrial genomes (mtDNA), are the most common group of inherited metabolic diseases and affect about 1 in 5,000 in the population (Stenton and Prokisch, 2020; Orsucci et al., 2021). High clinical and genetic heterogeneity, overlapping with neuromuscular diseases, is observed due to pathological changes in genes encoding complex energy metabolism pathways in organs with high energy requirements, such as muscles, the brain, and the heart. Clinical characteristics, biochemical and enzymatic analyses, brain imaging, and pathomorphological anomalies in muscles and fibroblasts are all assessed for clinical diagnosis (Rahman, 2020; Fernandez-Eulate et al., 2022).
Unique characteristics of the mitochondrial genome, such as heteroplasmy, maternal inheritance, and dynamic organelle structure involving more than 1,500 functional peptides encoded by nuclear DNA, complicate the molecular diagnosis of MDs (Schlieben and Prokisch, 2020). The underlying genetic origin may be secondary to pathogenic variants in one of the nuclear genes involved in the mitochondria’s transcription, translation, or function, concurrently small and gross pathogenic alterations in the mtDNA. The majority of the mitochondrial proteome is encoded by the nuclear genes. To date, 320 out of 1,136 MD-associated genes are proven to be disease-associated genes (Rath et al., 2021). A total of 96 disease-related variants are reported in 37 genes in the mitochondrial genome in pathogenic mutations confirmed using Mitomap (Lott et al., 2013).
Next-generation sequencing (NGS) technology allows whole-genome sequencing (WGS) or whole-exome sequencing (WES), including mtDNA. Based on clinical and genetic heterogeneity and unknown incidences of mitochondria-related diseases in different cohorts, non-targeted exome sequencing provides powerful access to molecular genetic diagnosis. NGS technology led to a paradigm shift in the differential diagnosis of MDs, altering the invasive function-to-gene approach to a non-invasive approach via the “genetics-first.” In parallel, since MDs are under the control of a dual genome, the classification of MDs has evolved from clinical and biochemical designations to a gene-based direction organized under the disease of mtDNA mutations and Mendelian mitochondrial disorders (Lightowlers et al., 2015). Despite numerous obstacles, motivation for providing molecular diagnosis in MDs is high because the clinical management of the disease, future care planning, and providing pre-implantation genetic diagnostic opportunities and relapse risk predictions are valuable for patients and their families. Moreover, definitive molecular diagnosis allows for replacing a critical cofactor for disease recovery or provides target-specific treatment such as restriction of a diet component. This study presents the results of dual-genome analysis toward diagnosing patients with a preliminary assessment of MDs with clinical findings, imaging, and biochemical values.
Materials and methods
Participants and clinical evaluations
The study group comprised 24 probands, six affected family members (families 1, 2, 6, 13, 16, and 18), and 29 healthy individuals [one healthy sibling from each of four families (families 5, 7, 8, and 9) and 25 healthy parents] from 24 unrelated families formed the study group. The participants provided consent to participate.
All affected individuals were suspected to have mitochondrial diseases (MDs) in the differential diagnosis according to clinical, radiological, and laboratory findings. Clinical genetic examination of patients is recorded, including radiological and laboratory findings with detailed medical history. Patients were evaluated at the genetic outpatient clinic, and a three-generation pedigree was drawn.
Radiological evaluations
Central nervous system findings, nonvascular distribution of stroke-like lesions, diffuse white matter damage, bilateral gray matter, basal ganglia, and brain stem involvement have been evaluated from the magnetic resonance imaging (MRI) findings of patients. Electromyography (EMG), electroencephalogram (EEG), and echocardiography (ECG) reports were examined.
Biochemical tests
The Carnitine–acylcarnitine profile was assessed by tandem mass spectrometry (MS/MS) and quantitative amino acid analyses, and organic acid analysis results were assessed by liquid and gas chromatography–mass spectrometry (LC and GC-MS). Blood lactate levels, alanine levels elevated secondary to the transamination of pyruvate, and increased excretion of urinary organic acids as tricarboxylic acid (TCA) cycle intermediates such as lactate and malate, succinate, 2-oxoglutarate, and fumarate were recorded. Creatine kinase (CK), aspartate aminotransferase (AST), and alanine aminotransferase (ALT) levels indicating muscle and liver involvement were documented. Ketone bodies like 3-OH-butyrate and acetoacetate were evaluated.
Histopathological tests
A microscopic examination of the tissue was performed. Muscle biopsy samples inside gauze pads were frozen in liquid nitrogen, and sections were obtained using a cryotome device at −80°C. The 8-μm sections were subjected to hematoxylin and eosin (HE), modified Gomori trichrome (MGT), PAS, oil Red O (ORO), myophosphorylase, and oxidative enzyme (NADH-TR, SDH, COX, and COX/SDH) stains for examination.
Genetic tests
Genomic DNA, together with mtDNA, was isolated from peripheral blood lymphocytes. A stepwise approach was used for genetic investigation. The initial test was mtDNA sequencing by an in-house developed fragmentation-based methodology. Patients with unidentified heteroplasmic or homoplasmic causative variant associations with mtDNA were underwent a second analysis step for exome sequencing with either single, paired, or trio investigation.
MtDNA sequencing
Four primer pairs, coupling on the flanking regions, were designed and used with the Thermo Scientific™ Phusion High-fidelity DNA Polymerase enzyme to amplify the mtDNA (16.569 bp) with four overlapping sizes, 5,077, 4,393, 5,465, and 5,540 bp. Amplified mtDNA samples are purified using the Roche High Pure PCR Product Purification Kit. The fragmentation process was performed using the Ion Xpress™ Plus Fragment Library Kit (Kat. No. 4471269). The library length was selected as 400 bp. Fragmentation was performed to maintain a median fragment size of 350–450 bp. The Ion 510 ™ Kit (Kat. No. A34461) was used to load samples to chips, the template was prepared in the Ion Chef ™ System, and sequencing was performed in the Ion S5 ™ System.
Exome sequencing
The Ion AmpliSeq™ Exome RDY Kit 1 × 8™ (Cat. No. A38262) was used to prepare exome libraries for sequencing genomic DNA. Barcoded libraries were formed using the Ion Xpress™ Barcode Adapter Kits™ (Cat. No. 4474517; whole adapter set between 1 and 96). Barcoded libraries were loaded in a single ion chip, and data were transferred to Torrent server software following sequencing.
Variant analysis
Data were automatically transferred to Ion Reporter™ software using Torrent Suite™ with the IonReporterUploader. The Browser Extensible Data (BED) file was added to the target and hotspot regions, and quality parameters were maintained in the recommended range. Each patient was analyzed with the analysis flow chart formed in the Ion Reporter™ System. Variants are viewed using the Integrative Genomics Viewer (IGV). The revised Cambridge Reference Sequence (rCRS) (GenBank access no: NC_012920.1), containing 16,569 base pairs, is used to determine mtDNA variants, and the Mitomap database was used to investigate the definitive pathogenic changes. Exome sequence variants with a minor allele frequency of <0.1% are classified according to the ACMG (Richards et al., 2015). In the data analysis, genes reported to be related to mitochondrial diseases (MitoCarta 3.0) are prioritized (Rath et al., 2021). Disease association of the exome variants is searched from the literature reports OMIM (Hamosh et al., 2005), ClinVar (Landrum et al., 2018), and GeneCards (Stelzer et al., 2016). Deleteriousness of the substitutions is scored by Combined Annotation-Dependent Depletion (CADD, 1.6) using the online server UCSC Browser for CADD scores in the phenotypes and literature track of the human gene (Rentzsch et al., 2019) and for deletion from the web server provided in the Mutation Significance Cutoff (MSC) of human genes (Itan et al., 2016). Domain/regions are defined according to UniProt (Dogan et al., 2016). The Human Splicing Finder is used to investigate the impact of alteration on exonic splicing silencer (ESS) and exonic splicing enhancer (ESE) sites (Desmet et al., 2009). Variants suspected to be pathogenic and clinically related are confirmed with by Sanger sequencing on ABI3500, followed by segregation in the families.
