 Yan Ma
Yan Ma Xiaohui Duan
Xiaohui Duan Xiaoxuan Liu
Xiaoxuan Liu Dongsheng Fan
Dongsheng Fan- 1Department of Neurology, Peking University Third Hospital, Beijing, China
- 2Beijing Key Laboratory of Biomarker and Translational Research in Neurodegenerative Diseases, Beijing, China
- 3Key Laboratory for Neuroscience, National Health Commission/Ministry of Education, Peking University, Beijing, China
- 4China-Japan Friendship Hospital, Beijng, China
Background: Charcot-Marie-Tooth disease (CMT) is the most common inherited neurological disorder suffered in childhood. To date, the disease features have not been extensively characterized in the Chinese paediatric population. In this study, we aimed to analyse the clinical profiles and genetic distributions of a paediatric CMT cohort in China.
Methods: A total of 181 paediatric CMT patients were enrolled. After preexcluding PMP22 duplication/deletion by multiplex ligation-dependent probe amplification (MLPA), Sanger sequencing, targeted next-generation sequencing (NGS) or whole-exome sequencing (WES) was performed to obtain a genetic diagnosis. Detailed information was collected to explore the spectrum of subtypes and genotype-phenotype correlations.
Results: Pathogenic mutations were identified in 68% of patients in this study; with PMP22 duplication, MFN2 and GJB1 were the most frequent disease-causing genes. Of note, respect to the higher prevalence worldwide, CMT1A (18.2%) was relatively lower in our cohort. Besides, the mean age at onset (8.3 ± 5.7 years) was significantly older in our series. In genotype-phenotype analyse, PMP22 point mutations were considered the most severe genotypes and were mostly de novo. In addition, the de novo mutations were identified in up to 12.7% of all patients, which was higher than that in other studies.
Conclusion: We identified a relatively lower detection rate of PMP22 duplication and a higher frequency of de novo variants among paediatric patients in China. We also identified the genetic and phenotypic heterogeneity of this cohort, which may provide clues for clinicians in directing genetic testing strategies for Chinese patients with early-onset CMT.
Introduction
Charcot-Marie-Tooth disease (CMT) is one of the most common inherited peripheral neuropathies with a prevalence of 1 in 2,500 individuals (Skre, 1974). As a collection of clinically and genetically heterogeneous disorders, CMT varies strikingly in terms of phenotypes and severity, especially in paediatric populations. In clinical practice, whereas CMT patients share common characteristics of progressive distal muscle weakness, sensory loss and deformity (Klein, 2020), they also have a clinically heterogeneous set of disorders, spanning a spectrum from mildly symptomatic forms to those resulting in severe disability. Based on median motor nerve conduction velocity (MNCV), CMT can be categorized as demyelinating type (median MNCV<38 m/s), axonal type (median MNCV>38 m/s) and intermediated type (median MNCV 25–45 m/s) (Pareyson and Marchesi, 2009). In each category, inheritance patterns may be autosomal dominant, autosomal recessive, or X-linked.
Recently, next-generation sequencing (NGS) and whole-exome sequencing (WES) have resulted in a rapid expansion of the genetic diagnosis of CMT, with over 100 genes identified and many more still to be discovered (Padilha et al., 2020). In comparison to CMT of all age groups, childhood-onset patients may present more phenotypic variability and mutation-specific manifestations (Cornett et al., 2016). In addition, it is worth noting that de novo cases are not rare for CMT, especially in the earlier-onset group (Landrieu and Baets, 2013; Ma et al., 2021). Therefore, owing to the clinical complexity and genetic diversity, the diagnosis of paediatric CMT is always difficult. Furthermore, previous published studies also illustrated that the distribution of subtypes varied in different geographical regions (Fridman et al., 2015). Thus, a comprehensive knowledge of the distribution of mutations within earlier-onset subgroup in different areas is important and challenging.
Paediatric CMT, with disease onset in the first 2 decades, is a group that deserves further attentions. First, the presentation of paediatric patients is quite different from that of adult patients in terms of disease severity and clinical features, as symptoms can be insidious and severely disabling. Besides, there are obvious delays in achieving motor milestones in earlier-onset patients, which may lead to a considerable socioeconomic burden (Baets et al., 2011). In addition, improved genetic diagnosis rate in paediatric patients may help facilitate clinical trials of the upcoming disease-modifying treatment. To date, studies on paediatric CMT have mainly been based on patients of European origins, with a reported diagnosis rate of 75.6%–92% (Cornett et al., 2016; Hoebeke et al., 2018; Argente-Escrig et al., 2021). However, the disease characteristics of paediatric CMT have not been extensively characterized in the Chinese population thus far, either in mutation spectrum or phenotypic analysis. Therefore, the clinical heterogeneity in patients of different origins, coupled with the expanding genetic diversity in earlier-onset CMT leads to a great need for in-depth studies on the mutational spectrum and detailed genotype-phenotype correlations in Chinese patients.
Thus, we conducted a study in a large paediatric CMT population in China, analysing the mutational distribution and clinical characteristics. In addition, we further explored the subtype frequencies and the genotype-phenotype correlations in this series, to provide some clues for clinicians in directing genetic testing strategies and selecting disease modifying therapies for early-onset CMT patients of Chinese origin.
