 Fengming Zhu
Fengming Zhu Yueqiang Li1
Yueqiang Li1 Ying Yao
Ying Yao Rui Zeng
Rui Zeng- 1Department of Nephrology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 2Department of Nutrition, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 3Key Laboratory of Organ Transplantation, Ministry of Education, NHC Key Laboratory of Organ Transplantation, Key Laboratory of Organ Transplantation, Chinese Academy of Medical Sciences, Wuhan, China
Background: Mutations in the collagen components of the glomerular basement membrane (GBM) often lead to hereditary glomerulonephritis. Previous studies have identified that autosomal dominant mutations of Col4A3, Col4A4 or Col4A5 are associated with thin basement membrane nephropathy (TBMN), Alport syndrome and other hereditary kidney diseases. However, the genetic mutations underlying other glomerulonephritis types have not been elucidated.
Methods: In this study, we investigated a Chinese family with hereditary nephritis using the methods of genetic sequencing and renal biopsy. Genomic DNA was extracted from peripheral blood of the proband and her sister, and subsequently was performed genetic sequencing. They were found to have the similar mutation sites. Other family members were then validated using Sanger sequencing. The proband and her sister underwent renal puncture biopsies, and experienced pathologists performed PAS, Masson, immunofluorescence, and immunoelectron microscopic staining of the kidney tissue sections.
Results: Through genetic sequencing analysis, we detected a novel heterozygous frameshift mutation c.1826delC in the COL4A4 (NM_000092.4) gene coding region, and 1 hybrid missense variation c.86G>A (p. R29Q) was also detected in the TNXB (NM_019105.6) gene coding region in several members of this Chinese family. Interestingly, we found that the same mutations caused different clinical features and distinct pathological changes in individual family members, which confirmed that pathological and genetic testing are crucial for the diagnosis and treatment of hereditary kidney diseases.
Conclusion: In this study, we found a novel heterozygous mutation in Col4A4 and co-mutations of the TNXB gene in this Chinese family. Our study indicated that the same Col4A4 mutated variants produced different pathological and clinical changes in different family members. This discovery may provide novel insights into the study of hereditary kidney disease. In addition, new genetic biology techniques and renal biopsy of individual family members are essential.
Introduction
Hereditary glomerulonephritis is one of the main causes of end-stage renal disease (ESRD). There are currently no effective treatments for this special disease. Therefore, an early and clear diagnosis through genetic testing may have substantial implications for the patient and his or her offspring, and under the proper guidance of a professional doctor, delay the deterioration of renal function.
The majority of hereditary nephritis is inextricably linked to genetic mutations (Mehta and Jim, 2017). Type 4 collagen is the most important component of the glomerular basement membrane and is secreted by podocytes. Mutations in the type 4 collagen gene (Col4) and its subunits result in the most common types of hereditary nephritis (Hudson, 2004; Deltas et al., 2013). Previous studies have demonstrated that the mutations in Col4 and its subunits cause thin basement membrane nephropathy (TBMN) (Cèlia Badenas et al., 2002; Mark Buzza et al., 2003) and Alport syndrome (Bárbara Tazón Vega, 2003), which are the common types of inherited nephritis. Autosomal dominant mutations of Col-4A3 and/or A4 lead to the development of TBMN (Hirabayashi et al., 2022). The Col4A4 gene encodes one of the six subunits of type IV collagen (E D Adamson SEA, 1979). Lemmink, et al. reported the first Col4A4 mutation in 1996 in TBMN patients (Smeets VVK et al., 1996). After that, more than 20 mutations at different sites of Col4A4 were described. However, the main mutation sites have not been clarified yet.
The TNXB gene encodes a member of the tenascin-X family of extracellular matrix glycoproteins, which is thought to function in collagen organization and extracellular matrix integrity during wound healing (Egging et al., 2007), and its deficiency or mutation has been related to the connective tissue disorder Ehlers‒Danlos syndrome (Schalkwijk et al., 2001; Merke et al., 2013). However, the relationship between TNXB mutation and kidney disease has not yet been elucidated.
In this study, we analyzed a Chinese family with hereditary glomerulonephritis and carried out gene mutation analyses using genetic sequencing. According to previous research, we suspected that the same genetic mutation may produce similar pathological changes, however, renal biopsy results challenged this view.
Materials and methods
Patients
The research subject is a Chinese family, including the proband, her parents, her son, her sister and the sister’s daughter and son. The proband (II1) is a 46-year-old woman who demonstrated continuous microscopic hematuria and microalbuminuria for more than 10 years and underwent a renal biopsy; chronic glomerulonephritis was confirmed without any deposition of fluorescent material. She then asked for genetic testing. Her mother died of uremia several years ago; therefore, renal biopsy and genetic testing could not be conducted for this individual. Her younger sister showed frequent urination, urgency, and dysuria for 5 years, and her renal biopsy results confirmed IgA nephropathy. Both the proband and her sister showed the mutations in Col-4A4 and TNXB through genetic testing. Her father, her son and her sister’s children did not have any clinical symptoms. We recommended them to take blood and urine samples for primary screening and found that the son of the proband had microscopic hematuria. Then, with the consent of the patient and her family members, we took blood samples from the family members and performed exon sequencing tests. We also advised her son to undergo renal biopsy, which was refused due to a busy work schedule. This study was approved by the Ethics Committee of Tongji Hospital affiliated with Tongji Medical of Huazhong University of Science and Technology. The proband and her family members provided written informed consent to participate in this study.