RNA investigation
The total RNA was extracted from a peripheral blood sample’s lymphocytes or a muscle biopsy using the TRIzol manufacturer protocol (Invitrogen). mRNA was converted to cDNA using 1 μg of the total RNA per reaction, using a reverse transcriptase protocol with a random hexamer (Bio-Rad iScript). Sequencing primer pairs are designed to couple flanking coding exons of the target.
Results
Study group
The demographic characteristics of 30 affected patients from 24 families, clinical, laboratory, radiology, and muscle histochemistry findings are given in Table 1. Of the cohort, four (13%; three females and one male) were adults (31, 7 ± 8, 9), and 26 (87%; 13 females and 13 males) were under 18 years of age (9, 2 ± 5, 7 years).
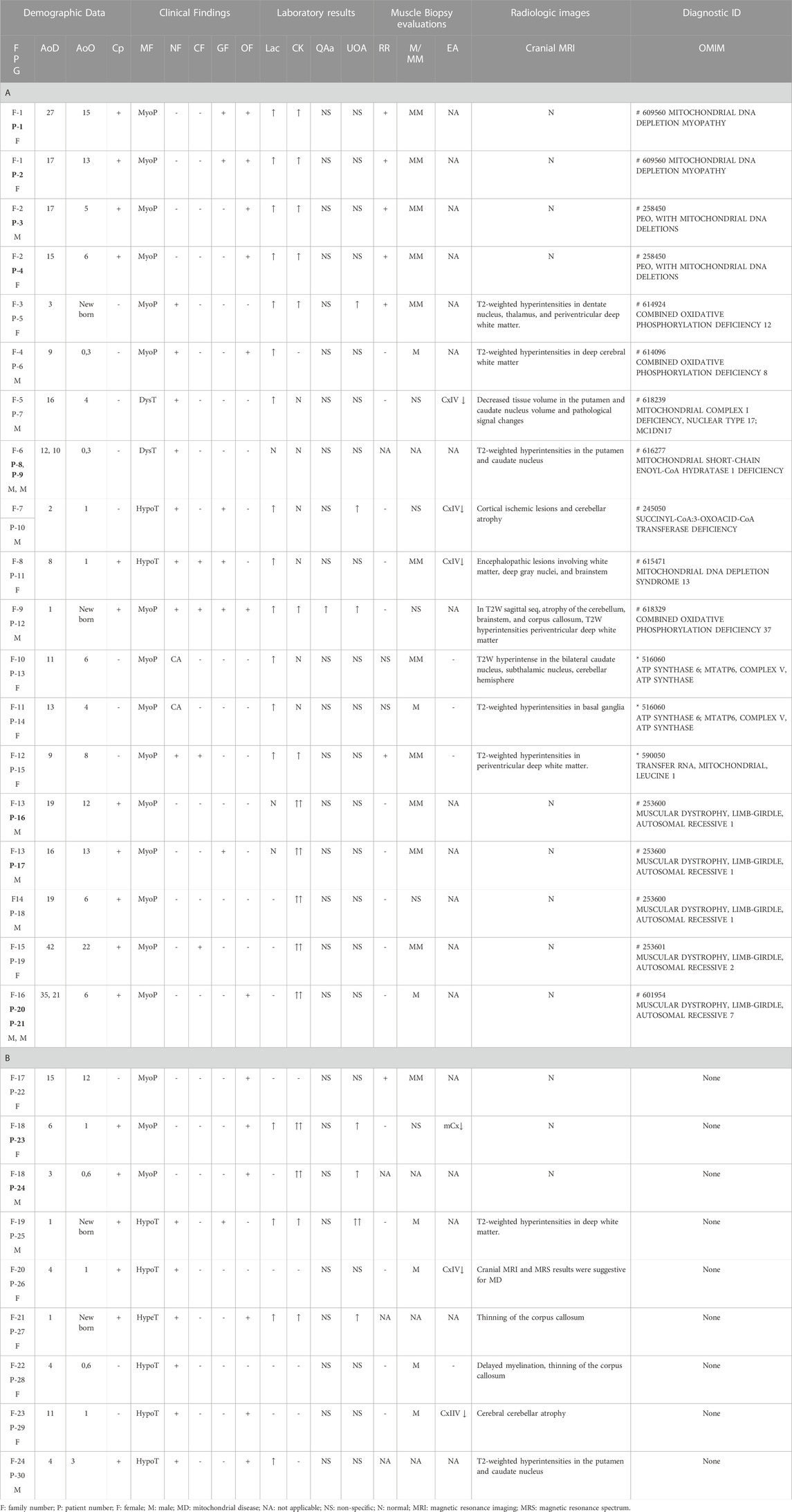
TABLE 1. Clinical and laboratory findings of patients (A and B). Patient numbers with a sibling relationship are shown in bold letters. A. Patients identified with pathogenic variants that are associated clinically. B. Patients without any associated variant.
The age of the onset of disease in the cohort varied from birth to 22 years. Consanguineous marriage was observed in 13 families (54%). Four patients had an affected sibling, and two had an affected cousin.
Clinical findings
Muscular system findings were revealed in all patients (100%), and central nervous system findings were present in 17 (17/30), patients. In addition, eleven patients had ptosis or progressive external ophthalmoplegia (11/30), seven patients had abnormal liver function or gastrointestinal system findings (7/30), and four patients had cardiomyopathy (4/30).
Laboratory findings
Of the 30 patients, 16 had elevated lactic acid levels (16/30, 53%), and 16 had elevated creatine kinase (CK) levels (16/30, 53%). Quantitative amino acid analysis revealed no specific findings except elevated alanine in one patient. Urine organic acid analysis revealed mitochondrial disease-related excretion in seven patients (7/30, 23%). Histopathological evaluation of the 24 muscular biopsy samples revealed mitochondrial myopathy (MM) findings in 13 patients (13/24, 54%), seven of whom had ragged red fibers (7/24, 29%). Others were reported to have mild myopathic changes (11/24, 46%). Complex enzyme activity was reported in only six patients from six families, isolated complex IV enzyme deficiencies in five (P7, 10, 11, 26, and 29), and multiple complex enzyme deficiencies in one (P23). Cranial MRI studies revealed pathological findings at specific sites consistent with mitochondrial disease in 17 patients (P5–15 and P25–30) (17/30, 57%), and no evidence of pathology was found in 13 patients (P1–4 and P16–24) (13/30, 43%) (mitochondrial depletion syndrome n = 4, LGMD n = 6, and three undiagnosed cases).
