 Shengping Zhong1,2*†
Shengping Zhong1,2*† Xiaowan Ma3†Yan Jiang2†Xujia Liu2,4Mengqing Zeng2Longyan Zhao1Lianghua Huang1Guoqiang Huang1Yongzhen Zhao5Ying Qiao3*Xiuli Chen5*
Xiaowan Ma3†Yan Jiang2†Xujia Liu2,4Mengqing Zeng2Longyan Zhao1Lianghua Huang1Guoqiang Huang1Yongzhen Zhao5Ying Qiao3*Xiuli Chen5*- 1Guangxi Key Laboratory of Marine Drugs, Institute of Marine Drugs, Guangxi University of Chinese Medicine, Nanning, China
- 2Guangxi Engineering Technology Research Center for Marine Aquaculture, Guangxi Institute of Oceanology Co., Ltd., Beihai, China
- 3Key Laboratory of Tropical Marine Ecosystem and Bioresource, Fourth Institute of Oceanography, Ministry of Natural Resources, Beihai, China
- 4Guangxi Key Laboratory of Marine Environmental Science, Guangxi Academy of Marine Sciences, Guangxi Academy of Sciences, Nanning, China
- 5Guangxi Key Laboratory of Aquatic Genetic Breeding and Healthy Aquaculture, Guangxi Academy of Fishery Sciences, Nanning, China
Introduction
Holothuroids, or sea cucumbers, are a diverse echinoderm group of economically significant marine benthic invertebrates (Han et al., 2016; Miller et al., 2017). Due to its large bode size and thick body wall, Stichopus monotuberculatus, a tropical holothuroid has become a popular tonic food in Indo-west pacific counties, where it is known as its delectable taste and high nutritional value (Chen et al., 2022). Because the body walls of holothuroids contain abundant and diverse biomedical compounds, such as sulfated polysaccharides, saponins, and peptides (Thimmappa et al., 2022) they have become the subject of increasing attention in pharmaceutical research as an important source of medicinal compounds (Xu et al., 2022).
Marine sulfated polysaccharides usually occur in echinoderms and brown algae, which are abundant and considered to be promising sources of medicinal compounds with significant pharmacological effects (Helbert, 2017; Lu et al., 2020). The marine sulfated polysaccharides are typically composed of sulfate groups and α-L-fucose. They are structurally diverse and the positions of their fucosylated and sulfonated features vary by marine species (Pomin, 2014; Wu et al., 2016). Sulfated fucans (SF) and fucosylated chondroitin sulfates (FCS) are the two most common types of sulfated polysaccharides occurring in marine animals, with SF occurring in echinoderms and brown algae (Helbert, 2017) and FCS being exclusive to echinoderms (Carvalhal et al., 2019). Fucosylated chondroitin sulfates, also called fucosylated glycosaminoglycan (FG), is typically composed of a chondroitin sulfate backbone attached to α-L-fucose branches, both of which are commonly sulfonated (Wu et al., 2016; Khotimchenko, 2018). The structural features of FCS and the pattern of sulfonation of the chondroitin sulfate backbone and the α-L-fucose branches varies across sea cucumber species (Yuan et al., 2022). It be important in maintaining the body wall integrity of sea cucumbers by preventing degradation by marine microorganisms (Xu et al., 2022). Studies of brown algae have detected duplication and expansion events at SF biosynthesis associated genes, such as the mannuronan C-5-epimerases (MC5Es) (Ye et al., 2015) and carbohydrate sulfotransferases (CHSTs), which increase the rigidity of the cell wall and provide genomic adaptions to different environments (Lu et al., 2020). A previous study on the structural diversity of holothurian FCS and the position and number of the fucosylated branches attached to the chondroitin sulfate backbone confirmed the species-specific patterns across holothuroids (Myron et al., 2014). These species-specific FCS structures may have significant impacts on various biological activities such as antithrombotic, antihyperlipidemic, and anticoagulant processes and wound healing (Mou et al., 2018). Studies of brown algae genomes have identified genes coding for key enzymes for sulfated polysaccharides (Nishitsuji et al., 2016). Moreover, the genes coding for downstream enzymes, such as sulfotransferases and fucosyltransferases have been revealed expanded in brown algae genome (Nishitsuji et al., 2019). However, despite the biological, ecological and economic importance of holothurian FCS, the key genes associated with its biosynthetic pathways and their evolution remain unknown.
Here in this report, in order to decipher the key genes associated FCS biosynthetic pathways in tropical sea cucumbers, S. monotuberculatus genome have been assembled and annotated. The genome comprised 168 contigs, with an overall length of 839.56 Mb. The contig N50 length was 11.51 Mb. In addition, our investigations of the evolution of the gene families revealed 1,011 significantly expanded genes and 408 genes exclusive to S. monotuberculatus. The gene families coding for key enzymes associated with FCS biosynthetic pathways, including fucosyltransferases and sulfotransferases, were also significantly expanded. The accessibility of first genomic sequences and annotations of S. monotuberculatus will provide a genomic approach to investigate the structural diversity of holothurian FCS, as well as novel perspectives into evolutionary adaptation of critical genes in holothurian FCS biosynthesis pathways.