Renal biopsy and pathological analysis
The proband and her sister underwent renal biopsy conducted by an experienced nephrologist using a Doppler ultrasound device in the year of 2017. The pathological tissue sections were subjected to Periodic Acid-Schiff (PAS) and Masson staining in our renal pathology laboratory referred to this literature for specific staining methods (Pei et al., 2019). After staining, an experienced pathologist examined the sections using light microscopy, immunofluorescence and electron microscopy.
Genetic mutation detection
Genomic DNA was extracted from the peripheral blood leukocytes of the proband and family members (Qiagen, Germany) according to the manufacturer’s instructions. The concentration and purity of DNA were measured using a NanoDrop 2000 ultra-microspectrophotometer (Thermo Scientific, United States). High-throughput sequencing was carried out at BGI Genomics (Wuhan, China) with an ABI 3730XL sequencer. Briefly, 1 µg of genomic DNA was used as a template, and specific primers were extended in the presence of DNA polymerase to obtain PCR products. The extension reaction was not terminated until the insertion of a dideoxynucleotide (ddNTP). The amplified products were subjected to 1% agarose gel electrophoresis. Then, the products were purified with a BigDye® XTerminator™ Purification Kit (ABI, United States) and subjected to sequencing with an ABI 3730XL sequencer using a BigDye® XTerminator™ Sequencing Kit (ABI, United States). A heterozygous frameshift mutation was detected in the coding region of the Col4A4 gene, and a missense mutation was also detected in the coding region of the TNXB gene of the proband. Then, the peripheral blood samples from the proband’s sister and other family members were subjected to Sanger sequencing to verify the genetic mutation condition. The pathogenicity of filtered polynucleotides was analyzed using a variety of bioinformatic analysis software (SIFT, (Sorting Intolerant From Tolerant) http://sift.jcvi.org; Mutation Taster, http://www.mutationtaster.org/).
Results
Clinical features of patients
The clinical information was acquired from 7 people in a Chinese family in Hubei Province. The renal biopsy confirmed that the proband and her younger sister had glomerulus nephritis. Her son, who refused to have a renal biopsy, was potentially also a patient according to his routine urine test results. The proband’s mother died of uremia. A family diagram for the group with glomerulonephritis is shown in Figure 1.
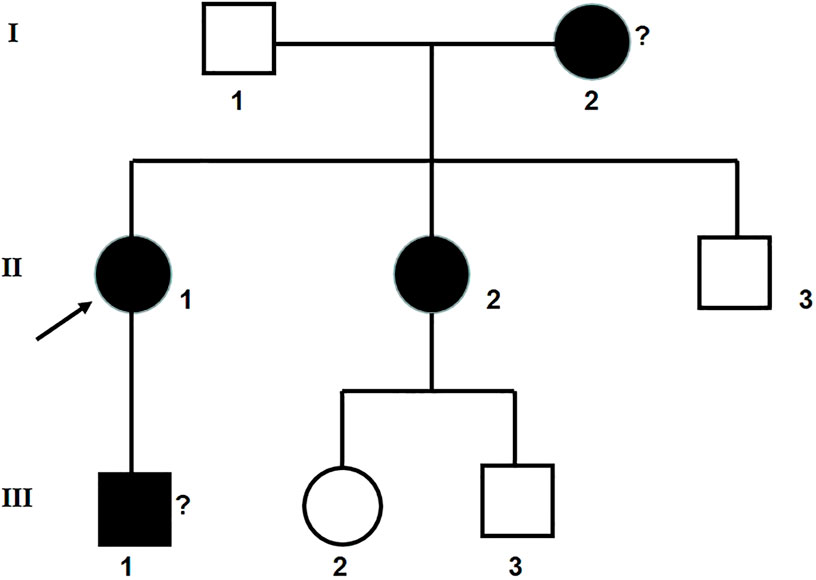
FIGURE 1. Family diagram for the group with glomerulonephritis. II 1: the proband.
The main clinical manifestations of the proband were continuous microscopic hematuria and microalbuminuria for more than 10 years. In the past 3 months, the proband presented with eyelid edema and nocturia (>3 times per night). Microscopic examination of the urine showed that erythrocytes in the urine were mixed, suggesting that the hematuria originated from the glomeruli. The glomerular filtration rate was decreased to 45.9 mL/min/1.73 m2, and the level of serum creatinine was 122 μmol/L. The proband’s son had no symptoms. Only laboratory tests revealed microscopic hematuria, and he refused to undergo a renal biopsy.
However, the proband’s younger sister showed different clinical features and was diagnosed with frequent urination, urgency, and dysuria for 5 years. Her routine urine tests showed hematuria and proteinuria, and she was positive for the left renal vein nutcracker syndrome. The routine urine test was negative for hematuria but still showed trace proteinuria after anti-inflammatory treatment. She underwent renal biopsy and was diagnosed with glomerulonephritis.