Molecular analysis
A total of 14 variants in nine nuclear genes (AARS2, EARS2, ECHS1, FBXL4, MICOS13, NDUFAF6, OXCT1, POLG, and TK2) in nine probands and two variants in two mtDNA genes (MT-ATP6 and MT-TL1) in three probands were disclosed (Table 2). The association of those genes in the related pathways involved in mitochondrial function are shown in Figure 1. The inheritance model was confirmed by parental DNA testing for all variants identified in the nuclear genome. Three healthy siblings from families 5, 7, and 9 did not carry the related variant, while the healthy sibling of P11 of family 8 was found to be a heterozygous carrier. Nine variants were associated with MDs in this study submitted to ClinVar, shown in bold letters in Table 2. The time for molecular diagnosis ranged between 1 year and 29 years (median of 9 years).
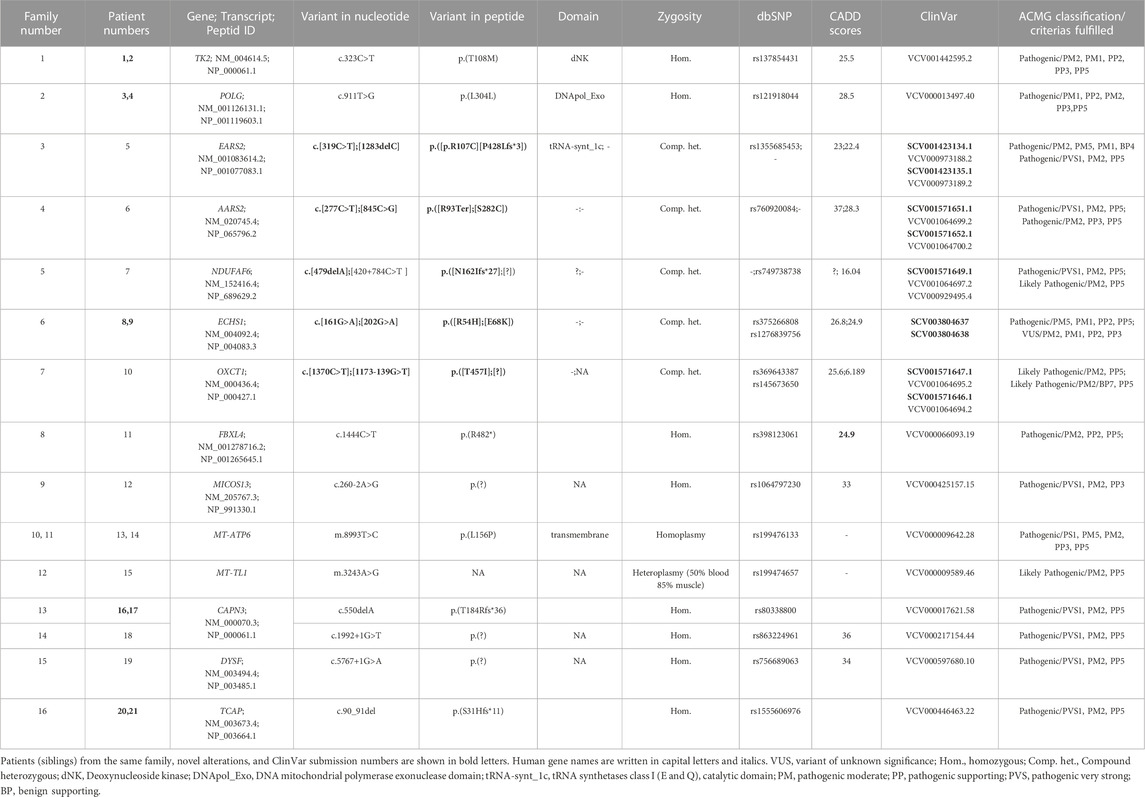
TABLE 2. Detail descriptions of the variants identified in patients. ACMG classifications of variants are performed by the Franklin website, accessed 21 April 2023 (https://franklin.genoox.com) based on population data, functional data, in-silico predictions, and reputable source data as availability.
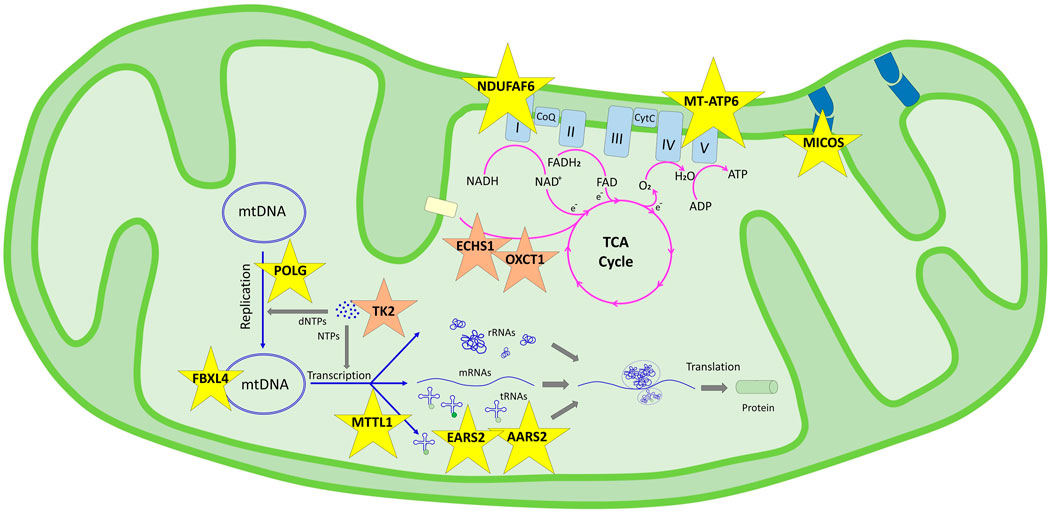
FIGURE 1. Schematic representation of the genes determined in this study and their pathways in the mitochondrial functions according to KEGG. Genes in the genetic information processing pathway are shown in yellow stars in two subgroups: translation/aminoacyl-tRNA biosynthesis (AARS2, EARS2, and MT-TL1) and mitochondrial biogenesis (FBXL4, NDUFAF6, MICOS13, POLG, and MT-ATP6). Genes in the metabolism pathway are shown in orange stars in three subgroups: nucleotide metabolism (TK2), carbohydrate metabolism (OXCT1), and fatty acid elongation (ECHS1).
In P22, a homozygous c.260C>T/p.(T87I) rs144000161 variant in the COX10 gene was identified and classified as a variant of unknown significance (VUS), and its association with the disease was excluded by detecting this change in the homozygous form in the healthy sibling. Electropherograms of the two different variants detected in the EARS2 gene on cDNA synthesized from the total RNA obtained from peripheral blood samples in P5 (II:1) and her parents (I:1 and I:2) showed that the inheritance of the allele with a frameshift to early stop underwent mRNA decay (Figure 2). The molecular genetic analysis showed MDs in 15 patients from 12 families. On identifying the relevant variants in the CAPN3, DYSF, and TCAP genes, the diagnosis was oriented toward limb-girdle muscular dystrophy (LGMD) in six patients from four families. The molecular basis in eight families (8/24, 33%) could not be determined (Table 1).