Materials and methods
Participants
A cohort of early-onset (age 0–20 years) CMT patients was enrolled consecutively in this study at Peking University Third Hospital and China-Japan Friendship Hospital from 2007 to 2021. The clinical criteria used for the diagnosis of CMT are well established in the literature (Bird, 1998). For all patients, clinical features, family history and the electrophysiological data were carefully collected and recorded. Experienced neurologists who specialized in inherited neuropathies evaluated the clinical data of all patients. Disease burden were assessed by Charcot-Marie-Tooth Paediatric Scale (CMTPedS) (Burns et al., 2012). On the basis of the disease burden scores, patients were classified as having mild, moderate, or severe disease (CMTPedS of ≤14, 15–29, or ≥30, respectively). Follow-up was carried out every 6 months through telephone calls or in-person interviews. The institutional ethics committee of Peking University Third Hospital approved this study. Patients or their legal guardians provided written, informed consents to participate in this study (2019-005-02).
Gene screening strategy and genetic analysis
Genomic DNA was collected and extracted from peripheral blood leukocytes by standard procedures according to the manufacturer’s instructions. From 2007 to 2013, the duplication/deletion mutation of PMP22 gene was pre-excluded in all clinically suspected demyelinating CMT patients by multiplex ligation-dependent probe amplification (MLPA) according to the guidelines. Then Sanger sequencing was used to detect missense mutation of PMP22 (NM_153321), GJB1 (NM_0001097642) and MPZ (NM_000530). In patients with axonal CMT, we investigated MFN2 (NM_014874), GJB1, MPZ, HSPB1 (NM_001540) and HSPB8 (NM_014365) by direct Sanger sequencing. From 2014 to 2018, a targeted NGS panel covering 165 genes associated with inherited neuropathies was introduced for all suspected cases after excluding PMP22 duplication/deletion mutation. Since 2019, an upgraded WES (Agilent Human All Exon V6) was performed on the index patients. The samples were sequenced on the HiSeq2500 and NEXTSEQ 500 (Illumina, San Diego, CA, United States) to discover causal genes. Identified variants by NGS or WES were further validated by Sanger sequencing. All previously reported pathogenic mutations were confirmed with reference to the Human Gene Mutation Database (HGMD) (http://www.hgmd.cf.ac.uk/ac/index.php). Moreover, novel variants were interpreted and classified according to the American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) standards and guidelines (Richards et al., 2015). A flow chart for gene screening strategy (Supplementary Figure S1) and a list of gene panel of NGS (Supplementary Table S1) are summarized in the Supplementary Materials.
Statistical analysis
All statistical analyses were performed using GraphPad Prism 7.0 (GraphPad Software, Inc., CA, United States) and SPSS V.23.0 software (IBM Corp., Armonk, United States). Descriptive statistics such as age at onset (AAO); electrophysiological parameters were expressed as mean ± SD (range).
Results
Characteristics of study participants
A total of 181 patients of Chinese descent from two clinical centres were recruited in this study. Of those patients, 41.4% (75/181) were classified as having demyelinating CMT, 47% (85/181) as having axonal CMT and 8.3% (15/181) as having intermediated CMT. In addition, six patients met the diagnostic criteria for hereditary neuropathy with liability to pressure palsies (HNPP). According to the inheritance patterns, 61 (33.7%) patients were categorized having autosomal dominant CMT, 30 (16.6%) as having autosomal recessive CMT and 12 (6.6%) as having X-linked CMT (CMTX). Furthermore, among 78 sporadic patients, 23 (12.7%) cases were identified to be of de novo origins. For all modes of inheritance, the median AAO was 8.3 ± 5.7 (0–19) years, with a mean diagnostic age of 14.3 ± 6.0 (0.5–19.5) years. In general, most patients first noticed symptoms such as weakness, falls and pes cavus at disease onset. Additionally, according to disease severity, PMP22 point mutation and MPZ were prone to cause the most severe phenotypes. The detailed CMTPedS based on different genotypes were summarized in the Supplementary Table S2.
Distribution of CMT subtypes
Among the different CMT subtypes, patients with demyelinating CMT presented with an earlier onset age (5.9 ± 5.5 years). Although most were associated with classic demyelinating phenotypes (55/75, 73.3%), there was considerable phenotypic heterogeneity such as prominent deep sensory disturbances (SH3TC2, PRX) and scoliosis (SH3TC2, MPZ, and PMP22). On electrophysiological examination, motor conduction velocity (MCV) values were uniformly decreased, with some even below 10 m/s. For the patients with axonal CMT, the AAO was higher (7.8 ± 5.6 years) than that in patients with demyelinating forms. Clinical presentation showed an absence of upper limb involvement in approximately half of the patients. Mutation-specific manifestations were also obvious, with GARS mutation manifesting as predominant upper limbs involvement. Moreover, 12 patients reported clinically pure motor involvement with only slight sensory impairment on electrophysiological examination. Furthermore, the disease severity also varied in axonal CMT according to different pathological mutations. In contrast, patients of intermediate CMT mainly had the classic phenotype, with a relatively benign disease course of late-onset (10.2 ± 4.5 years) and mild peripheral neuropathy (Table 1).

TABLE 1. Comparisons of clinical and electrophysiological data in pediatric CMT.