Materials and methods
DNA and RNA sequencing
An individual S. monotuberculatus (body weight 5.92 g) was collected from Guangxi Province, China (21.484,179 N, 109.720,886 E), and kept in an aerated water tank before tissue sampling. The intestine, tentacles, muscle, and tube-feet were dissected out and flash-frozen using liquid nitrogen. Genomic DNA was isolated from a muscle sample using a QIAamp DNA Mini Kit (QIAGEN, Hilden, Germany). A Nanopore 20 kb insert library was constructed using approximately 1 µg of genomic DNA and Blue Pippin electrophoresis (Sage Science, Inc., Beverly, CA, United States) was used to select DNA fractions larger than 20 kb. A DNA sequencing library for S. monotuberculatus was prepared by Grandomics Biosciences Co., Ltd (Wuhan, China) using an SQK-LSK109 ligation kit (Oxford NanoporeTechnologies, Oxford, UK). In addition, a 350 bp insert size paired-end DNA sequencing library with 2 × 150 bp lengths was prepared using a MGISEQ-2000 platform (Wuhan, China) in accordance with the manufacturer’s instructions. Finally, S. monotuberculatus RNA was isolated from the intestine, tentacle, and tube-feet samples using an RNAiso kit (TaKaRa, Tokyo, Japan) and then a MGISEQ-2000 system was used to prepare three S. monotuberculatus paired-end RNA sequencing libraries with lengths of 2 × 150 bp.
Genome assembly
The 2 × 150 bp paired-end DNA sequences were used to evaluate the S. monotuberculatus genome length and polymorphisms. After using fastp (v. 0.23.2) (Chen et al., 2018) to trim the DNA sequences, Jellyfish (v. 2.3.0) (Marçais and Kingsford, 2011), and Genomescope (v, 2.0) (Ranallo-Benavidez et al., 2020) were used to conduct a k-mer analysis with a length setting of 19. The correct-then-assemble strategy of NextDenovo (v. 2.5.0) (github.com/Nextomics/NextDenovo) was used to assemble the preliminarily S. monotuberculatus genome using Nanopore sequencing data. NextPolish (v. 1.4.1) (Hu et al., 2020) was used to fix the contig errors in the preliminarily S. monotuberculatus genome using S. monotuberculatus paired-end DNA sequences. Purge_Dups (v. 1.2.6) (Guan et al., 2020) was then used to remove redundant sequences. Finally, the completeness of the S. monotuberculatus genome was assessed using the metazoa_odb10 data in the BUSCO software (v. 5.4.4) (Manni et al., 2021).
Prediction of transposable elements, gene structure, and functional annotation
Initially, we used EDTA (v. 2.1.0) (Ou et al., 2019) to apply an ab initio strategy to evaluate the transposable elements (TEs) in the S. monotuberculatus genome. A homology-based strategy was then applied using the predicted TEs library and RepeatMasker (v. 4.1.2) (www.repeatmasker.org) to further detect any remaining comprehensive TEs sequences. The protein-coding gene structures were annotated using a combination of transcript-based, homology-based, and ab initio prediction strategies. For the transcript-based annotation, the RNA sequencing data from the intestine, tentacles, and tube-feet samples were used to assembly de novo transcript sequences using Trinity (v. 2.14) (Grabherr et al., 2011). Afterwards, the genome guided-transcripts were assembled using StringTie (v. 2.2.1) (Shumate et al., 2022). Finally, the transcript-based predicted proteins in the S. monotuberculatus genome were obtain by aligning all transcript sequences to the S. monotuberculatus genomic sequences using PASA (v. 2.5.2) (Campbell et al., 2006) with default parameters. For the homology-based method, the protein-coding genes in the S. monotuberculatus genome were validated using GeMoMa (v. 1.9) (Hoff, 2019) with default settings obtained from the echinoderm protein data in Genbank, including those for Strongylocentrotus purpuratus (GCF_000002235.5), Patiria miniata (GCF_015706575.1), Lytechinus variegatus (GCF_018143015.1), Apostichopus japonicus (GCF_002754855.1), Holothuria glaberrima (GCF_009936505.2), Asterias rubens (GCF_902459465.1), Anneissia japonica (GCF_011630105.1), and Chiridota heheva (GCF_020152595.1). For the ab initio method, the RNA sequencing data of S. monotuberculatus and the echinoderm protein data were applied to predict the coding genes using BRAKER2 (v. 2.1.6) (Brůna et al., 2021) in the GeneMark-ETP mode. Finally, the predictions of the three strategies were merged and evaluated using EvidenceModeler (v. 1.1.1) (Haas et al., 2008), and the evaluated results were functionally annotated using DIAMOND (v. 2.1.3) (Buchfink et al., 2021) with default parameters to search several protein databases, and well as the Gene Ontology (GO), Swiss-Prot, UniProtKB-TremBL, and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases, using an E-value limit for homologous annotation of 1e-5. The completeness of the prediction of the protein-coding genes in the S. monotuberculatus genome was further validated using BUSCO (v. 5.4.4) with the metazoa_odb10 data. The identification of transfer RNAs (tRNAs) and ribosomal RNAs (rRNAs) in the S. monotuberculatus genome was accomplished using tRNAscan-SE (v. 2.0.6) (Lowe and Chan, 2016) and RNAmmer (v. 1.2) (Huang et al., 2009), respectively. The identification of microRNAs and small nuclear RNAs (snRNAs) was performed using Infernal (v. 1.1.2) (Nawrocki et al., 2009) to search the Rfam database with default parameters.