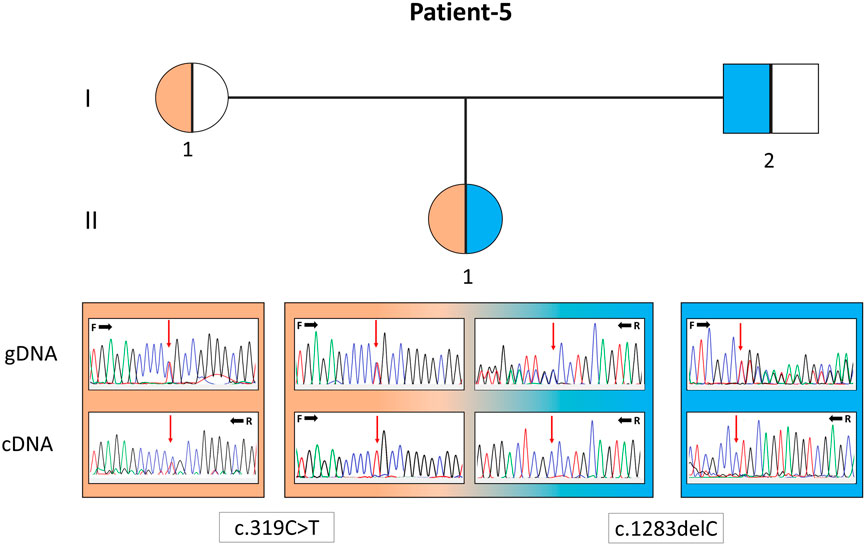
FIGURE 2. Pedigree of patient 5 with the EARS2 gene variants. Electropherograms of the sequences from the genomic DNA (gDNA) and complementary DNA (cDNA) are presented. The presence of only the allele carrying the missense variant in the patient and only the normal allele in her father indicates that the deletion-type mutation was subjected to mRNA decay.
Discussion
The involvement of diverse organs and various ages of onset and severity complicate the molecular pathologies underlining the MDs. Serious obstacles resulting from the impaired interactions among the molecules required for cellular homeostasis and metabolism can be grouped under complex pathways involved in mitochondrial function and integrity due to pathogenic changes in genes encoded by either mtDNA or genomic DNA, resulting in severe diseases with separately grouped organ involvements, age of onset, and heterogeneous phenotype. These interactions are classified under manifolds of pathways (Kyoto Encyclopedia of Genes and Genomes, KEGG) like metabolism, genetic information processing, environmental information processing, cellular processing, organizational systems, and drug interactions (Kanehisa et al., 2023).
In our cohort, mitochondrial pathway associations were defined for 11 genes; nine were genomic and two were mitochondrial DNA genes, enlightening accurate diagnosis in 12 of our families. Although extensive interactions overlap in pathways, we discuss the patient’s findings under those pathway involvements (Figure 1).
Genetic information processing/translation/aminoacyl-tRNA biosynthesis (AARS2, EARS2, and MT-TL1)
Human diseases caused by mitochondrial translation defects may arise from pathogenic variations in mitochondrial tRNA, mitochondrial aminoacyl-tRNA synthetase, mitochondrial rRNA, and mitochondrial ribosomal protein-encoding genes (Webb et al., 2020). Most infantile-onset mitochondrial encephalopathies that are genetically unidentified may have prominent biochemical characteristics related to multiple respiratory chain complex defects, its subunits encoded either by the mtDNA or, for the majority, by the nuclear DNA. Aminoacyl-tRNA synthetases (ARSs) are a group of structures recently reported to be involved in the peripheral nervous system, either with loss- or gain-of-function mutations (Boczonadi et al., 2018). AARS2 and EARS2 encode mitochondrial aminoacyl-tRNA synthetases (mt-ARSs), essential for protein synthesis in the mitochondria and generation of oxidative phosphorylation (OXPHOS) system components. Combined OXPHOS deficiency (COXPD) is the most common of biallelic diseases associated with mitochondrial-ARSs (Turvey et al., 2022). MT-TL1 is one of the 22 mt-tRNAs encoded by the mitochondrial genome and is primarily responsible for delivering amino acids to the nascent polypeptide chain during mitochondrial protein translation. In our cohort, P6, 5, and 15 were associated with pathogenic alteration in the AARS2, EARS2, and MT-TL1 genes. P6 was a 9-year-old male with seizures, myopathy and ophthalmological findings whose findings started at the postnatal third month and carried a compound heterozygous variant in the AARS2 gene. P5, with compound heterozygous pathogenic variants in the EARS2 gene, was a 1.5-year-old baby girl with early-onset hypoglycemic seizures, inability to hold her head, hypotonia, epilepsy, imaging, and laboratory findings. Cranial MRI revealed symmetrical T2A pathological signal intensity increase in bilateral dentate nuclei, thalamus, and periventricular deep white matter. The patient and, as a carrier, her father showed mRNA decay of the deletion allele (c.1283delC). In a study with 12 patients with various EARS2 gene mutations, all had similar MRI findings (Steenweg et al., 2012), suggesting that cranial MRI may be more accountable evidence than standard biochemical studies for the discrimination of ARS-associated diseases. Although several MRI findings highly suggest specific ARS mutations, no MRI pattern is common to all patients (Roux et al., 2021). P15 was a 9-year-old female with the most prevalent MELAS (Mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes) variant in the MT-TL1 gene (m.3243A>G), found in 50% and 85% heteroplasmic cells in her blood and muscle cells, respectively. Among MDs caused by defects in mt-tRNAs, m.3243A>G is the most common and accounts for ∼80% of all mt-tRNA-related MDs, and the relationship between pathogenic mt-tRNA levels and the clinical phenotype is not correlated (Richter et al., 2021). Mammalian mt-tRNA genes have accumulated more mutations than their cytoplasmic counterparts during evolution. It has been proposed that the induced adaptation of cognate mitochondrial ARSs compensates for the structural complexity in mammalian mt-tRNAs. This interaction is critical for understanding the disease and severity of mt-tRNA mutations.
Genetic information processing/mitochondrial biogenesis (FBXL4, NDUFAF6, MICOS13, POLG, and MT-ATP6)
The FBXL4 gene encodes a subunit of modular E3 ubiquitin ligase as a regulator of mitochondrial biogenesis, and NDUFAF6 encodes a subunit of the complex-1 factor of the mitochondrial respiratory chain complex assembly factors grouped under major mitochondrial quality control factors (KEGG). P11 has homozygous missense variation (c.1444C>T/p.(R482W)) in the FBXL4 gene and the variants that were previously described (Gai et al., 2013). Clinical findings attributed to this variant in the literature were highly heterogeneous in the three affected male siblings; the first child died due to a severe lactic acidosis attack at 4 years of age; the second was alive at 9 years of age and had mild facial dysmorphism with severe intellectual disability, intermittently elevated plasma lactate levels, and exercise intolerance; and the third was 6 months old with neonatal depression at delivery and attacks of severe lactic acidosis. However, growth parameters and facial appearance were normal. P11, at the 26th gestational age, presented developmental delay. In her follow-ups at 15 months of age, she had lactic acidosis and elevated blood alanine levels, and organic acid analysis revealed excretion consistent with mitochondrial disease. Muscle biopsy analysis revealed OXPHOS enzyme deficiency, and histopathology was reported as lipid storage myopathy. She was under follow-up of cardiology due to hypertrophic cardiomyopathy. She is currently 9 years old. Her Denver developmental screening test showed a 50% delay in the developmental milestones, her verbal ability was restricted, and she can only say a few sentences with speech therapy. She also had mild dysmorphic facial characteristics.