Genotypes distribution characteristics
Among all 181 index patients, pathogenic mutations were identified in 123 patients, with a diagnostic rate of 68% (123/181). In all CMT subtypes, the leading causes were CMT1A/PMP22 duplication (18.2%; 33/181), CMT2A/MFN2 mutation (7.7%; 14/181) and CMTX1/GJB1 mutation (6.6%; 12/181). In addition, casual mutation were identified in the following genes: GDAP1 (5%; 9/181), PMP22 point mutation (4.4%; 8/181), IGHMBP2 (3.3%; 6/181), MORC2 (3.3%; 6/181), MPZ (2.2%; 4/181), SORD (1.7%; 3/181) and SH3TC2(1.7%; 3/181). Furthermore, mutations in the remaining CMT-related genes (i.e., HSPB1, PRX, BSCL2, DYNC1H1, HINT1, GARS, AARS, MARS, HK1, TRPV4, KIF5A, EGR2, FGD4, MTMR2, SPG11, NEFL, DHTKD1) were each responsible for <1% of all CMT cases. According to the clinical subtypes, PMP22 duplication, MFN2 and GJB1 were the most common causative genes in demyelinating CMT, axonal CMT and CMTX respectively.
Of the patients with demyelinating CMT, 76% (57/75) carried a genetic mutation, with the most frequent genetic causes being PMP22 duplication (44%, 33/75), PMP22 point mutation (10.7%, 8/75) and GDAP1 mutation (5.3%, 4/75), accounting for 60% of all demyelinating CMT patients with an identified mutation. For patients with axonal CMT, 60% had a genetically confirmed diagnosis (51/85), which was lower than that for patients with the demyelinating subtype. Moreover, mutational screening showed marked genetic heterogeneity, with mutations in MFN2 (16.5%; 14/85), IGHMBP (7.1%; 6/85) and MORC2 (7.1%; 6/85) being the three most frequent causes. For intermediated CMT, specific genetic mutations were identified in 80% (12/15) of patients. Of note, up to 12.7% (23/181) of patients were found to have de novo mutations with the following distribution (11 mutations in PMP22, three in MFN2, three in MPZ, and three in MORC2), which was not rare. Genotype distribution details are summarized in Figure 1.
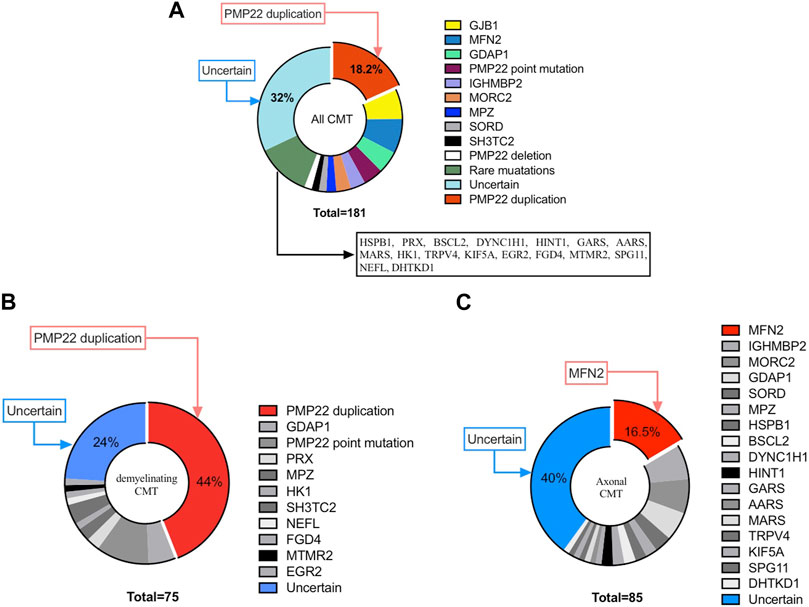
FIGURE 1. Distribution of Chinese paediatric CMT patients in our cohort. (A) Distribution of CMT. (B) Distribution of demyelinating CMT. (C) Distribution of axonal CMT.
Genotype-phenotype correlations
In terms of AAO, CMT can be further categorized into infantile-onset (<3 years), childhood-onset (3–10 years) and adolescent-onset (11–20 years) subtypes. In general, childhood-onset (38.1%; 69/181) was the most common subtype, with a successful genetic diagnosis rate of 66.7% (46/69). In this group, mutations in PMP22, MFN2, GJB1, and GDAP1 were the most common causal mutations. Significantly, patients with infantile-onset CMT had the highest mutation detection rate of 79.6% (39/49), among whom mutations in PMP22, MFN2, and IGHMBP2 were the top three common causes of pathologies. In the adolescent-onset group, the diagnosis rate was 60.3% (38/63), with mutations in PMP22 duplication, GJB1, MFN2, and SORD being the major aetiologies. The detailed genetic distribution according to AAO is shown in Figure 2.
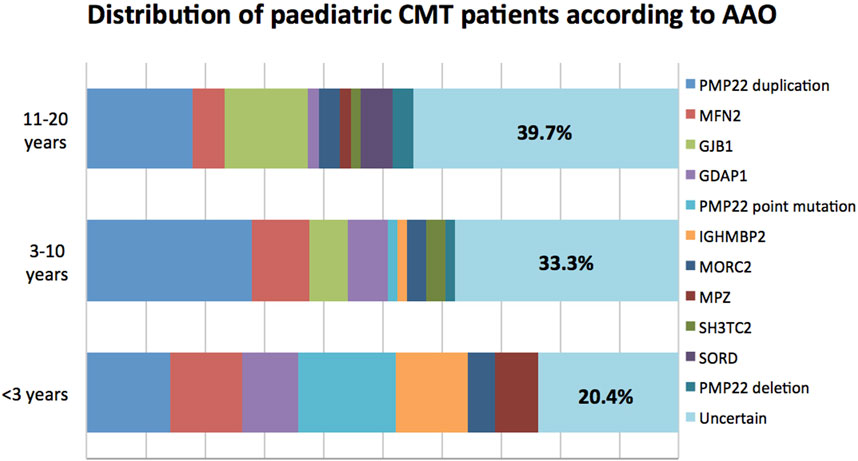
FIGURE 2. Distribution of paediatric CMT patients according to AAO.