The test results for rheumatism, immune system and other related factors of the proband and her sister were negative, excluding secondary kidney damage.
The specific clinical characteristics and urine test results of the proband and their relatives are shown in Table 1.
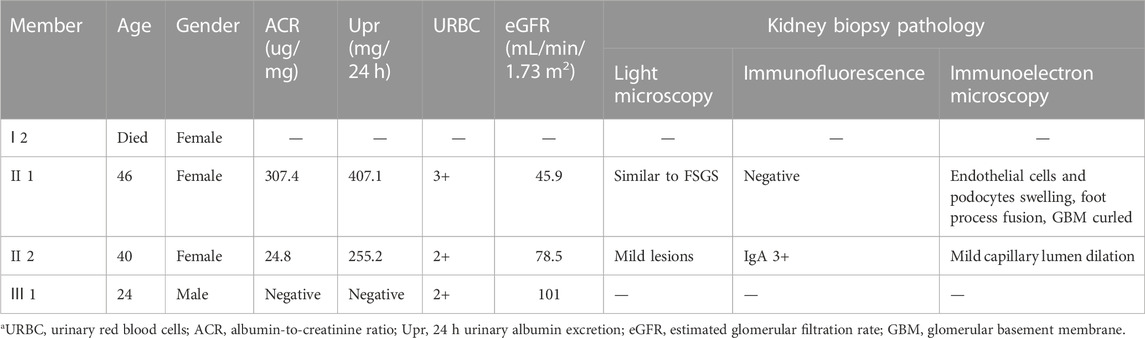
TABLE 1. Clinical features, urine/blood tests, kidney biopsy results of proband and carriers in the year of 2017.
Pathological features of the proband
Light Microscopy: 3 large fibrocystic crescents with microthrombi, 1 small fibrous-cellular crescent formation, 1 ischemic sclerosis and 3 glomerular sclerosis formations of 11 glomeruli were seen in the renal tissue. Mild and segmental proliferation of mesenchymal cells and stroma were observed in the remaining glomeruli. Several endothelial cells in the glomeruli developed segmental proliferation. Some of the tubules were detached from the tubular base membrane, and some showed multifocal atrophy. Mononuclear cells and lymphocytes infiltrated the renal interstitium multifocally, accompanied by fibrosis. Immunofluorescence: Negative. Immunoelectron microscopy: Capillary loops were irregularly arranged with partial cavity collapse. Endothelial cells showed swelling without obvious proliferation. Podocytes were swollen, with partial vacuolar degeneration and foot process fusion. Some renal tubular epithelial cells were necrotic and exfoliated, and the residual basement membrane was curled. The renal interstitium was scattered with infiltration of inflammatory cells and deposition of collagen fibers. Pathological diagnosis: Focal glomerular lesion with partial crescent formation.
Pathological features of the proband’s younger sister
In contrast, the proband’s younger sister showed completely different pathological features. Light Microscopy: 5 of 17 glomeruli showed spherical sclerosis. The capillaries were obviously shrunken. The walls of the capsules were significantly thickened with stratification. The remaining glomeruli were not uniform in size, with a small increase in volume and slight hyperplasia in mesangial cells. Tubulointerstitial lesions were mild-to-moderate, with tubular epithelial cell edema, granular cell degeneration, focal tubular atrophy, basement membrane thickening, and epithelial cell flattening. A small amount of fibrosis and mononuclear cell infiltration and several foam cells were observed in the renal interstitium. Immunofluorescence showed that IgA (+++) was deposited diffusely in the glomerular mesangial area. Immunoelectron microscopy: part of the capillary lumen was dilated. The basement membrane was normal in morphology, and no electron-dense deposits were observed. Most of the foot processes were fused. Pathological diagnosis: IgA nephropathy with mild mesangial proliferative lesions with glomerular sclerosis (5/17), as shown in Table 1; Figure 2.
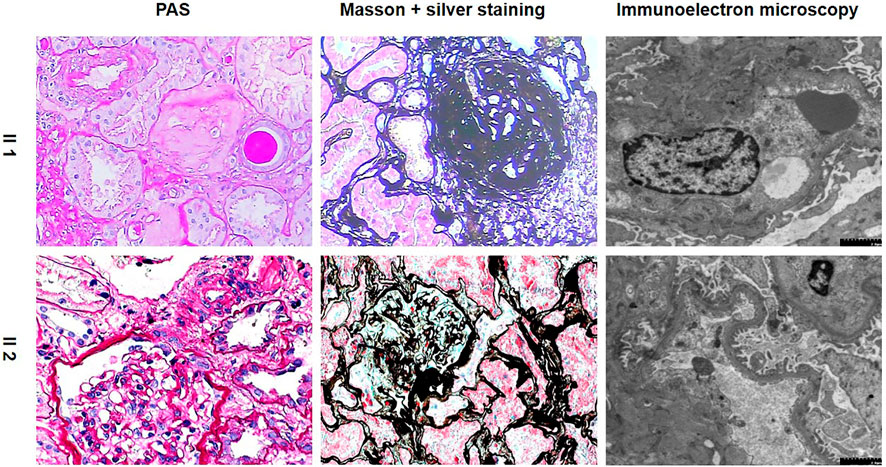
FIGURE 2. Pathological features of the proband and her younger sister. (PAS, periodic acid-schiff.)