P7 with compound heterozygous variants in the NDUFAF6 gene-encoding factor 6 of the NADH dehydrogenase complex I was predicted to have dysfunctional highly preserved mitochondrial proteins among eukaryotes that play an essential role in the early combining stage of the mitochondrial respiratory chain. NDUFAF6 pathologies in the literature are related to a type of Leigh syndrome without cognitive involvement or seizures, manifesting with isolated cerebellar or extrapyramidal symptoms depending on the affected structures that onset with the loss of abilities in childhood between 1 and 5 years of age and staying stable or progresses slowly (Catania et al., 2018). P7’s clinical findings were similar to those reported in the literature. He showed a loss of abilities developed after 3 years of age, inability to walk and talk, dystonic contractures, scoliosis, and contracture in his hands.
P12 was a 1-year-old male identified with homozygous acceptor splice site alteration (c.260–2A>G) in intron 3 of the MICOS13 gene. The MICOS13 gene encodes a component of the MICOS complex, a large protein complex of the mitochondrial inner membrane that forms contact with the outer membrane. Before his accurate diagnosis, he had been treated in the neonatal intensive care unit for 1 month with a presumptive diagnosis of metabolic disease and symptoms of lack of sucking on the third postnatal day, respiratory distress, jaundice, severe metabolic acidosis, and hyperammonemia findings. He passed away at 15 months of age due to severe metabolic acidosis and respiratory failure. The MICOS13 variant identified was previously associated with combined oxidative phosphorylation deficiency in a baby girl whose initial signs were observed later than ours at 6 months. mRNA investigation obtained from her fibroblast culture cells showed two aberrant splice sites, one with 21 bp reduced size of exon 4, and the second was a frameshift by the deletion of 44 bp of exon 4 with a premature stop codon (Godiker et al., 2018).
MT-ATP6, encoding the ATP synthetase (complex V) subunit, and POLG, encoding the catalytic subunit of polymerase gamma, are essential for mitochondrial biogenesis through OXPHOS and DNA replication mechanisms, respectively. In the complex V subunit encoded by MT-ATP6, the pathogenic variants at m.8993 are involved in nearly 50% of the reported MT-ATP6 disease cases. The m.8993T>C variant causes a milder disease phenotype than m.8993T>G (Ganetzky et al., 2019). The MT-ATP6 gene variant (m.8993T>C) was identified in P13, 11 years old, and P14, 13 years old. Both were found to be 99% homoplasmic in their blood cells and shared mild phenotypes; also, the onset of disease and laboratory and imaging findings were similar. Both cases are Leigh-like phenotype with post-infectious acute onset ataxia at 6–7 years, loss of muscle strength, and basal ganglia involvement in cranial MRI. Their symptoms were alleviated with mitochondrial support treatments, and they were followed up in the metabolism outpatient clinic.
The POLG mutations, which are clinically among the mitochondrial depletion syndromes, are characterized by quantitative abnormalities of the mitochondrial DNA, causing the impairment of energy production in single or multiple organ systems (Rahman and Poulton, 2009). Myopathy may cause symptoms from the infantile period to early childhood, and children are primarily brought with complaints of skeletal muscle weakness. The most common symptoms of mitochondrial muscle disease are lethargy, fatigue, exercise intolerance, and pain. Myopathy is generally a part of a complex multisystem disorder in childhood; isolated muscle involvement is a more common characteristic of adult-onset disease. Outer eye muscles are frequently involved in the pediatric group when myopathy is the only finding; myopathy is proximal rather than distal. Many patients with chronic progressive external ophthalmoplegia (CPEO) develop myopathy. P3 and 4, the two affected siblings carrying the POLG variant (c.911T>G), initially presented with progressive myopathy that started with the involvement of ophthalmic muscles at 6 years of age in both average growth and neuromotor development. Unlike in the literature data, symptoms were of the early-onset and progressive type. It has been reported that environmental factors such as immunity dysfunction, viral infection, mitochondrial toxins, and genetic modifiers have consequences on the severity of the disease and complicate foresight for patients (Rahman and Copeland, 2019). Both patients 3 and 4 passed away at the age of 17 due to respiratory failure.
Metabolism/nucleotide metabolism (TK2)
Another mitochondrial DNA (mtDNA) depletion syndrome is associated with the TK2 gene. Thymidine kinase-2 is the first enzyme in the deoxypyrimidine salvage pathway within mitochondria that phosphorylates thymidine and deoxycytidine. These are subsequently converted to deoxynucleoside triphosphates required for mtDNA replication and maintenance. Most patients with early onset before 2 years of age show severe and rapid progression. The less severe forms begin in childhood through adulthood and exhibit slower progression; bulbar, proximal limb, and respiratory weakness can be severe, causing the inability to walk or breathe independently. Early and accurate diagnosis is critical since a specific treatment is under development (Dominguez-Gonzalez et al., 2019; Dominguez-Gonzalez et al., 2022). Two affected siblings from the late-onset group, patient 1, young adult, and patient 2, adolescent, with severe neuromuscular weakness, including diaphragmatic weakness, requiring respiratory support, have, with previously reported homozygous c.323C>T/p.(T108M), a variant in the TK2 gene. They were able to reach early initiation of medication. The beneficial effects of the therapy were verified by functional tests, including the 6-min walk test, forced vital capacity, and maximal inspiratory pressure, with mean changes that appear to be clinically significant.
Metabolism/carbohydrate metabolism (OXCT1)
The OXCT1 gene encodes a critical mitochondrial enzyme essential for ketone body metabolism in all extrahepatic tissues (Grunert et al., 2021). P10 with the compound heterozygous variant in the OXCT1 gene (c.[1173-139G>T];[1370C>T]/p.([?];[T457I]) was a 1.5-year-old baby boy, had normal development until the 11th month, except for lack of postnatal crying and 8 days of admittance to the neonatal intensive care unit. He had his first metabolic attack during an upper respiratory tract infection in the 11th month and two more episodes after gastroenteritis. He had metabolic acidosis; organic acidemia and keratolysis were considered, but a definitive diagnosis could not be achieved. He had arrest during the second attack and responded to resuscitation. He was discharged with a tracheostomy. Metabolic attacks following infection, ketone positivity in blood and urine, and asymptomatic state between attacks all supported succinyl-CoA 3-ketoacid-CoA transferase (SCOT) deficiency, and the compound heterozygosity for the OXCT1 gene complied with the autosomal recessive inheritance (Sasai et al., 2017). The OXCT1 gene is associated with SCOT deficiency and causes mitochondrial dysfunction secondary to ketone body catabolism disorder. Manifestation is characterized by clinical findings that depend on mutation intensity and residual enzyme activity, episodic or permanent ketosis.