Phylogenomics and gene family investigation
To investigate the comparative phylogenomics and gene families of S. monotuberculatus, OrthoFinder (v. 2.5.4) (Emms and Kelly, 2019) was used to verify the orthologous gene clusters of S. monotuberculatus and 14 related species including Acanthaste planci (GCF_001949145.1), Hemicentrotus pulcherrimus (GCF_003118195.1), Holothuria scabra (GCF_026123075.1), Pisaster ochraceus (GCF_010994315.2), Plazaster borealis (GCF_021014325.1), Saccoglossus kowalevskii (GCF_000003605.2), S. purpuratus, P. miniata, L. variegatus, Anneissia japonicus, H. glaberrima, A. rubens, Apostichopus japonica, and C. heheva. The protein sequences of the 14 related species were retrieved from Genbank and Echinobase (www.echinobase.org), and the longest protein sequence was chosen to identify the orthologous protein. To investigate the evolutionary status of S. monotuberculatus, the single-copy protein sequences were first obtained from the orthologous OrthoFinder results and then MUSCLE (v. 3.8.31) was used to align those sequences. The phylogenetic tree was constructed from the continuous super protein sequences combined from all of the aligned single-copy sequences using RAxML-avx (v. 8.2.9) (Stamatakis, 2014) under LG4M amino acid substitution model with 1,000 bootstrap replicates. The evolutionary times of H. scabra and A. japonicus were retrieved from the TimeTree website (www.timetree.org) and calibrated using the r8s software (v. 1.71) to evaluate the evolutionary time of S. monotuberculatus and the 14 closely related species. In order to evaluate the evolutionary history of the S. monotuberculatus gene families, the likelihood analysis method was performed using CAFE (v. 5.0) (Mendes et al., 2020) with p < 0.05. In addition, functional enrichment analyses, including KEGG and GO, were employed using TBtools (Chen et al., 2020) to evaluate the biological functions of the S. monotuberculatus species-specific and expanded gene families.
Results and discussions
Genome assembly
Both the Nanopore and MGI platforms were used to assemble the S. monotuberculatus genome. The former produced 51.53 Gb sequences with an average length of 22,615 bp, while the latter produced 64.72 Gb sequences with a Q20 quality level of 97.96%. The genomic features of the S. monotuberculatus genome were estimated using the MGI platform data in Genomescope to perform k-mer-based estimations. The results showed a predicted size of the S. monotuberculatus genome of 784.77 Mb, as well as a repetitive sequence ratio of 31.63% and a high heterozygous rate of 1.59% (Figure 1B; Supplementary Table S2). The heterozygous rate of the S. monotuberculatus genome was close to that of A. japonicus, while its genome size was slightly smaller (Zhang et al., 2017). Due to the high heterozygosity of the S. monotuberculatus genome, the long reads data were used in NextDenovo to assemble its complex regions. After a first round of assembly, 292 contigs with 10.52 Mb in contig N50 were achieved. To resolve the redundancy of heterozygous regions, Purge_Dups was applied after correcting contigs errors using NextPolish. Eventually, 168 contigs with 839.56 Mb in size, 11.51 Mb in contig N50, and a longest contig of 52.05 Mb were established in the S. monotuberculatus genome (Supplementary Table S1). This is the largest reported contig N50 for any holothurian genome to date (Luo et al., 2022). Based on the metazoa_odb10 lineages, the BUSCO analysis with genome mode was performed to assess the genome integrity of S. monotuberculatus. The result showed that it covered 97.9% BUSCOs, including 94.2% complete and 3.7% fragmented, while only 2.1% were classified as missing (Supplementary Table S3). Our assembled genome has the greatest level of completeness of any reported holothurian genome to date, including H. scabra (Luo et al., 2022), H. glaberrima (Medina-Feliciano et al., 2021), C. heheva (Zhang et al., 2022), and A. japonicus (Li et al., 2018). In addition, 95.98% of the short read data can be confidently mapped to the S. monotuberculatus final genome using BWA v. 0.7.17. These results indicate that the genome completeness and integrity are high in the final assembled S. monotuberculatus genome.