There were some mutation-specific manifestations among the CMT subtypes. PMP22 point mutations were found in patients with the most severe demyelinating CMT subtypes (CMTPedS = 32.2 ± 6.5). Among these patients, 87.5% (7/8) developed the disease before the age of 3 years and 62.5% (5/8) were severely affected. Moreover, de novo variants at specific amino acid positions, e.g., p. S72W, p. S79P, and p. G150V caused the severe phenotypes of Dejerine-Sottas disease (DSS), which manifests as earlier onset, delayed motor development, hypotonia and profoundly slowed MCV (<15 m/s) (Agrahari et al., 2015). The second most severe paediatric CMT subtype in the cohort was caused by MPZ mutation. Certain de novo variants, such as p. R98C, p. S233Rfs*18 and the novel p. L174Rfs*66 were associated with clinical features of weakness, atrophy, deformity and motor retardation. Furthermore, patients with SH3TC2 variants also presented with a moderate to severe phenotype, which usually manifested as severe weakness, sensory ataxia and scoliosis. In particular, for patients who developed symptoms before 10 years of age, the disease seemed to be more severe than that of the other subgroups. In addition, disease progression gradually stabilized with increasing age.
With regard to disease severity, PMP22 duplication and MFN2 mutations were the most frequent causes of moderate phenotypes. In addition, mutations in GJB1 and GDAP1 were prone to cause mild phenotypes. The phenotypes of PMP22 point mutations were confirmed to be severe clinical features, accounting for 29.4% (5/17) of the severe cases in total, followed by MPZ and SH3TC2 mutations.
Discussion
In this study, we identified genotype and phenotype distributions of paediatric CMT patients in a large Chinese cohort. We also performed an in-depth genotype-phenotype correlation study, which was the largest study focused on paediatric CMT patients in China thus far. In our findings, the mean age when parents first noticed symptoms was 8.3 ± 5.7 years, which was significantly older than that in the French study (4.1 years) (Hoebeke et al., 2018). Similarly, the diagnostic age was also significantly different between these two studies (14.3 years in the Chinese patients vs. 8.3 years in the French cohort). It should be noted that the demyelinating CMT (AAO = 5.9 ± 5.5 years) in our series only accounted for 41.4% (75/181) of all CMT compared to that of 61.3% in the French cohort, which may partly explain the differences in onset age. Besides, the longer diagnostic delay in our study may largely due to the feasibility to access to our specialized clinics rather than disease severity.
Based on clinical subtypes, demyelinating CMT manifests with earlier disease onset than other CMT subtypes. Clinically, the majority (73.3%) of demyelinating CMT patients presented with typical phenotypes, whereas axonal CMT patients varied strikingly in terms of clinical features due to specific genotypes. In addition to the classic features of progressive distal muscle weakness, CMT2A patients may present with complex phenotypes, including tremor, optic atrophy and pyramidal signs. Additionally, 14.1% of axonal CMT patients are characterized as having motor-predominant neuropathy, which is difficult to distinguish from distal hereditary motor neuropathies (dHMN) (Liu et al., 2020). For CMT1X, males were more affected than females, with a mild to moderate phenotype of adolescent-onset and intermediate slowing in electrophysiological examination, which was in consistent with previous reports (Panosyan et al., 2017).
Overall, a confirmed genetic diagnosis was achieved in 123 of 181 patients (68%), with a higher detection rate in patients with the demyelinating forms (57/75; 76%) than in those with the axonal forms (51/85; 60%). Compared among patients of all ages, the genetic confirmation rate in our paediatric cohort was similar to that reported in South China (70%) (Xie et al., 2021) and Taiwan (73.1%) (Hsu et al., 2019). However, this rate was higher than that worldwide (60.4%) (Fridman et al., 2015), suggesting that paediatric patients has a higher genetic diagnosis rate than those of all age groups. Nevertheless, this result is somewhat lower than that in other series when comparing within the paediatric groups (Cornett et al., 2016; Hoebeke et al., 2018; Argente-Escrig et al., 2021) (Table 2). To date, the largest paediatric series based on eight sites reported a diagnosis rate of 75.6% (Cornett et al., 2016). In another study from France, up to 92% of patients received a genetic diagnosis (Hoebeke et al., 2018). Compared with these studies, the reasons for our low mutation detection rate may be partly due to the small proportion of patients carrying PMP22 duplication (18.2% of patients in our study versus 37.4%–61.3% in the aforementioned studies). It is worth noting that in other studies based on Chinese populations, CMT1A also only accounted for 19.5%–29.5% of CMT cases (Liu et al., 2020; Xie et al., 2021), with a similar rate in Japan and Korea (15%–26.3%) (Choi et al., 2004; Abe et al., 2011). The difference in the prevalence of PMP22 duplication might be attributed to the heterogeneity among patients of different origins as well as the underestimation of mild patients in our country who did not receive detailed examination at hospitals. Therefore, this result indicates that although some asymptomatic or mild CMT1A cases may be missed in diagnosis, there is indeed distributional heterogeneity among patients of different origins.