Metabolism/fatty acid elongation (ECHS1)
The ECHS1 gene encodes an enzyme to hydrate medium- and short-chained fatty enoyl-CoA thioesters from four carbons long to up to C16, resulting in fatty acid oxidation disorder when defective (Catania et al., 2018). P10 and P11, 12 years and ten years old brothers were offsprings from a nonconsanguineous marriage; both were followed up for ten years without a diagnosis. Their symptoms were a progressive neuromotor developmental delay, spasticity, truncal hypotonia and basal ganglion involvement in cranial MRI. Metabolic and molecular tests were not informative. Two different nonsynonymous variants (c.202G>A/p.(E68K) and c.161G>A/p.(R54H) in the ECHS1 gene have been identified in these siblings in compound heterozygous form. The c.161G>A alteration was reported in a patient with mitochondrial short-chain enoyl-CoA hydratase-1 deficiency (Fitzsimons et al., 2018). However, the c.202G>A (rs1276839756) variant was not associated before and was reported as very rare in populations (0.0008%) with a “pathogenic” classification. The association of the very first ECHS1 deficiency in MD was presented in a study of 435 individuals with impaired mitochondrial energy metabolism, revealing ten patients with 13 ECHS1 gene variants, 12 being missense type, suggesting bi-allelic truncating variants may embryonically be lethal. Furthermore, the study suggested that patients may benefit from dietary therapy (Haack et al., 2015).
Limb-girdle muscular dystrophy (LGMD) is a disorder that must be highly considered in the differential diagnosis of MDs due to clinical overlaps. Since not only disrupted mechano-signaling components but also functional insufficiencies of mitochondria play a role, all are rare forms with different inheritance patterns and are genetically quite heterogeneous. The fine detail is the presence of myopathy in the histopathological analysis of the muscle biopsy material and the overlapping in neuromuscular findings in clinical evaluation (Wang et al., 2018). Similar to those observed in mitochondrial depletion, in LGMD, the age of onset varies from childhood to adolescence, and the clinical course is generally progressive (Nigro and Savarese, 2014). An example of its similarities with mitochondrial pathology was revealed in a cohort study composed of dysferlinopathy patients who reported a decrease in the mtDNA copy number, cytochrome c oxidase (COX) deficiency, and a decrease in complex I and IV activities (Liu et al., 2016). Out of the 12 families, 4 (25%) were diagnosed with LGMD.
Accurate diagnosis in clinical science is the fundamental component of timely and effective care and is the basis for improving the patient’s health and providing proper counseling to the families. The discovery of new genes that play a role in mitochondrial pathogenesis increases over time and improves knowledge of cellular homeostasis and metabolism pathways. Non-invasive, bi-genomic sequencing in a single test method has changed the algorithm investigating mitochondrial diseases as the step before an invasive procedure. Today, biochemical and pathologic studies requiring muscle biopsy are carried out when the diagnosis is indistinct, following high-throughput analysis of the nuclear and mitochondrial genome analysis. In the last decade, the concept of MDs has been re-visited, and a new classification is recommended that focuses on genetic origin rather than biochemical deficiency (Ferreira et al., 2021). The mitochondrial and exome sequencing of the patients in our study achieved 67% (16/24) diagnostic success and eight of our families remained undiagnosed. Recent reports with similar approaches have reported different rates of diagnoses, revealing it to be between 39% and 77% (Taylor et al., 2014; Wortmann et al., 2015; Pronicka et al., 2016; Puusepp et al., 2018; Theunissen et al., 2018; Kerr et al., 2020; Kose et al., 2021) (Table 3). In these studies, we observe that the diagnosis rate increases when the MD criterion scale is used. Nevertheless, unknown functional effects of genes or new variants may result in unexplored patients remaining undiagnosed.
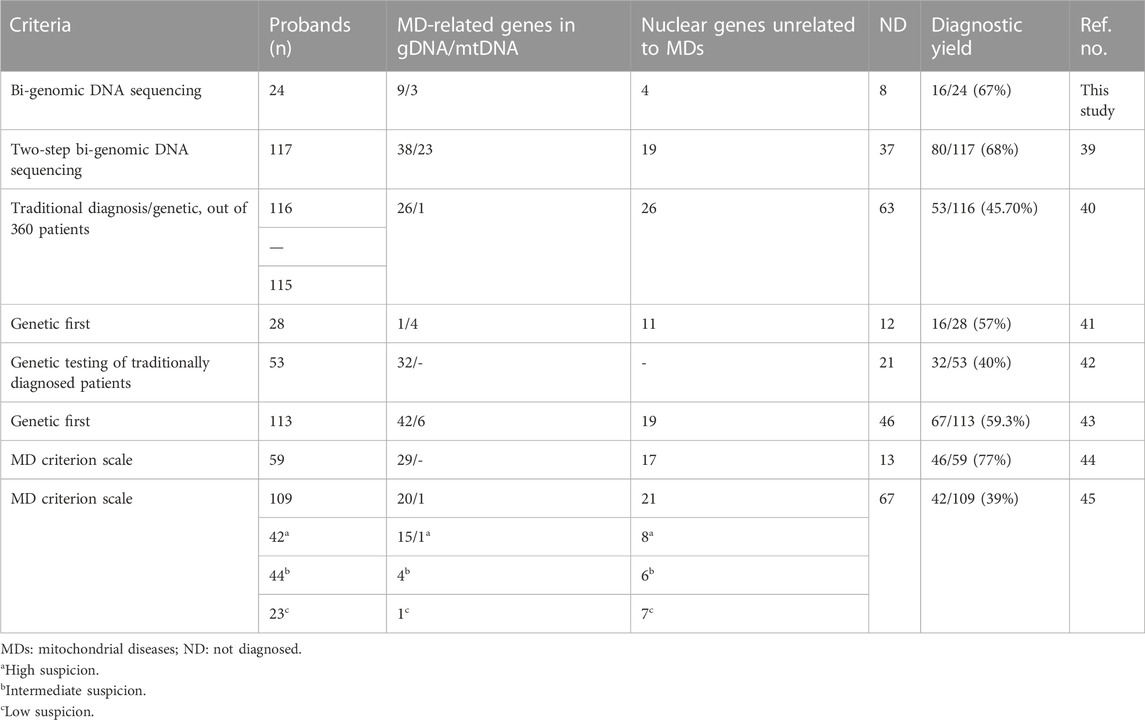
TABLE 3. Overview of the diagnostic rates of mitochondrial disease by exome sequencing in our study and in other published data.
The most efficient approach for diagnosing MDs today is sequencing the nuclear genome and mtDNA if harbored in a single test procedure; however, as evidenced by our study, nuclear gene analysis must be prioritized for diagnostic utility. Investigation of the non-coding regions of the genome by whole-genome sequencing, pathogenic copy number variation in the nuclear genome, and gross deletions or duplications in the mtDNA are the investigations that could not be performed in our study. Furthermore, since mtDNA investigation was conducted on isolated peripheral blood sample cells (P15 with the MT-TL1 mutation was investigated in both muscle and blood samples), mtDNA variants of low heteroplasmy was not diagnosed. The discipline of mitochondrial medicine has significantly progressed by determining more than 400 genes in the nuclear and mtDNA genomes related to MDs; however, reasons for clinical variations in individuals with the same genetic deficits and tissue specificity remain unanswered.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/snp/, SCV001423134.1 https://www.ncbi.nlm.nih.gov/snp/, SCV001423135.1 https://www.ncbi.nlm.nih.gov/snp/, SCV001571651.1 https://www.ncbi.nlm.nih.gov/snp/, SCV001571652.1 https://www.ncbi.nlm.nih.gov/snp/, SCV001571649.1 https://www.ncbi.nlm.nih.gov/snp/, SCV003804637 https://www.ncbi.nlm.nih.gov/snp/, SCV003804638 https://www.ncbi.nlm.nih.gov/snp/, SCV001571647.1 https://www.ncbi.nlm.nih.gov/snp/, and SCV001571646.1.