Sequencing of COL4A4 and TNXB
According to the high-throughput sequencing results of the proband in the early stage, a heterozygous frameshift mutation of c.1826delC was detected in the COL4A4 (NM_000092.4) gene coding region (the corresponding amino acid was p.Pro609Glnfs*44), and 1 hybrid missense variation of c.86G>A (p. R29Q) was also detected in the TNXB (NM_019105.6) gene coding region (the corresponding amino acid was p.R29Q). Sanger sequencing of the above sites was performed on the proband and her sister. They possessed the same genetic mutations at the same sites, as shown in Figure 3; Table 2. Her son had only the Col4A4 mutation. The proband’s father and brother had only a nonsense mutation at the splice site of c.86G>A of the TNXB gene. Genetic mutations were not detected in other living family members, as shown in Figure 3; Table 2. The screened nucleotides were subjected to gene mutation analysis (Mutation Taster; SIFT) indicted that the c.1826delC mutation of Col4A4 was predicted to be pathogenic and the c.86G>A locus of TNXB was predicted to be polymorphism (probable harmless) (https://www.mutationtaster.org/MT69/MutationTaster69.cgi). Further enquiries for genetic sequencing data can be directed to the link (https://ngdc.cncb.ac.cn/gsa-human/submit/hra/subHRA006673/finishedOverview).
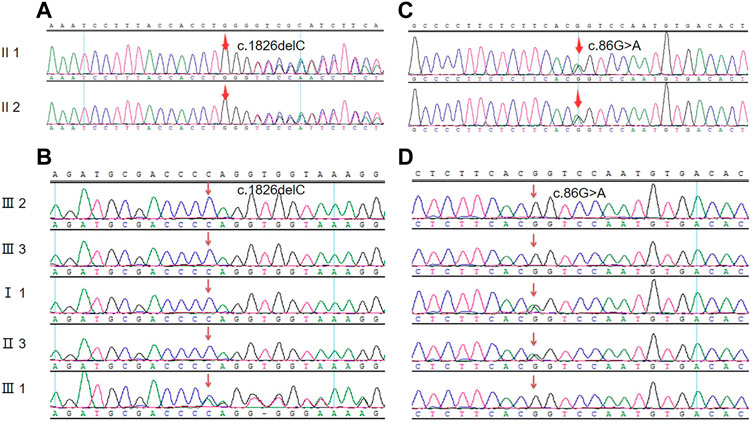
FIGURE 3. Mutated nucleic acid sequence for the genes of Col4A4 and TNXB. (A) Genetic sequencing of the proband and her sister at the splice site of c.1826delC of the Col4A4 gene. (B) Sanger sequencing peak map of other family members at the splice site of c.1826delC of the Col4A4 gene. (C) Genetic sequencing of the proband and her sister at the splice site of c.86G>A of the TNXB gene. (D) Sanger sequencing peak map of other family members at the splice site of c.86G>A of the TNXB gene.

TABLE 2. Genetic screening results of proband and her sister at the splice sites of COL4A4 and TNXB genes.
Follow-up
Prior to receiving the genetic test results, the proband and her sister were given glucocorticoids and immunosuppressants (cyclophosphamide, CTX, cumulative dose of 3 g) treatment for 3 times. The levels of creatinine and urea nitrogen did not obviously change; thus, the intensive treatment was stopped until the genetic test results returned. We then treated the proband with an angiotensin converting enzyme inhibitor (ACEI) to reduce proteinuria, an alpha keto acid to supplement kidney essential amino acids, an oxidized starch with a covered aldehyde group to eliminate small molecule toxins, and Chinese patent medicine in the form of Huangkui capsules and Huaiqihuang granules to reduce the level of albuminuria and hematuria. To date, the eGFR level of the proband has remained stable for 4 years, without substantial anemia, while blood pressure and blood glucose and lipid levels have remained within normal ranges. The proteinuria and hematuria level of the proband’s younger sister were effectively relieved, and the level of creatinine returned to normal. Then, she was treated with an ACEI, Chinese patent medicine in the form of Huangkui capsules, Huaiqihuang granules and Kunxian capsules and other treatments for 5 years. Similar to the proband, her younger sister maintained normal levels of kidney function and urine protein by taking only Candesartan tablets, a type of angiotensin II AT1 receptor antagonist, for 4 years.
The proband and her sister are reviewed every 2-3 months at our outpatient clinic. Treatment regimen has been adjusted according to renal function and urinalysis results. After 5 years, the renal function of both the proband and her younger sister deteriorated, as shown in Figure 4. Moreover, the proband began to develop mild anemia and elevated blood pressure. We adjusted the treatment plan in hopes of slowing the deterioration rate of renal function.