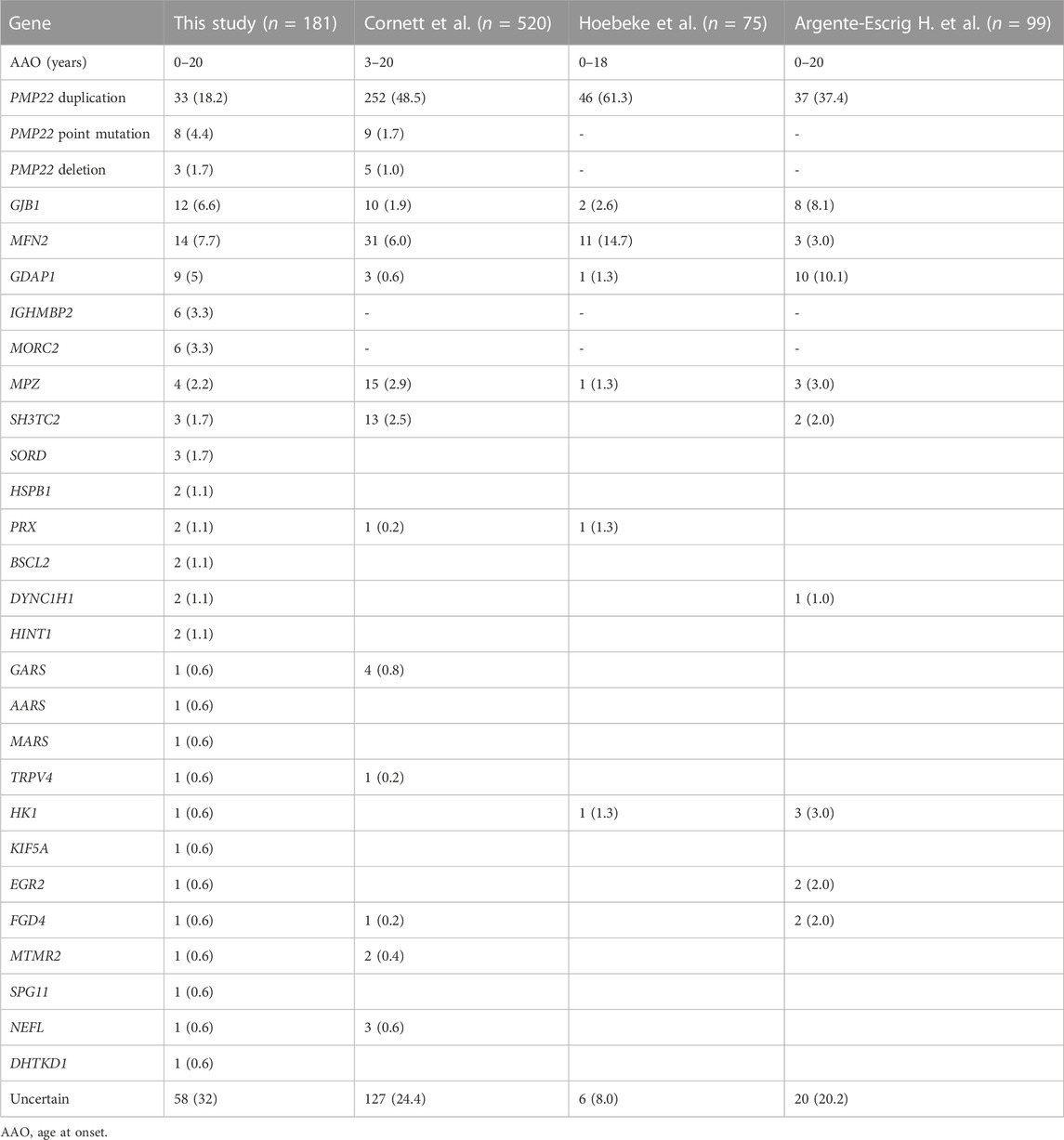
TABLE 2. Gene distributions compared with previous studies.
In our current series, the most frequent disease-causing genes were PMP22 duplication, followed by MFN2 and GJB1. This distribution of genetic subtypes in our patients was similar to those reported previously in Chinese patients. A study focused on South Chinese CMT patients identified PMP22 (19.1%–29%), GJB1 (13.5%–13.8%) and MFN2 (6.5%–10.1%) as the most frequent causative genes (Chen et al., 2019; Xie et al., 2021). In another study, PMP22 (48.7%), GJB1 (9.4%) and MFN2 (3.3%) also ranked as the top three disease-causing genes in a Taiwanese CMT population (Hsu et al., 2019). Likewise, compared with Caucasian paediatric populations, the genetic spectrum was partly different in our cohort. Except for the remarkably lower detection rate of PMP22 duplication in our study, mutations in IGHMBP2, MORC2, and SORD, manifesting as predominantly motor involvement, were also first discussed in pediatric CMT. In contrast, the proportions of the other frequent causative genes in our study, such as GJB1, MFN2 and MPZ were similar to those reported in studies of European ancestry (Cornett et al., 2016; Hoebeke et al., 2018; Pipis et al., 2020; Argente-Escrig et al., 2021). Among rare genes, GDAP1 was also reported in most paediatric CMT series, with a distribution of 0.6%–10.1% in Caucasian patients and 5% in our cohort.