Ethics statement
The studies involving human participants were reviewed and approved by the Faculty of Medicine Ethics Council of Istanbul University (08.06.2018/11). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
GG, MB, MK, DG, SG, and MD contributed to clinical and metabolic examinations and data collection. AG and AA performed a clinical genetic examination and genetic counseling. AG, GT, and CG designed in-house mtDNA sequencing and performed exome analysis. GU performed the histopathological evaluations. SB and BK conducted the cell culture. AG, GT, CG, and OU performed and evaluated the molecular investigation. AG and OU drafted and wrote the manuscript and designed the tables and figures with the assistance of CG. The manuscript was critically reviewed by SB, GG, AG, and OU. All authors listed made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This study was funded by the Scientific Research Projects Coordination Unit of Istanbul University, Project No. TDK-2018-32544.
Acknowledgments
The authors also thank the patients and their families for participating in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Boczonadi, V., Jennings, M. J., and Horvath, R. (2018). The role of tRNA synthetases in neurological and neuromuscular disorders. FEBS Lett. 592 (5), 703–717. doi:10.1002/1873-3468.12962
Catania, A., Ardissone, A., Verrigni, D., Legati, A., Reyes, A., Lamantea, E., et al. (2018). Compound heterozygous missense and deep intronic variants in NDUFAF6 unraveled by exome sequencing and mRNA analysis. J. Hum. Genet. 63 (5), 563–568. doi:10.1038/s10038-018-0423-1
Desmet, F.O., Hamroun, D., Lalande, M., Collod-Beroud, G., Claustres, M., and Beroud, C. (2009). Human splicing finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 37 (9), e67. doi:10.1093/nar/gkp215
Dogan, T., MacDougall, A., Saidi, R., Poggioli, D., Bateman, A., O'Donovan, C., et al. (2016). UniProt-DAAC: Domain architecture alignment and classification, a new method for automatic functional annotation in UniProtKB. Bioinformatics 32 (15), 2264–2271. doi:10.1093/bioinformatics/btw114
Dominguez-Gonzalez, C., Fernandez-Torron, R., Moore, U., de Fuenmayor-Fernandez de la Hoz, C.P., Velez-Gomez, B., Cabezas, J.A., et al. (2022). Muscle MRI characteristic pattern for late-onset TK2 deficiency diagnosis. J. Neurol. 269 (7), 3550–3562. doi:10.1007/s00415-021-10957-0
Dominguez-Gonzalez, C., Madruga-Garrido, M., Mavillard, F., Garone, C., Aguirre-Rodriguez, F.J., Donati, M.A., et al. (2019). Deoxynucleoside therapy for thymidine kinase 2-deficient myopathy. Ann. Neurol. 86 (2), 293–303. doi:10.1002/ana.25506
Fernandez-Eulate, G., Carreau, C., Benoist, J. F., Lamari, F., Rucheton, B., Shor, N., et al. (2022). Diagnostic approach in adult-onset neurometabolic diseases. J. Neurol. Neurosurg. Psychiatry 93 (4), 413–421. doi:10.1136/jnnp-2021-328045
Ferreira, C.R., Rahman, S., Keller, M., Zschocke, J., and Group, I.A. (2021). An international classification of inherited metabolic disorders (ICIMD). J. Inherit. Metab. Dis. 44 (1), 164–177. doi:10.1002/jimd.12348
Fitzsimons, P. E., Alston, C.L., Bonnen, P.E., Hughes, J., Crushell, E., Geraghty, M. T., et al. (2018). Clinical, biochemical, and genetic features of four patients with short-chain enoyl-CoA hydratase (ECHS1) deficiency. Am. J. Med. Genet. A 176 (5), 1115–1127. doi:10.1002/ajmg.a.38658
Gai, X., Ghezzi, D., Johnson, M.A., Biagosch, C.A., Shamseldin, H. E., Haack, T. B., et al. (2013). Mutations in FBXL4, encoding a mitochondrial protein, cause early-onset mitochondrial encephalomyopathy. Am. J. Hum. Genet. 93 (3), 482–495. doi:10.1016/j.ajhg.2013.07.016
Ganetzky, R.D., Stendel, C., McCormick, E.M., Zolkipli-Cunningham, Z., Goldstein, A.C., Klopstock, T., et al. (2019). MT-ATP6 mitochondrial disease variants: Phenotypic and biochemical features analysis in 218 published cases and cohort of 14 new cases. Hum. Mutat. 40 (5), 499–515. doi:10.1002/humu.23723
Godiker, J., Gruneberg, M., DuChesne, I., Reunert, J., Rust, S., Westermann, C., et al. (2018). QIL1-dependent assembly of MICOS complex-lethal mutation in C19ORF70 resulting in liver disease and severe neurological retardation. J. Hum. Genet. 63 (6), 707–716. doi:10.1038/s10038-018-0442-y
Grunert, S.C., Foster, W., Schumann, A., Lund, A., Pontes, C., Roloff, S., et al. (2021). Succinyl-CoA:3-oxoacid coenzyme A transferase (SCOT) deficiency: A rare and potentially fatal metabolic disease. Biochimie 183, 55–62. doi:10.1016/j.biochi.2021.02.003
Haack, T.B., Jackson, C.B., Murayama, K., Kremer, L.S., Schaller, A., Kotzaeridou, U., et al. (2015). Deficiency of ECHS1 causes mitochondrial encephalopathy with cardiac involvement. Ann. Clin. Transl. Neurol. 2 (5), 492–509. doi:10.1002/acn3.189
Hamosh, A., Scott, A.F., Amberger, J. S., Bocchini, C.A., and McKusick, V.A. (2005). Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 33, D514–D517. doi:10.1093/nar/gki033
Itan, Y., Shang, L., Boisson, B., Ciancanelli, M. J., Markle, J.G., Martinez-Barricarte, R., et al. (2016). The mutation significance cutoff: Gene-level thresholds for variant predictions. Nat. Methods 13 (2), 109–110. doi:10.1038/nmeth.3739
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M., and Ishiguro-Watanabe, M. (2023). KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51 (D1), D587–D592. doi:10.1093/nar/gkac963
Kerr, M., Hume, S., Omar, F., Koo, D., Barnes, H., Khan, M., et al. (2020). MITO-FIND: A study in 390 patients to determine a diagnostic strategy for mitochondrial disease. Mol. Genet. Metab. 131 (1-2), 66–82. doi:10.1016/j.ymgme.2020.08.009
Kose, M., Isik, E., Aykut, A., Durmaz, A., Kose, E., Ersoy, M., et al. (2021). The utility of next-generation sequencing technologies in diagnosis of Mendelian mitochondrial diseases and reflections on clinical spectrum. J. Pediatr. Endocrinol. Metab. 34 (4), 417–430. doi:10.1515/jpem-2020-0410
Landrum, M. J., Lee, J.M., Benson, M., Brown, G.R., Chao, C., Chitipiralla, S., et al. (2018). ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46 (D1), D1062-D1067–D7. doi:10.1093/nar/gkx1153
Lightowlers, R. N., Taylor, R.W., and Turnbull, D.M. (2015). Mutations causing mitochondrial disease: What is new and what challenges remain? Science 349 (6255), 1494–1499. doi:10.1126/science.aac7516
Liu, F., Lou, J., Zhao, D., Li, W., Zhao, Y., Sun, X., et al. (2016). Dysferlinopathy: Mitochondrial abnormalities in human skeletal muscle. Int. J. Neurosci. 126 (6), 499–509. doi:10.3109/00207454.2015.1034801
Lott, M.T., Leipzig, J.N., Derbeneva, O., Xie, H.M., Chalkia, D., Sarmady, M., et al. (2013). mtDNA variation and analysis using mitomap and mitomaster. Curr. Protoc. Bioinforma. 44 (123), 1 23 1–26. doi:10.1002/0471250953.bi0123s44
Nigro, V., and Savarese, M. (2014). Genetic basis of limb-girdle muscular dystrophies: The 2014 update. Acta Myol. 33 (1), 1–12.