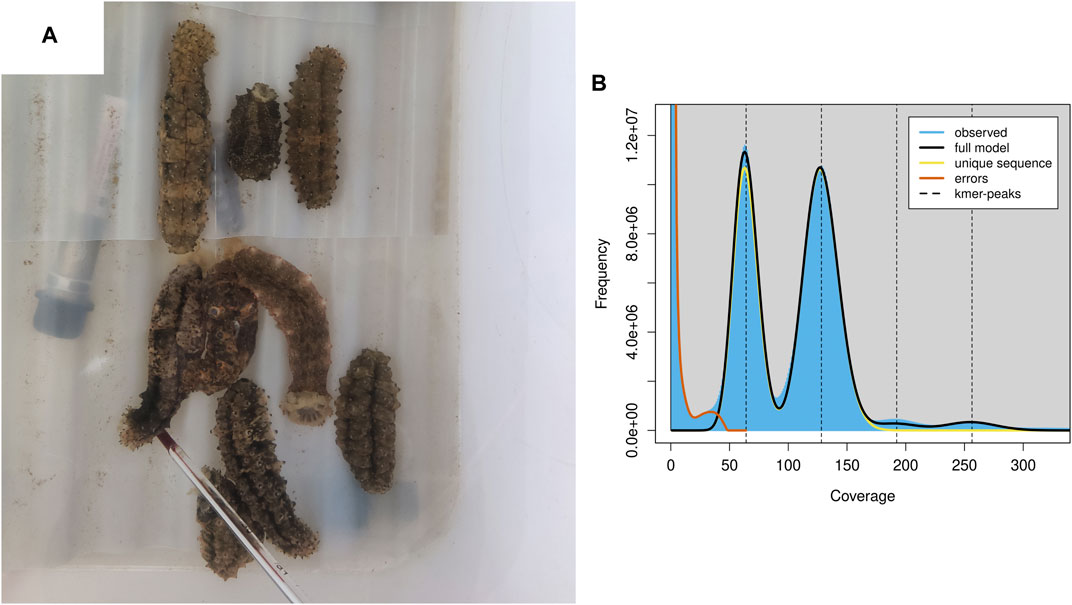
FIGURE 1. S. monotuberculatus and the genomics feature. (A) S. monotuberculatus reared in water tank. (B) The genomics feature analysis of S. monotuberculatus.
Genome annotation
The integrated results of the predicted TEs analysis showed that a total length of 298.36 Mb (35.54%) of the S. monotuberculatus genome could be identified as TEs, which is the highest among the published Stichopodidae genomes, including A. japonicus (26.68%) and Parastichopus parvimensis (25.02%). However, the proportion of TEs in the S. monotuberculatus genome was much smaller than in other deep-sea holothurian genomes, such as C. heheva (56.40%) (Zhang et al., 2022) and Paelopatides sp. Yap (73.93%) (Shao et al., 2022). The S. monotuberculatus genome was found to be richest in terminal inverted repeats, which made up 19.41% of the genome sequences, while long terminal repeats (6.88%) and tandem repeats (5.67%) were the next two richest types (Supplementary Table S4). Using EvidenceModeler to combine protein-coding gene predictions, the S. monotuberculatus genome was confirmed to have 36,422 protein-coding genes (Supplementary Table S1). The protein-coding gene number of S. monotuberculatus was a bit higher than that of A. japonicus (30,305), but the protein-coding gene length of S. monotuberculatus (12,278 bp on average, Supplementary Table S1) was a bit longer than that of A. japonicus (7,787 bp on average) (Zhang et al., 2017). Compared to S. monotuberculatus and A. japonicus, H. scabra genome has a much longer protein-coding gene length (25,967 bp on average) but much less protein-coding gene number (16,642) (Luo et al., 2022). In the comparison study of three A. japonicus genome assemblies, Jo’s assembly has the lowest protein-coding gene number (21,771) and the shortest protein-coding gene length (5,388 bp on average). However, as the quality of the assembly has improved, the gene annotation completeness has also increased significantly, resulting in the longest protein-coding gene length (8,918 bp on average) and the highest protein-coding gene number around thirty-thousand (Li et al., 2018). The improvement of S. monotuberculatus assembly quality may also result in an increase in the number of predicted protein-coding genes. The BUSCO evaluation based on metazoa_odb10 lineages showed that the verified genes covered 99.1% of metazoa_odb10 genes, including 98.6% classified as complete and 0.5% classified as fragmented (Supplementary Table S3). The completeness of the gene predictions in the S. monotuberculatus genome is the greatest of any reported holothurian genome to date, indicating that the quality of the gene structure prediction is sufficiently high for downstream analyses, such as comparative phylogenomics analysis. In addition, 79.31% of the S. monotuberculatus genes were functionally annotated in the various protein databases, including UniProtKB-TremBL (79.08%), Swiss-Prot (49.96%), KEGG (38.25%), and GO (36.82%) (Supplementary Table S5). Moreover, a total length of 0.34 Mb in the S. monotuberculatus genome was predicted to be non-coding RNAs, including 1,101 tRNAs, 1,452 microRNAs, 181 rRNAs, and 208 snRNAs (Supplementary Table S6).
Phylogenomics and gene family investigation
The orthologous genes shared between S. monotuberculatus and other Echinodermata species were identified using OrthoFinder. A total of 30,979 genes (85.1%) were identified as orthologous gene clusters in the S. monotuberculatus protein-coding gene set, of which 2,793 genes (7.7%) were found to be species-specific orthologous genes. In addition, all Echinodermata share 6,191 orthogroups, along with 422 single-copy orthogroups (Supplementary Table S7). RAxML used 73,344 amino acid sites of these single-copy orthogroups to build the Echinodermata phylogenetic tree with the hemichordate S. kowalevskii rooted as an outgroup species. The phylogenetic results showed that S. monotuberculatus initially clustered with A. japonicus, and that Stichopodidae and Holothuriidae were clustered as sister groups to form the Aspidochirotida clade. Apodida (C. heheva) and Aspidochirotida were clustered together within the Holothuroidea, with Apodida being the basal taxon of Holothuroidea (Supplementary Figure S1). Our phylogenetic results indicated that Stichopodidae and Holothuriidae have a close sister relationship within Aspidochirotida, and that Apodida occupies the basal branch in the class Holothuroidea. These results were consistent with the phylogenetic analyses of Holothuroidea inferred by morphological (KERR and KIM, 2008) and phylogenomic analyses (Zhang et al., 2022).