There was marked genetic heterogeneity according to AAO in our findings. In the infantile-onset group, the diagnosis rate ranked the highest at 79.6%, with mutations in the PMP22, MFN2, and IGHMBP2 genes being the three most frequent causes. As age increased, the diagnosis rate was gradually decreased, with 66.6% in the childhood-onset group and 60.3% in the adolescent-onset group. Following the three most frequent genes, the remaining common causative genes of childhood-onset CMT were GDAP1, SH3TC2, and MORC2, while HSPB1 and SORD were mostly implicated in adolescent-onset CMT. In addition, disease severity was correlated with particular genotypes. Generally, earlier onset was the most predictive marker of significant disease severity for most CMT subtypes, while for CMT1A, the disease worsened consistently throughout childhood and adolescence, which was in line with previous studies (Cornett et al., 2016). For all subtypes, symptoms related to disease severity including decreased hand dexterity and weakness in ankle dorsiflexion. In comparison to patients with other genotypes, patients with PMP22 point mutation developed a severe phenotype with significant disability, accounting for 29.4% of the severe cases in total. In the CMT2A subgroup, patients with an AAO before 10 years tended to have more severe disease than those in the late-onset subgroup.
The most common phenotypes for infantile-onset patients were DSS in our findings. Genetically, most cases are caused by a dominant or de novo mutation in the PMP22, MPZ or EGR2 genes (Gargaun et al., 2016; Grosz et al., 2019; Yoshimura et al., 2019). In addition to PMP22 mutations, variants in rare genes, e.g., EGR2, PRX, and FGD4 were found more frequently due to the introduction of NGS and WES, which was consistent with previous studies (Yoshimura et al., 2019).
Interestingly, the presence of de novo variants was identified in 12.7% of all patients and thus was not rare. Certain PMP22 variants, such as p. S72W, p. S79P and p. G150V, were mainly observed to occur de novo. In addition, patients carrying de novo variants of MPZ (p. R98C, p. S233Rfs*18 and p. L174Rfs*66) were also uncommon, with a severe DSS phenotype of motor retardation, weakness and foot deformity. In previous studies, de novo variants for MFN2 were not rare. In some series of CMT2A patients, a frequency of de novo MFN2 variants has been reported of 14.4%–35.0% (Ma et al., 2021; Verhoeven et al., 2006; Kijima et al., 2005). In pediatric group, the frequency may be higher, with up to 45.5% (5/11) being reported of de novo in a French CMT2A cohort (Hoebeke et al., 2018). In literature, de novo variants are common disease mechanism in some childhood onset inherited diseases; it is associated with a reduced life expectancy and reduced reproductive fitness. As de novo variants are usually too deleterious to be passed on in evolution, it is more common in pediatric patients rather than other age groups (Mohiuddin et al., 2022). Compared with European paediatric CMT patients, in whom de novo variants were identified in only 6%–6.5% (Yoshimura et al., 2019; Argente-Escrig et al., 2021), the frequency in our cohort was to some extent higher. Except for the possible geographic differences, the reason that less awareness of disease might deceive mild non-de novo individuals to access to our specialized clinics may also contribute to the lower frequency in our cohort. Considering that the prevalence of de novo variants was highest in early-onset and severe cases, genetic screening should be performed in patients with early-onset and severe peripheral neuropathy regardless of family history.
In our cohort, variants in IGHMBP2, MORC2, and SORD accounted for a certain proportion of paediatric CMT patients, which was not reported in previous studies from Western countries. Recently, the SORD gene has been considered one of the most frequent causative genes for autosomal recessive axonal CMT or dHMN (Cortese et al., 2020), which share a phenotype of motor-predominant peripheral neuropathy. In this study, we identified three patients carrying either a homozygous or a compound heterozygous c.757delG (p. Ala253GlnfsTer27) variant, which was consistent with the literature (Liu et al., 2021). However, MORC2 variants are clinically heterogeneous with phenotypes that can be characterized by congenital or early-onset spinal muscular atrophy like or pure motor axonal neuropathy (Sevilla et al., 2016). Therefore, genetic screening according to the clinical phenotype should be conducted in paediatric patients in whom CMT is suspected especially for autosomal recessive inheritance or newly discovered genes.
In summary, this study represented a major effort to investigate paediatric CMT characteristics in a Chinese population. In-depth analysis highlighted the relatively lower detection rate of PMP22 duplication and higher frequency of de novo variants among paediatric patients in this specific geographic region. In addition, we illustrated genetic heterogeneity according to AAO, disease severity and clinical features. Indeed, since patients were recruited from two clinic centres, the results of the study could not represent the whole Chinese pediatric population. In the future, longitudinal studies and multi-centre studies would yield more information and analytical results.
Data availability statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving human participants were reviewed and approved by the Institutional Ethics Committee of Peking University Third Hospital (2019-005-02). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
XL and DF conceptualized and designed the study. YM, XD, and XL performed data analysis. YM wrote the manuscript. XD, XL, and DF revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Natural Science Foundation of China under Grant (No. 81873784, No. 82071426), Clinical Cohort Construction Program of Peking University Third Hospital (No. BYSYDL2019002, No. BYSYDL2021007) and Clinical Medicine Plus X-Youth Scholars Project, Peking University, the Fundamental Research Funds for the Central Universities (PKU2021LCXQ019).