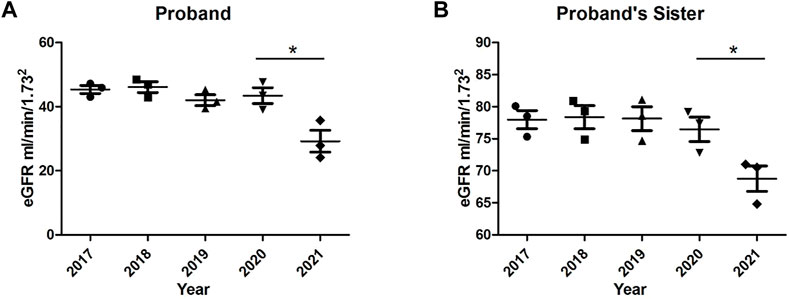
FIGURE 4. Trends in glomerular filtration rates (eGFR) of the proband and her younger sister over 5 years. (A) eGFR of the proband; (B) eGFR of the proband’s younger sister. *p < 0.05, n = 3–5.
Discussion
In this study, we analyzed a Chinese family with hereditary kidney disease through renal biopsy and genetic sequencing technology. We identified a novel variant, Col4A4 c.1826delC, and a hybrid missense variation in TNXB c.86G>A. Interestingly, we found that the same genetic mutations manifested different renal pathological changes through examination of pathological specimens from renal biopsies, in which the proband showed focal glomerular lesions with partial crescent formation, while her sister showed IgA nephropathy with glomerular sclerosis.
Type 4 collagen, which is secreted by endothelial cells and epithelial cells into the extracellular matrix and then self-assembled into a polygonal network structure, is the main component of the GBM extracellular matrix and is encoded by 6 genes (Col4A1-A6) [(E D Adamson SEA, 1979); B V Howard et al. (1976);(Khoshnoodi et al., 2008; Deng et al., 2016). Among them, the mutation of the COL4A4 gene produces an abnormal and incorrectly bound α4 (IV) chain, which enters the triple helix structure of type IV collagen, leading to the instability of the upper molecular structure (Khoshnoodi et al., 2008). Numerous studies have confirmed that mutations in different subtypes of Col4A3, A4, and A5 can lead to Col4-associated glomerulopathy (Bárbara Tazón Vega, 2003), including Alport syndrome (AS) (Korstanje et al., 2014; Guo et al., 2017; Du et al., 2022), thin basement membrane nephropathy (TBMN) (Xu et al., 2016; Hirabayashi et al., 2022), and focal segmental glomerulosclerosis (FSGS) (Xie et al., 2014; Wu et al., 2016; Fan et al., 2020).
However, unlike previous studies, we found genetic mutations of Col4A4 in some members of this family, but neither the proband nor her relatives had any pathological and clinical manifestations of the related diseases such as AS and TBMN. According to the electron microscopy results, the GBM of the proband and her sister only presented minor lesions. In genetic sequencing, we detected an autosomal recessive mutation in the Col4A44 gene. This mode of inheritance usually requires two pathogenic mutations to cause the clinical phenotype. Therefore, we speculate that the single pathogenic locus mutation detected in this study only results in a small fragment base abnormality or an in-frame 3-fold shift, which may form partially retained functional truncated proteins, which are not yet sufficient to cause abnormalities in the morphology and structure of the triple helix of type IV collagen. According to the light microscopy results, the proband’s lesions tended to be focal glomerular lesions with crescents but without obvious sclerosis. Her sister showed IgA nephropathy with mild mesangial proliferative lesions with glomerular sclerosis. The presence of microscopic hematuria in the son of the proband does not exclude the possibility of IgA nephropathy. None of the family members presented sensorineural hearing abnormalities, deafness or early-onset renal failure. We speculate that this paradox may be due to the fact that heterozygous mutations trigger a relatively mild phenotype than homozygous mutations because normal α4 (IV) chains are still produced.
We found that the proband and her sister had different renal pathological changes. There was no deposition of fluorescent substances in the kidney tissue of the proband, while IgA deposition was detected in the mesangial area of her younger sister. Thus, we assumed that the different fluorescence deposition states represented different kidney pathological changes. Stapleton and others found candidate variants in 10 families and identified a likely pathogenic variant in COL4A5 in one family and a variant of unknown significance (VUS) in COL4A3 in another (Stapleton et al., 2020), which confirmed the heterogeneity of IgA nephropathy (Stapleton et al., 2020) and provided evidence that while a proportion of patients with renal IgA deposits may carry pathogenic variants of known kidney disease-related genes such as the Col4 gene, others may not (Stapleton et al., 2020). In our study, we found that Col4 mutation can lead to focal segmental glomerulosclerosis, which is consistent with previous literature reports (Xie et al., 2014; Wu et al., 2016).