An evolutionary investigation of gene families was conducted comparing S. monotuberculatus with other Echinodermata species. The results showed that 693 gene families in the S. monotuberculatus genome were unique and that 857 gene families had undergone significant expansion, whereas only 167 gene families had undergone significant contraction (Figure 2A). The structural features of FCS from holothuroids are species-specific, and mainly due to the different positions of the sulfonated branches on the chondroitin sulfate backbone and to the different fucosylated branch chains across the holothuroids (Xu et al., 2022). Studies of S. monotuberculatus have shown that these species-specific FCS structural features improve anticoagulant activity (Yuan et al., 2022). In this study, gene families coding for key enzymes involved in FCS biosynthesis, including carbohydrate sulfotransferases (CHSTs, EC 2.8.2) (Supplementary Figure S2) which were identified as significantly expanded gene families and fucosyltransferases (FUTs, EC 2.4.1) (Supplementary Figure S3) which were identified as species-specific to S. monotuberculatus. In previous studies on brown algae, the sulfotransferases and fucosyltransferases genes were also expanded independently, and the copies of these genes varied among brown algae genomes, which may be important in the efficient biosynthesis of SF (Nishitsuji et al., 2019). Compared with other Echinodermata species, 13 copies of the carbohydrate sulfotransferase 11 (CHST11, EC 2.8.2.5) gene, 10 copies of the carbohydrate sulfotransferase 13 (CHST13, EC 2.8.2.5) gene, and 22 copies of the carbohydrate sulfotransferase (CHST15, EC 2.8.2.33) gene were identified as significantly expanded in the S. monotuberculatus genome. Meanwhile, The KEGG enrichment analyses of significantly expanded S. monotuberculatus gene families showed that these genes were significantly enriched in 62 pathways, as well as those involved in FCS biosynthesis, such as Glycosaminoglycan biosynthesis - chondroitin sulfate, Pentose and glucuronate interconversions, Glycan biosynthesis and metabolism, and Glycosaminoglycan binding proteins (Figure 2B). Significant expansions of gene families coding for key enzymes associated with FCS biosynthesis in the S. monotuberculatus genome may play crucial roles in the biosynthesis of complex and diverse FCS and may be important for maintaining body wall integrity in S. monotuberculatus. Furthermore, eight copies of the fucosyltransferase 4 (FUT4, EC 2.4.1.-) gene, nine copies of the fucosyltransferase 7 (FUT7, EC 2.4.1.-) gene, and one copy of the fucosyltransferase 3 (FUT3, EC 2.4.1.65) gene were identified as unique to the S. monotuberculatus genome. In previous studies on brown algae, N. decipiens genome also has unique fucosyltransferase and extracellular matrix genes which may facilitate sulfated fucan biosynthesis in Nemacystus decipiens (Nishitsuji et al., 2019). Compared to other species of Aspidochirotida, these unique fucosyltransferase genes may be critical for S. monotuberculatus producing FCS more efficiently to maintain body wall integrity. GO enrichment analyses also showed that these unique genes were significantly enriched in the Glycosaminoglycan biosynthesis pathway, such as the polysaccharide binding (GO:0030247), proteoglycan binding (GO:0043394), fucosyltransferase activity (GO:0008417), and α-1,3-fucosyltransferase activity (GO:0046920) pathways (Figure 2C). In summary, the species specificity of key fucosyltransferase genes may play an important role in the biosynthesis of unique FCS in S. monotuberculatus, indicating the complex genomic adaptations for FCS biosynthesis in sea cucumbers.