Acknowledgments
We thank the patients for their consent and participation in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1188361/full#supplementary-material
References
Abe, A., Numakura, C., Kijima, K., Hayashi, M., Hashimoto, T., and Hayasaka, K. (2011). Molecular diagnosis and clinical onset of Charcot-Marie-Tooth disease in Japan. J. Hum. Genet. 56, 364–368. doi:10.1038/jhg.2011.20
Agrahari, A., and George Priya Doss, C. (2015). Impact of I30T and I30M substitution in MPZ gene associated with dejerine-sottas syndrome type B (dssb): A molecular modeling and dynamics. J. Theor. Biol. 382, 23–33. doi:10.1016/j.jtbi.2015.06.019
Argente-Escrig, H., Frasquet, M., Vázquez-Costa, J. F., Millet-Sancho, E., Pitarch, I., Tomás-Vila, M., et al. (2021). Pediatric inherited peripheral neuropathy: A prospective study at a Spanish referral center. Ann. Clin. Transl. Neurol. 8 (9), 1809–1816. doi:10.1002/acn3.51432
Baets, J., Deconinck, T., De Vriendt, E., Zimoń, M., Yperzeele, L., Van Hoorenbeeck, K., et al. (2011). Genetic spectrum of hereditary neuropathies with onset in the first year of life. Brain. 134(Pt 9): 2664–2676. doi:10.1093/brain/awr184
Bird, T. D. (1998). “Charcot-marie-tooth hereditary neuropathy overview,” in GeneReviews®. M. P. Adam, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, L. J. Bean, K. W. Grippet al. (Seattle (WA): University of Washington, Seattle).
Burns, J., Ouvrier, R., Estilow, T., Shy, R., Laurá, M., Pallant, J. F., et al. (2012). Validation of the Charcot-Marie-Tooth disease pediatric scale as an outcome measure of disability. Ann. Neurol. 71 (5), 642–652. doi:10.1002/ana.23572
Chen, C. X., Dong, H. L., Wei, Q., Li, L. X., Yu, H., Li, J. Q., et al. (2019). Genetic spectrum and clinical profiles in a southeast Chinese cohort of Charcot-Marie-Tooth disease. Clin. Genet. 96 (5), 439–448. doi:10.1111/cge.13616
Choi, B. O., Lee, M. S., Shin, S. H., Hwang, J. H., Choi, K. G., Kim, W. K., et al. (2004). Mutational analysis of PMP22, MPZ, GJB1, EGR2 and NEFL in Korean Charcot-Marie-Tooth neuropathy patients. Hum. Mutat. 24 (2), 185–186. doi:10.1002/humu.9261
Cornett, K. M., Menezes, M. P., Bray, P., Halaki, M., Shy, R. R., Yum, S. W., et al. (2016). Phenotypic variability of childhood charcot-marie-tooth disease. JAMA Neurol. 73 (6), 645–651. doi:10.1001/jamaneurol.2016.0171
Cortese, A., Zhu, Y., Rebelo, A. P., Negri, S., Courel, S., Abreu, L., et al. (2020). Biallelic mutations in SORD cause a common and potentially treatable hereditary neuropathy with implications for diabetes. Nat. Genet. 52 (5), 473–481. doi:10.1038/s41588-020-0615-4
Fridman, V., Bundy, B., Reilly, M. M., Pareyson, D., Bacon, C., Burns, J., et al. (2015). CMT subtypes and disease burden in patients enrolled in the inherited neuropathies consortium natural history study: A cross-sectional analysis. J. Neurol. Neurosurg. Psychiatry 86 (8), 873–878. doi:10.1136/jnnp-2014-308826
Gargaun, E., Seferian, A. M., Cardas, R., Le Moing, A. G., Delanoe, C., Nectoux, J., et al. (2016). EGR2 mutation enhances phenotype spectrum of Dejerine-Sottas syndrome. J. Neurol. 263 (7), 1456–1458. doi:10.1007/s00415-016-8153-9
Grosz, B. R., Golovchenko, N. B., Ellis, M., Kumar, K., Nicholson, G. A., Antonellis, A., et al. (2019). A de novo EGR2 variant, c.1232A > G p. Asp411Gly, causes severe early-onset Charcot-Marie-Tooth Neuropathy Type 3 (Dejerine-Sottas Neuropathy). Sci. Rep. 9 (1), 19336. doi:10.1038/s41598-019-55875-4
Hoebeke, C., Bonello-Palot, N., Audic, F., Boulay, C., Tufod, D., Attarian, S., et al. (2018). Retrospective study of 75 children with peripheral inherited neuropathy: Genotype-phenotype correlations. Arch. Pediatr. 25 (8), 452–458. doi:10.1016/j.arcped.2018.09.006
Hsu, Y. H., Lin, K. P., Guo, Y. C., Tsai, Y. S., Liao, Y. C., and Lee, Y. C. (2019). Mutation spectrum of charcot-marie-tooth disease among the han Chinese in taiwan. Ann. Clin. Transl. Neurol. 6 (6), 1090–1101. doi:10.1002/acn3.50797
Kijima, K., Numakura, C., Izumino, H., Umetsu, K., Nezu, A., Shiiki, T., et al. (2005). Mitochondrial GTPase mitofusin 2 mutation in Charcot-Marie-Tooth neuropathy type 2A. Hum. Genet. 116 (1-2), 23–27. doi:10.1007/s00439-004-1199-2