Previous studies have detected Col4A3/A4/A5 mutations in patients with familial IgA nephropathy (Savige et al., 2021). In a recent study, Li Zhu et al. detected 31.1% of genotypic mutations in the Col4A3/A4/A5 genes in patients with IgA nephropathy with thinned GBM lesions (IgAN-tGBM) and in 0.19% patients with sporadic IgA nephropathy (Yuan et al., 2023). In this study, the pathological diagnosis of the proband’s sister (II-2) confirmed IgA nephropathy, and genetic testing suggested a mutation in the Col4A4 and TNXB gene. However, no significant lesions were seen in the glomerular basement membrane. Other family members do not match the diagnostic criteria for IgA nephropathy, which was considered that II-2 might be a patient with sporadic IgA nephropathy accompanied by Col4 gene mutation without tGBM.
Previous research showed that mutations in the COL4A4 gene can cause benign familial hematuria (OMIM# 141200) (Cèlia Badenas et al., 2002), which is autosomal dominant; in addition, mutations in the TNXB gene (encoding tenascin-X, an extracellular matrix protein) can cause vesicoureteral reflux type 8 (OMIM#: 615,963) (Gbadegesin et al., 2013), which is autosomal dominant as well. In our study, the proband and her sister demonstrated benign familial hematuria, the same variation in Col4A4 was identified in both individuals, which was consistent with known reports (Bárbara Tazón Vega, 2003; Yang et al., 2019). Furthermore, we detected a hybrid missense variation in TNXB c.86G>A in four members of this family. However, none of them showed abnormalities during kidney color Doppler ultrasound, such as bladder ureteral reflux. Dr. Deborah P. Merke and others demonstrated that biallelic TNXB variants caused Ehlers‒Danlos syndrome in patients with congenital adrenal hyperplasia (Merke et al., 2013; Watanabe et al., 2021; Marino et al., 2022). All of the patients had skin hyperextensibility and significant joint hypermobility. Joint laxity was extreme and some patients had a history of joint dislocations, chronic arthralgias, and chronic tendinitis and/or bursitis. There were no clinical manifestations of skin hyperextensibility or significant joint hypermobility in the proband and her family members in this study. The severity of the disease depended on the carrying capacity of the TNXB variant, as evidenced by the allelic heterogeneity of the cohort (Chen et al., 2016). Combined with this finding, we speculated that the TNXB mutation identified in our study may be a monoallelic hybrid missense mutation, and whether it is common or unique in frequency is unknown at this time. Further research is needed to investigate this mutation and its manifestations.
A large number of studies have shown that patients with hereditary kidney diseases do not undergo the active intensive treatment because of their genetic mutations (Mehta and Jim, 2017; Boyer et al., 2021). In this study, as the renal biopsy report showed partial crescent formation, the proband underwent intensive treatment with glucocorticoids + CTX in the early stages of disease, aiming to suppress immune and inflammatory responses, and thereby reducing the formation of crescents. According to the results of the 5-year follow-up observation, the proband’s renal function remained stable without obvious anemia, which was attributed to the early intensive treatment. The immunofluorescence kidney biopsy results of the proband’s sister, who also received early glucocorticoid + CTX treatment showed a large number of IgA deposits in the glomeruli. Renal function was stable, which was due to the intensive and powerful immunosuppressive therapy during the early stage. After 3 cycles of treatment, hematuria and albuminuria were obviously relieved. At this point, genetic testing confirmed hereditary glomerulonephritis, and glucocorticoid and immunosuppressive therapy was discontinued.
The limitations of this study are as follows: 1. the son of the proband did not undergo renal biopsy; 2. Immunofluorescence staining was not finished at the renal specimens for different Col4 subtypes. In the future, we will continue to suggest the son of the proband to undergo a renal biopsy and use the frozen sections to conduct immunofluorescence staining for Col4 subtypes as a way to confirm the conclusion that the collagen mutation in this study is pathogenic. There is currently no effective treatment for hereditary kidney disease. In the future, kidney function may continue to deteriorate in the proband, and her family members may pass on their Col4A4 mutation to subsequent generations. We will continue to search for new treatments for them. The proband’s son is now of child-bearing age, and we suggested him to consider selecting healthy embryos through new reproductive technologies to avoid the transmission of mutated genes from generation to generation (Bergmann, 2017).
In conclusion, we identified a Chinese family with similar genetic mutations but different pathological changes and clinical phenotype by genetic testing and renal puncture biopsy pathology. This study enriches the genetic spectrum of hereditary kidney diseases and also provides new ideas for the diagnosis and treatment of these diseases.
Data availability statement
The data presented in the study are deposited in the Genome Sequence Archive (GSA)-OMIX repository, accession number OMIX004157.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee of Tongji Hospital affiliated to Tongji Medical College of Huazhong University of Science and Technology. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual (s), and minor (s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
FZ was responsible for article writing, data collection, revision, and photo production. YL and YW were responsible for pathology examination, photo revision and proofreading. YY and RZ was responsible for the review of the paper. All authors contributed to the article and approved the submitted version.
Funding
This project was supported by the National Natural Science Foundation of China (Project Nos 82100742, 81770681, 81974086, and 81800608).