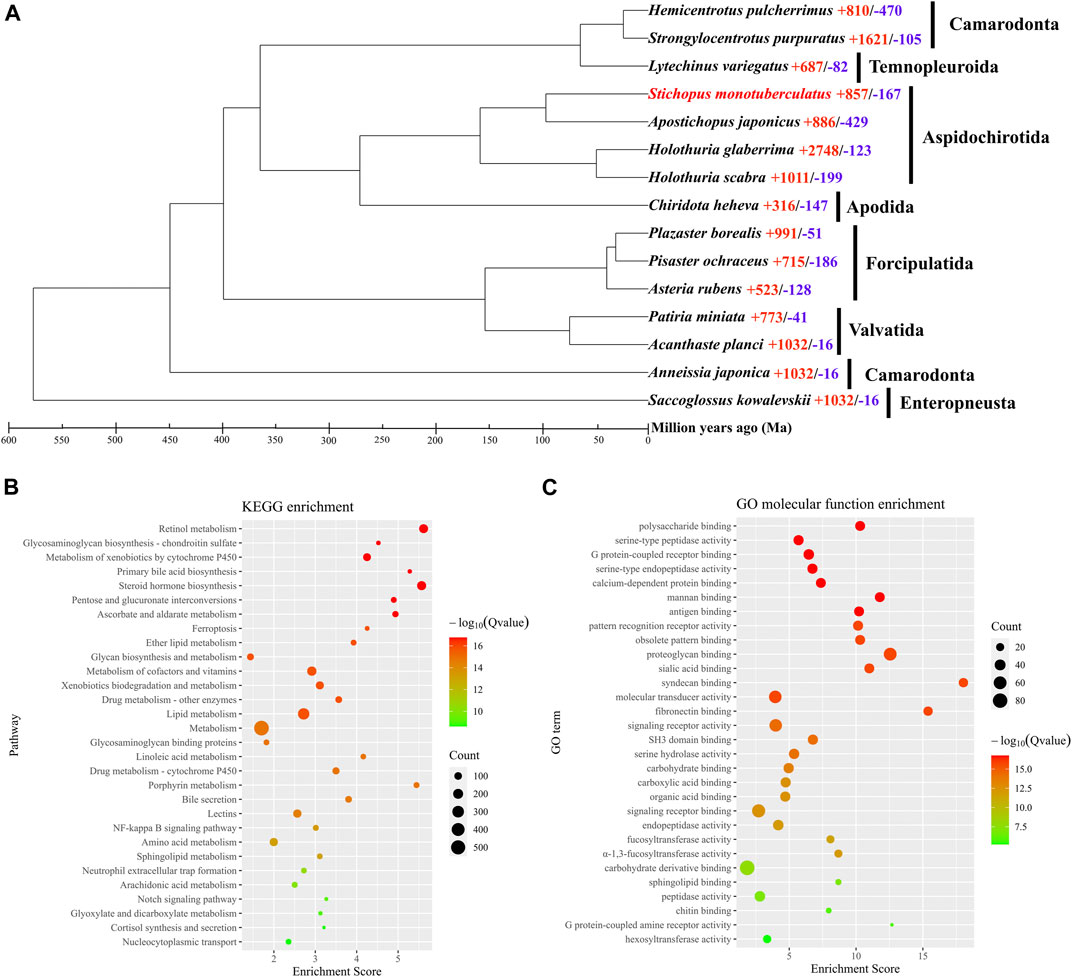
FIGURE 2. Phylogenomics and enrichment analyses. (A) Comparative phylogenomics analysis. Red numbers, indicates significantly expansion genes; while blue numbers, indicates significantly contracted genes. (B) KEGG enrichment analysis of significantly expansion genes. (C) GO enrichment analysis of species-specific genes.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the https://figshare.com/, https://doi.org/10.6084/m9.figshare.22177898.v1; https://www.ncbi.nlm.nih.gov/, PRJNA938157, SRR23615351, SRR23610520-SRR23610521 and SRR23604910-SRR23604915.
Ethics statement
The studies involving animals were reviewed and approved by Committee on Animal Research and Ethics of Guangxi University of Chinese Medicine.
Author contributions
SZ, XM, and YJ: conceived study. SZ, XM, and YQ: bioinformatics analysis. XL, MZ and LZ: collected samples. LH, GH, and YZ: investigation samples. SZ, YQ, and XC: wrote manuscript. All authors approved the final submission. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by National Natural Science Foundation of China (31960225), China Postdoctoral Science Foundation (2021M701798), Guangxi University of Chinese Medicine “GuiPai Traditional Chinese Medicine inheritance and innovation team” Project (2022A007), Development Program of High-level Talent Team under Qihuang Project of Guangxi University of Chinese Medicine (2021004), Guangxi Natural Science Foundation (2021GXNSFAA196074, 2021GXNSFAA220030, 2022GXNSFBA035166, 2020GXNSFBA297126, AD19245135, and AD19245161), Fund of Hainan Provincial Key Laboratory of Tropical Maricultural Technologies (TMTOF202204), the Open Fund of Guangxi Key Laboratory of Aquatic Genetic Breeding and Healthy Aquaculture (GXKEYLA2022-03) and PhD research startup foundation of Guangxi University of Chinese Medicine (2019BS018).
Conflict of interest
Guangxi Institute of Oceanology Co., Ltd. was the employer of authors Shengping Zhong, Yan Jiang, Xujia Liu, and Mengqing Zeng.