Klein, C. J. (2020). Charcot-Marie-Tooth disease and other hereditary neuropathies. Contin. (Minneap Minn) 26 (5), 1224–1256. doi:10.1212/CON.0000000000000927
Landrieu, P., and Baets, J. (2013). Early onset (childhood) monogenic neuropathies. Handb. Clin. Neurol. 115, 863–891. doi:10.1016/B978-0-444-52902-2.00049-7
Liu, X., Duan, X., Zhang, Y., and Fan, D. (2020a). Clinical and genetic diversity of PMP22 mutations in a large cohort of Chinese patients with charcot-marie-tooth disease. Front. Neurol. 11, 630. doi:10.3389/fneur.2020.00630
Liu, X., Duan, X., Zhang, Y., Sun, A., and Fan, D. (2020b). Molecular analysis and clinical diversity of distal hereditary motor neuropathy. Eur. J. Neurol. 27 (7), 1319–1326. doi:10.1111/ene.14260
Liu, X., He, J., Yilihamu, M., Duan, X., and Fan, D. (2021). Clinical and genetic features of biallelic mutations in SORD in a series of Chinese patients with charcot-marie-tooth and distal hereditary motor neuropathy. Front. Neurol. 12, 733926. doi:10.3389/fneur.2021.733926
Ma, Y., Sun, A., Zhang, Y., Fan, D., and Liu, X. (2021). The genotype and phenotype features in a large Chinese MFN2 mutation cohort. Front. Neurol. 12, 757518. doi:10.3389/fneur.2021.757518
Mohiuddin, M., Kooy, R. F., and Pearson, C. E. (2022). De novo mutations, genetic mosaicism and human disease. Front. Genet. 13, 983668. doi:10.3389/fgene.2022.983668
Padilha, J. P. D., Brasil, C. S., Hoefel, A. M. L., Winckler, P. B., Donis, K. C., Brusius-Facchin, A. C., et al. (2020). Diagnostic yield of targeted sequential and massive panel approaches for inherited neuropathies. Clin. Genet. 98 (2), 185–190. Epub 2020 Jun 29. doi:10.1111/cge.13793
Panosyan, F. B., Laura, M., Rossor, A. M., Pisciotta, C., Piscosquito, G., Burns, J., et al. (2017). Cross-sectional analysis of a large cohort with X-linked Charcot-Marie-Tooth disease (CMTX1). Neurology 89 (9), 927–935. doi:10.1212/WNL.0000000000004296
Pareyson, D., and Marchesi, C. (2009). Diagnosis, natural history and management of Charcot-Marie-Tooth disease. Lancet. Neurology 8 (7), 654–667. doi:10.1016/S1474-4422(09)70110-3
Pipis, M., Feely, S. M. E., Polke, J. M., Skorupinska, M., Perez, L., Shy, R. R., et al. (2020). Natural history of charcot-marie-tooth disease type 2A: A large international multicentre study. Brain 143 (12), 3589–3602. doi:10.1093/brain/awaa323
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of medical genetics and Genomics and the association for molecular Pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Sevilla, T., Lupo, V., Martínez-Rubio, D., Sancho, P., Sivera, R., Chumillas, M. J., et al. (2016). Mutations in the MORC2 gene cause axonal Charcot-Marie-Tooth disease. Brain 139, 62–72. doi:10.1093/brain/awv311
Skre, H. (1974). Genetic and clinical aspects of Charcot-Marie-Tooth's disease. Clin. Genet. 6 (2), 98–118. doi:10.1111/j.1399-0004.1974.tb00638.x
Verhoeven, K., Claeys, K. G., Züchner, S., Schröder, J. M., Weis, J., Ceuterick, C., et al. (2006). MFN2 mutation distribution and genotype/phenotype correlation in Charcot-Marie-Tooth type 2. Brain. 129(Pt 8), 2093–2102. doi:10.1093/brain/awl126
Xie, Y., Lin, Z., Liu, L., Li, X., Huang, S., Zhao, H., et al. (2021). Genotype and phenotype distribution of 435 patients with Charcot-Marie-Tooth disease from central south China. Eur. J. Neurol. 28 (11), 3774–3783. doi:10.1111/ene.15024
Keywords: paediatric Charcot-Marie-Tooth disease, inherited peripheral neuropathy, genetic distribution, genotype-phenotype correlations, de novo variants
Citation: Ma Y, Duan X, Liu X and Fan D (2023) Clinical and mutational spectrum of paediatric Charcot-Marie-Tooth disease in a large cohort of Chinese patients. Front. Genet. 14:1188361. doi: 10.3389/fgene.2023.1188361
Received: 17 March 2023; Accepted: 03 July 2023;
Published: 13 July 2023.
Edited by:
Jordi Pérez-Tur, Spanish National Research Council (CSIC), SpainReviewed by:
Hiroshi Takashima, Kagoshima University, JapanFiore Manganelli, University of Naples Federico II, Italy
Copyright © 2023 Ma, Duan, Liu and Fan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoxuan Liu, bHVjeWFuX2xpdUBiam11LmVkdS5jbg==; Dongsheng Fan, ZHNmYW5Ac2luYS5jb20=
†These authors have contributed equally to this work and share first authorship