Acknowledgments
We thank all members of the proband and her family. We are also grateful to our pathologists and laboratory technicians and laboratory staff.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Bárbara Tazón Vega, C. B., Ars, E., Lens, X., Milà, M., Darnell, A., and Torra, R. (2003). Autosomal recessive Alport's syndrome and benign familial hematuria are collagen type IV diseases. Am. J. Kidney Dis. 42 (5), 952–959. doi:10.1053/S0272-6386(03)01058-8
Bergmann, C. (2017). Advances in renal genetic diagnosis. Cell. Tissue Res. 369 (1), 93–104. doi:10.1007/s00441-017-2636-6
Boyer, O., Schaefer, F., Haffner, D., Bockenhauer, D., Hölttä, T., Bérody, S., et al. (2021). Management of congenital nephrotic syndrome: Consensus recommendations of the ERKNet-ESPN working group. Nat. Rev. Nephrol. 17 (4), 277–289. doi:10.1038/s41581-020-00384-1
Cèlia Badenas, M. P., Tazón, B., Heidet, L., Arrondel, C., Armengol, A., Andres, A., et al. (2002). Mutations in the COL4A4 and COL4A3 genes cause familial benign hematuria. J. Am. Soc. Nephrol. 13 (5), 1248–1254. doi:10.1681/ASN.V1351248
Chen, W., Perritt, A. F., Morissette, R., Dreiling, J. L., Bohn, M. F., Mallappa, A., et al. (2016). Ehlers-danlos syndrome caused by biallelic TNXB variants in patients with congenital adrenal hyperplasia. Hum. Mutat. 37 (9), 893–897. doi:10.1002/humu.23028
Deltas, C., Pierides, A., and Voskarides, K. (2013). Molecular genetics of familial hematuric diseases. Nephrol. Dial. Transpl. 28 (12), 2946–2960. doi:10.1093/ndt/gft253
Deng, S., Xu, H., Yuan, J., Xiao, J., Yuan, L., Deng, X., et al. (2016). Identification of a novel collagen type IV alpha-4 (COL4A4) mutation in a Chinese family with autosomal dominant Alport syndrome using exome sequencing. Indian J. Med. Res. 144 (2), 200–205. doi:10.4103/0971-5916.195026
Du, R., Liu, J., Hu, Y., Peng, S., Fan, L., Xiang, R., et al. (2022). Novel heterozygous mutation in COL4A4 responsible for Alport syndrome in a Chinese family. Front. Genet. 13, 899006. doi:10.3389/fgene.2022.899006
E D Adamson SEA (1979). The localization and synthesis of some collagen types in developing mouse embryos. Cell. 16 (4), 953–965. doi:10.1016/0092-8674(79)90110-7
Egging, D., van Vlijmen-Willems, I., van Tongeren, T., Schalkwijk, J., and Peeters, A. (2007). Wound healing in tenascin-X deficient mice suggests that tenascin-X is involved in matrix maturation rather than matrix deposition. Connect. Tissue Res. 48 (2), 93–98. doi:10.1080/03008200601166160
Fan, L. L., Liu, L., Luo, F. M., Du, R., Wang, C. Y., Dong, Y., et al. (2020). A novel heterozygous variant of the COL4A4 gene in a Chinese family with hematuria and proteinuria leads to focal segmental glomerulosclerosis and chronic kidney disease. Mol. Genet. Genomic Med. 8 (12), e1545. doi:10.1002/mgg3.1545
Gbadegesin, R. A., Brophy, P. D., Adeyemo, A., Hall, G., Gupta, I. R., Hains, D., et al. (2013). TNXB mutations can cause vesicoureteral reflux. J. Am. Soc. Nephrol. 24 (8), 1313–1322. doi:10.1681/ASN.2012121148
Guo, L., Li, D., Dong, S., Wan, D., Yang, B., and Huang, Y. (2017). Mutation analysis of COL4A3 and COL4A4 genes in a Chinese autosomal-dominant Alport syndrome family. J. Genet. 96 (2), 389–392. doi:10.1007/s12041-017-0786-7
Hirabayashi, Y., Katayama, K., Mori, M., Matsuo, H., Fujimoto, M., Joh, K., et al. (2022). Mutation analysis of thin basement membrane nephropathy. Genes. (Basel). 13 (10). doi:10.3390/genes13101779
Howard, B. V., Gunson, D., and Kefalides, N. A. (1976). Characterization of the collagen synthesized by endothelial cells in culture. Proc. Natl. Acad. Sci. U. S. A. 73 (7), 2361–2364. doi:10.1073/pnas.73.7.2361
Hudson, B. G. (2004). The molecular basis of goodpasture and Alport syndromes: Beacons for the discovery of the collagen IV family. J. Am. Soc. Nephrol. 15 (10), 2514–2527. doi:10.1097/01.ASN.0000141462.00630.76
Khoshnoodi, J., Pedchenko, V., and Hudson, B. G. (2008). Mammalian collagen IV. Microsc. Res. Tech. 71 (5), 357–370. doi:10.1002/jemt.20564