The remaining authors state that no business or financial relationships that may be considered as a potential conflict of interest existed during the study.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1182002/full#supplementary-material
References
Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M., and Borodovsky, M. (2021). BRAKER2: Automatic eukaryotic genome annotation with GeneMark-ep+ and AUGUSTUS supported by a protein database. NAR Genomics Bioinforma. 3 (1), lqaa108. doi:10.1093/nargab/lqaa108
Buchfink, B., Reuter, K., and Drost, H.-G. (2021). Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18 (4), 366–368. doi:10.1038/s41592-021-01101-x
Campbell, M. A., Haas, B. J., Hamilton, J. P., Mount, S. M., and Buell, C. R. (2006). Comprehensive analysis of alternative splicing in rice and comparative analyses with Arabidopsis. BMC Genomics 7 (1), 327. doi:10.1186/1471-2164-7-327
Carvalhal, F., Cristelo, R. R., Resende, D. I. S. P., Pinto, M. M. M., Sousa, E., and Correia-da-Silva, M. (2019). Antithrombotics from the sea: Polysaccharides and beyond. Mar. Drugs 17 (3), 170. doi:10.3390/md17030170
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34 (17), i884–i890. doi:10.1093/bioinformatics/bty560
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13 (8), 1194–1202. doi:10.1016/j.molp.2020.06.009
Chen, M., Sun, S., Xu, Q., Gao, F., Wang, H., and Wang, A. (2022). Influence of water temperature and flow velocity on locomotion behavior in tropical commercially important sea cucumber Stichopus monotuberculatus. Front. Mar. Sci. 9, 931430. doi:10.3389/fmars.2022.931430
Emms, D. M., and Kelly, S. (2019). OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 20 (1), 238. doi:10.1186/s13059-019-1832-y
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29 (7), 644–652. doi:10.1038/nbt.1883
Guan, D., McCarthy, S. A., Wood, J., Howe, K., Wang, Y., and Durbin, R. (2020). Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36 (9), 2896–2898. doi:10.1093/bioinformatics/btaa025
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., Orvis, J., et al. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9 (1), R7. doi:10.1186/gb-2008-9-1-r7
Han, Q., Keesing, J. K., and Liu, D. (2016). A review of sea cucumber aquaculture, ranching, and stock enhancement in China. Rev. Fish. Sci. Aquac. 24 (4), 326–341. doi:10.1080/23308249.2016.1193472
Helbert, W. (2017). Marine polysaccharide sulfatases. Front. Mar. Sci. 4. doi:10.3389/fmars.2017.00006
Hoff, K. J. (2019). MakeHub: Fully automated generation of UCSC genome browser assembly hubs. Genomics, Proteomics Bioinforma. 17 (5), 546–549. doi:10.1016/j.gpb.2019.05.003
Hu, J., Fan, J., Sun, Z., and Liu, S. (2020). NextPolish: A fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36 (7), 2253–2255. doi:10.1093/bioinformatics/btz891
Huang, Y., Gilna, P., and Li, W. (2009). Identification of ribosomal RNA genes in metagenomic fragments. Bioinformatics 25 (10), 1338–1340. doi:10.1093/bioinformatics/btp161
Kerr, A. M., and Kim, J. (2008). Phylogeny of Holothuroidea (Echinodermata) inferred from morphology. Zoological J. Linn. Soc. 133 (1), 63–81. doi:10.1111/j.1096-3642.2001.tb00623.x
Khotimchenko, Y. (2018). Pharmacological potential of sea cucumbers. Int. J. Mol. Sci. 19 (5), 1342. doi:10.3390/ijms19051342
Li, Y., Wang, R., Xun, X., Wang, J., Bao, L., Thimmappa, R., et al. (2018). Sea cucumber genome provides insights into saponin biosynthesis and aestivation regulation. Cell. Discov. 4 (1), 29. doi:10.1038/s41421-018-0030-5
Lowe, T. M., and Chan, P. P. (2016). tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44 (W1), W54–W57. doi:10.1093/nar/gkw413
Lu, C., Shao, Z., Zhang, P., and Duan, D. (2020). Genome-wide analysis of the Saccharina japonica sulfotransferase genes and their transcriptional profiles during whole developmental periods and under abiotic stresses. BMC Plant Biol. 20 (1), 271. doi:10.1186/s12870-020-02422-3
Luo, H., Huang, G., Li, J., Yang, Q., Zhu, J., Zhang, B., et al. (2022). De novo genome assembly and annotation of Holothuria scabra (Jaeger, 1833) from nanopore sequencing reads. Genes. and Genomics 44 (12), 1487–1498. doi:10.1007/s13258-022-01322-0
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A., and Zdobnov, E. M. (2021). BUSCO update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38 (10), 4647–4654. doi:10.1093/molbev/msab199
Marçais, G., and Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27 (6), 764–770. doi:10.1093/bioinformatics/btr011
Medina-Feliciano, J. G., Pirro, S., García-Arrarás, J. E., Mashanov, V., and Ryan, J. F. (2021). Draft genome of the sea cucumber Holothuria glaberrima, a model for the study of regeneration. Front. Mar. Sci. 8, 603410. doi:10.3389/fmars.2021.603410
Mendes, F. K., Vanderpool, D., Fulton, B., and Hahn, M. W. (2020). CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics 36 (22-23), 5516–5518. doi:10.1093/bioinformatics/btaa1022