Orsucci, D., Caldarazzo Ienco, E., Rossi, A., Siciliano, G., and Mancuso, M. (2021). Mitochondrial syndromes revisited. J. Clin. Med. 10 (6), 1249. doi:10.3390/jcm10061249
Pronicka, E., Piekutowska-Abramczuk, D., Ciara, E., Trubicka, J., Rokicki, D., Karkucinska-Wieckowska, A., et al. (2016). New perspective in diagnostics of mitochondrial disorders: Two years' experience with whole-exome sequencing at a national paediatric centre. J. Transl. Med. 14 (1), 174. doi:10.1186/s12967-016-0930-9
Puusepp, S., Reinson, K., Pajusalu, S., Murumets, U., Oiglane-Shlik, E., Rein, R., et al. (2018). Effectiveness of whole exome sequencing in unsolved patients with a clinical suspicion of a mitochondrial disorder in Estonia. Mol. Genet. Metab. Rep. 15, 80–89. doi:10.1016/j.ymgmr.2018.03.004
Rahman, S., and Copeland, W.C. (2019). POLG-related disorders and their neurological manifestations. Nat. Rev. Neurol. 15 (1), 40–52. doi:10.1038/s41582-018-0101-0
Rahman, S. (2020). Mitochondrial disease in children. J. Intern Med. 287 (6), 609–633. doi:10.1111/joim.13054
Rahman, S., and Poulton, J. (2009). Diagnosis of mitochondrial DNA depletion syndromes. Arch. Dis. Child. 94 (1), 3–5. doi:10.1136/adc.2008.147983
Rath, S., Sharma, R., Gupta, R., Ast, T., Chan, C., Durham, T.J., et al. (2021). MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 49 (D1), D1541–D1547. doi:10.1093/nar/gkaa1011
Rentzsch, P., Witten, D., Cooper, G.M., Shendure, J., and Kircher, M. (2019). Cadd: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47 (D1), D886–D94. doi:10.1093/nar/gky1016
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Richter, U., McFarland, R., Taylor, R.W., and Pickett, S.J. (2021). The molecular pathology of pathogenic mitochondrial tRNA variants. FEBS Lett. 595 (8), 1003–1024. doi:10.1002/1873-3468.14049
Roux, C.J., Barcia, G., Schiff, M., Sissler, M., Levy, R., Dangouloff-Ros, V., et al. (2021). Phenotypic diversity of brain MRI patterns in mitochondrial aminoacyl-tRNA synthetase mutations. Mol. Genet. Metab. 133 (2), 222–229. doi:10.1016/j.ymgme.2021.04.004
Sasai, H., Aoyama, Y., Otsuka, H., Abdelkreem, E., Naiki, Y., Kubota, M., et al. (2017). Heterozygous carriers of succinyl-CoA:3-oxoacid CoA transferase deficiency can develop severe ketoacidosis. J. Inherit. Metab. Dis. 40 (6), 845–852. doi:10.1007/s10545-017-0065-z
Schlieben, L. D., and Prokisch, H. (2020). The dimensions of primary mitochondrial disorders. Front. Cell Dev. Biol. 8, 600079. doi:10.3389/fcell.2020.600079
Steenweg, M. E., Ghezzi, D., Haack, T., Abbink, T. E., Martinelli, D., van Berkel, C.G., et al. (2012). Leukoencephalopathy with thalamus and brainstem involvement and high lactate 'LTBL' caused by EARS2 mutations. Brain 135 (5), 1387–1394. doi:10.1093/brain/aws070
Stelzer, G., Rosen, N., Plaschkes, I., Zimmerman, S., Twik, M., Fishilevich, S., et al. (2016). The GeneCards suite: From gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinforma. 54, 1 30 1–1 3. doi:10.1002/cpbi.5
Stenton, S.L., and Prokisch, H. (2020). Genetics of mitochondrial diseases: Identifying mutations to help diagnosis. EBioMedicine 56, 102784. doi:10.1016/j.ebiom.2020.102784
Taylor, R.W., Pyle, A., Griffin, H., Blakely, E.L., Duff, J., He, L., et al. (2014). Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA 312 (1), 68–77. doi:10.1001/jama.2014.7184
Theunissen, T.E.J., Nguyen, M., Kamps, R., Hendrickx, A.T., Sallevelt, S., Gottschalk, R.W.H., et al. (2018). Whole exome sequencing is the preferred strategy to identify the genetic defect in patients with a probable or possible mitochondrial cause. Front. Genet. 9, 400. doi:10.3389/fgene.2018.00400
Turvey, A.K., Horvath, G.A., and Cavalcanti, A.R.O. (2022). Aminoacyl-tRNA synthetases in human health and disease. Front. Physiol. 13, 1029218. doi:10.3389/fphys.2022.1029218
Wang, L., Zhang, V.W., Li, S., Li, H., Sun, Y., Li, J., et al. (2018). The clinical spectrum and genetic variability of limb-girdle muscular dystrophy in a cohort of Chinese patients. Orphanet J. Rare Dis. 13 (1), 133. doi:10.1186/s13023-018-0859-6
Webb, B.D., Diaz, G.A., and Prasun, P. (2020). Mitochondrial translation defects and human disease. J. Transl. Genet. Genom 4, 71–80. doi:10.20517/jtgg.2020.11
Keywords: mitochondrial diseases, mtDNA, bi-genomic sequencing, exome sequencing, differential diagnosis
Citation: Gedikbasi A, Toksoy G, Karaca M, Gulec C, Balci MC, Gunes D, Gunes S, Aslanger AD, Unverengil G, Karaman B, Basaran S, Demirkol M, Gokcay GF and Uyguner ZO (2023) Clinical and bi-genomic DNA findings of patients suspected to have mitochondrial diseases. Front. Genet. 14:1191159. doi: 10.3389/fgene.2023.1191159
Received: 21 March 2023; Accepted: 02 May 2023;
Published: 12 June 2023.
Edited by:
Lawrence Todd Reiter, University of Tennessee Health Science Center (UTHSC), United StatesReviewed by:
Xiaonan Zhao, Baylor College of Medicine, United StatesFrederic Tort, Hospital Clinic of Barcelona, Spain
Copyright © 2023 Gedikbasi, Toksoy, Karaca, Gulec, Balci, Gunes, Gunes, Aslanger, Unverengil, Karaman, Basaran, Demirkol, Gokcay and Uyguner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zehra Oya Uyguner, by51eWd1bmVyQGlzdGFuYnVsLmVkdS50cg==