Korstanje, R., Caputo, C. R., Doty, R. A., Cook, S. A., Bronson, R. T., Davisson, M. T., et al. (2014). A mouse Col4a4 mutation causing Alport glomerulosclerosis with abnormal collagen alpha3alpha4alpha5(IV) trimers. Kidney Int. 85 (6), 1461–1468. doi:10.1038/ki.2013.493
Marino, R., Moresco, A., Perez Garrido, N., Ramirez, P., and Belgorosky, A. (2022). Congenital adrenal hyperplasia and ehlers-danlos syndrome. Front. Endocrinol. (Lausanne) 13, 803226. doi:10.3389/fendo.2022.803226
Mark Buzza, H. D., Wang, Y., Wilson, D., Babon, J. J., Cotton, R. G., and Savige, J. (2003). Mutations in the COL4A4 gene in thin basement membrane disease. Kidney Int. 63 (2), 447–453. doi:10.1046/j.1523-1755.2003.00780.x
Mehta, L., and Jim, B. (2017). Hereditary renal diseases. Semin. Nephrol. 37 (4), 354–361. doi:10.1016/j.semnephrol.2017.05.007
Merke, D. P., Chen, W., Morissette, R., Xu, Z., Van Ryzin, C., Sachdev, V., et al. (2013). Tenascin-X haploinsufficiency associated with Ehlers-Danlos syndrome in patients with congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 98 (2), E379–E387. doi:10.1210/jc.2012-3148
Pei, G., Yao, Y., Yang, Q., Wang, M., Wang, Y., Wu, J, et al. (2019). Lymphangiogenesis in kidney and lymph node mediates renal inflammation and fibrosis. Sci. Adv. 5 (6). doi:10.1126/sciadv.aaw5075
Savige, J., Storey, H., Watson, E., Hertz, J. M., Deltas, C., Renieri, A., et al. (2021). Consensus statement on standards and guidelines for the molecular diagnostics of Alport syndrome: Refining the ACMG criteria. Eur. J. Hum. Genet. 29 (8), 1186–1197. doi:10.1038/s41431-021-00858-1
Schalkwijk, J., Steijlen, P. M., Dean, W. B., Taylor, G., van Vlijmen, I. M., van Haren, B, et al. (2001). A recessive form of the Ehlers-Danlos syndrome caused by tenascin-X deficiency. N. Engl. J. Med. 345 (16), 1167–1175. doi:10.1056/NEJMoa002939
Smeets Vvk, H. J., van de Heuvel, L. P., Lemmink, H. H., Schröder, C. H., and Monnens, L. A. (1996). Hereditary disorders of the glomerular basement membrane. Pediatr. Nephrol. 10 (6), 779–788. doi:10.1007/s004670050217
Stapleton, C. P., Kennedy, C., Fennelly, N. K., Murray, S. L., Connaughton, D. M., Dorman, A. M., et al. (2020). An exome sequencing study of 10 families with IgA nephropathy. Nephron 144 (2), 72–83. doi:10.1159/000503564
Watanabe, S., Ito, Y., Samura, O., Nakano, H., Sawamura, D., Asahina, A., et al. (2021). Novel gross deletion mutation c.-105_4042+498del in the TNXB gene in a Japanese woman with classical-like ehlers-danlos syndrome: A case of uneventful pregnancy and delivery. J. Dermatol 48 (5), e227–e228. doi:10.1111/1346-8138.15837
Wu, Y., Hu, P., Xu, H., Yuan, J., Yuan, L., Xiong, W., et al. (2016). A novel heterozygous COL4A4 missense mutation in a Chinese family with focal segmental glomerulosclerosis. J. Cell. Mol. Med. 20 (12), 2328–2332. doi:10.1111/jcmm.12924
Xie, J., Wu, X., Ren, H., Wang, W., Wang, Z., Pan, X., et al. (2014). COL4A3 mutations cause focal segmental glomerulosclerosis. J. Mol. Cell. Biol. 6 (6), 498–505. doi:10.1093/jmcb/mju040
Xu, Y., Guo, M., Dong, H., Jiang, W., Ma, R., Liu, S., et al. (2016). A novel COL4A4 mutation identified in a Chinese family with thin basement membrane nephropathy. Sci. Rep. 6, 20244. doi:10.1038/srep20244
Yang, C., Song, Y., Chen, Z., Yuan, X., Chen, X., Ding, G., et al. (2019). A nonsense mutation in COL4A4 gene causing isolated hematuria in either heterozygous or homozygous state. Front. Genet. 10. doi:10.3389/fgene.2019.00628
Keywords: COL4A4, TNXB, heterozygous gene mutations, hereditary nephritis, renal biopsy
Citation: Zhu F, Li Y, Wang Y, Yao Y and Zeng R (2023) The same heterozygous Col4A4 mutation triggered different renal pathological changes in Chinese family members. Front. Genet. 14:1180149. doi: 10.3389/fgene.2023.1180149
Received: 07 March 2023; Accepted: 18 May 2023;
Published: 31 May 2023.
Edited by:
Xu-Jie Zhou, Peking University, ChinaReviewed by:
Fujun Lin, Xin Hua Hospital affiliated to Shanghai Jiao Tong University School of Medicine, ChinaYang Li, Peking University, China
Copyright © 2023 Zhu, Li, Wang, Yao and Zeng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rui Zeng, emVuZ3J1aUB0amgudGptdS5lZHUuY24=