Miller, A. K., Kerr, A. M., Paulay, G., Reich, M., Wilson, N. G., Carvajal, J. I., et al. (2017). Molecular phylogeny of extant Holothuroidea (Echinodermata). Mol. Phylogenetics Evol. 111, 110–131. doi:10.1016/j.ympev.2017.02.014
Mou, J., Li, Q., Qi, X., and Yang, J. (2018). Structural comparison, antioxidant and anti-inflammatory properties of fucosylated chondroitin sulfate of three edible sea cucumbers. Carbohydr. Polym. 185, 41–47. doi:10.1016/j.carbpol.2018.01.017
Myron, P., Siddiquee, S., and Al Azad, S. (2014). Fucosylated chondroitin sulfate diversity in sea cucumbers: A review. Carbohydr. Polym. 112, 173–178. doi:10.1016/j.carbpol.2014.05.091
Nawrocki, E. P., Kolbe, D. L., and Eddy, S. R. (2009). Infernal 1.0: Inference of RNA alignments. Bioinformatics 25 (10), 1335–1337. doi:10.1093/bioinformatics/btp157
Nishitsuji, K., Arimoto, A., Iwai, K., Sudo, Y., Hisata, K., Fujie, M., et al. (2016). A draft genome of the Brown alga, Cladosiphon okamuranus, S-strain: A platform for future studies of ‘mozuku’ biology. DNA Res. 23 (6), 561–570. doi:10.1093/dnares/dsw039
Nishitsuji, K., Arimoto, A., Higa, Y., Mekaru, M., Kawamitsu, M., Satoh, N., et al. (2019). Draft genome of the Brown alga, Nemacystus decipiens, Onna-1 strain: Fusion of genes involved in the sulfated fucan biosynthesis pathway. Sci. Rep. 9 (1), 4607. doi:10.1038/s41598-019-40955-2
Ou, S., Su, W., Liao, Y., Chougule, K., Agda, J. R. A., Hellinga, A. J., et al. (2019). Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 20 (1), 275. doi:10.1186/s13059-019-1905-y
Pomin, V. H. (2014). Holothurian fucosylated chondroitin sulfate. Mar. Drugs 12 (1), 232–254. doi:10.3390/md12010232
Ranallo-Benavidez, T. R., Jaron, K. S., and Schatz, M. C. (2020). GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11 (1), 1432. doi:10.1038/s41467-020-14998-3
Shao, G., He, T., Mu, Y., Mu, P., Ao, J., Lin, X., et al. (2022). The genome of a hadal sea cucumber reveals novel adaptive strategies to deep-sea environments. iScience 25 (12), 105545. doi:10.1016/j.isci.2022.105545
Shumate, A., Wong, B., Pertea, G., and Pertea, M. (2022). Improved transcriptome assembly using a hybrid of long and short reads with StringTie. PLOS Comput. Biol. 18 (6), e1009730. doi:10.1371/journal.pcbi.1009730
Stamatakis, A. (2014). RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30 (9), 1312–1313. doi:10.1093/bioinformatics/btu033
Thimmappa, R., Wang, S., Zheng, M., Misra, R. C., Huang, A. C., Saalbach, G., et al. (2022). Biosynthesis of saponin defensive compounds in sea cucumbers. Nat. Chem. Biol. 18 (7), 774–781. doi:10.1038/s41589-022-01054-y
Wu, N., Zhang, Y., Ye, X., Hu, Y., Ding, T., and Chen, S. (2016). Sulfation pattern of fucose branches affects the anti-hyperlipidemic activities of fucosylated chondroitin sulfate. Carbohydr. Polym. 147, 1–7. doi:10.1016/j.carbpol.2016.03.013
Xu, H., Zhou, Q., Liu, B., Chen, F., and Wang, M. (2022). Holothurian fucosylated chondroitin sulfates and their potential benefits for human health: Structures and biological activities. Carbohydr. Polym. 275, 118691. doi:10.1016/j.carbpol.2021.118691
Ye, N., Zhang, X., Miao, M., Fan, X., Zheng, Y., Xu, D., et al. (2015). Saccharina genomes provide novel insight into kelp biology. Nat. Commun. 6 (1), 6986. doi:10.1038/ncomms7986
Yuan, Q., Li, H., Wang, Q., Sun, S., Fang, Z., Tang, H., et al. (2022). Deaminative-cleaved S. monotuberculatus fucosylated glycosaminoglycan: Structural elucidation and anticoagulant activity. Carbohydr. Polym. 298, 120072. doi:10.1016/j.carbpol.2022.120072
Zhang, X., Sun, L., Yuan, J., Sun, Y., Gao, Y., Zhang, L., et al. (2017). The sea cucumber genome provides insights into morphological evolution and visceral regeneration. PLOS Biol. 15 (10), e2003790. doi:10.1371/journal.pbio.2003790
Keywords: Stichopus monotuberculatus, fucosylated chondroitin sulfates, biosynthetic pathway, genomic adaptation, Echinodermata
Citation: Zhong S, Ma X, Jiang Y, Liu X, Zeng M, Zhao L, Huang L, Huang G, Zhao Y, Qiao Y and Chen X (2023) The draft genome of the tropical sea cucumber Stichopus monotuberculatus (Echinodermata, Stichopodidae) reveals critical genes in fucosylated chondroitin sulfates biosynthetic pathway. Front. Genet. 14:1182002. doi: 10.3389/fgene.2023.1182002
Received: 08 March 2023; Accepted: 03 May 2023;
Published: 12 May 2023.
Edited by:
Mehar S. Khatkar, The University of Sydney, AustraliaReviewed by:
Abigail Elizur, University of the Sunshine Coast, AustraliaJie Gong, Nantong University, China
Copyright © 2023 Zhong, Ma, Jiang, Liu, Zeng, Zhao, Huang, Huang, Zhao, Qiao and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shengping Zhong, c2hwemhvbmdAZm94bWFpbC5jb20=; Ying Qiao, cWlhb3lpbmcwNjE4QGhvdG1haWwuY29t; Xiuli Chen, Y2hlbnhpdWxpMjAwMUAxNjMuY29t
†These authors have contributed equally to this work