 Yi-Dan Liu
Yi-Dan Liu Dan-Dan Tan
Dan-Dan Tan Dan-Yu Song1
Dan-Yu Song1 Yan-Bin Fan
Yan-Bin Fan Wei Wei
Wei Wei Hui Xiong
Hui Xiong- 1Department of Pediatrics, Peking University First Hospital, Beijing, China
- 2Beijing Kangso Medical Inspection Co., Ltd., Beijing, China
POMGNT1, encoding protein O-mannose beta-1,2-N-acetylglucosaminyltransferase 1, is one of the genes responsible for dystroglycanopathy (DGP), which includes multiple phenotypes such as muscle-eye-brain disease (MEB), congenital muscular dystrophy with intellectual disability, and limb-girdle muscular dystrophy Here, we report a case of MEB that is the result of a homozygous variant of POMGNT1 that is revealed through uniparental disomy (UPD). An 8-month-old boy was admitted with mental and motor retardation, hypotonia, esotropia, early onset severe myopia, and structural brain abnormalities. A panel testing of genetic myopathy-related genes was used to identify a homozygous c.636C>T (p.Phe212Phe) variant in exon 7 of POMGNT1 in the patient, a heterozygous c.636C>T variant in the father, and the wild type in the mother. Quantitative polymerase chain reaction (q-PCR) revealed no abnormal copy numbers in exon 7. Trio-based whole-exome sequencing (trio-WES) revealed a possible paternal UPD on chromosome 1 of the patient. Chromosomal microarray analysis (CMA) revealed a 120,451 kb loss of heterozygosity (LOH) on 1p36.33-p11.2, encompassing POMGNT1, and a 99,319 kb loss of heterozygosity on 1q21.2-q44, which indicated UPD. Moreover, RNA sequencing (RNA-seq) verified that the c.636C>T variant was a splice-site variant, leading to skipping of exon 7 (p.Asp179Valfs*23). In conclusion, to the best of our knowledge, we present the first case of MEB caused by UPD, providing valuable insights into the genetic mechanisms underlying this condition.
Introduction
Dystroglycanopathy (DGP) is a group of autosomal recessive muscular dystrophies caused by O-glycosylation defects in alpha-dystroglycans. It has three phenotypes based on severity: type A (severe forms of congenital muscular dystrophy [CMD] with brain and eye abnormalities, including Walker-Warburg syndrome [WWS], muscle-eye-brain disease [MEB], and Fukuyama CMD [FCMD]), type B (CMD with or without intellectual disability), and type C (a mild form of limb-girdle muscular dystrophy [LGMD]) (Song et al., 2021).
MEB (OMIM 253280) was first described by Santavuori et al. (1977) in Finland in 1977. Typical clinical features include profound hypotonia at birth, motor and cognitive developmental delays, and ocular abnormalities. In 2001, the first gene responsible for MEB, POMGNT1, was identified by Yoshida et al. (2001) in Japan. In recent years, we have reported novel POMGNT1 variants that cause MEB in Chinese patients and identified a novel copy number variation, g.6668-8257del of POMGNT1, as a founder mutation (Jiao et al., 2013; Fu et al., 2017). In our clinical and genetic analysis of a large cohort of Chinese patients with DGP, POMGNT1 is the most common gene responsible for MEB (Song et al., 2021).
Homozygous variants or compound heterozygous variants lead to autosomal recessive inherited diseases, and disease-causing variants conventionally originate from both parents. However, in rare cases, the variant from one parent is the only cause, such as in uniparental disomy (UPD). UPD is defined as two copies of a whole or partial chromosome derived from one parent. These two copies can be maternal or paternal. Maternal UPD is approximately twice as common as paternal UPD (Benn, 2021). Here, we report a case of MEB due to a homozygous variant in POMGNT1 revealed by paternal UPD.
Case description
Clinical data
An 8-month-old boy was admitted to the pediatric clinic of our hospital because of a “mental and motor developmental delay.”
He had a delay in motor development since birth and unsteadily raised his head at 4 months. He was unable to roll over, sit without support, or crawl at 8 months. He was unable to grip items on his own. He had delayed intellectual development. He could smile but could not babble. The parents had a non-consanguineous marriage, and no related disease was observed in the family history.
Physical examination revealed a head circumference of 43 cm, esotropia, hypotonia, and muscle weakness with limb muscle strength between level 3 and level 3+, predominantly proximal. The bilateral patellar tendon reflexes could not be induced. No pathological reflection of the Babinski’s sign was observed.
Serum creatine kinase (CK) levels were elevated (1799 IU/L). Brain magnetic resonance imaging (MRI) showed cerebellar and brain stem hypoplasia with a characteristic flattening, pachygyria-polymicrogyria of frontotemporal lobes (type “cobblestone pavement”), white matter lesions, and multiple subcortical cerebellar cysts (Figure 1A), which were typical brain changes in DGP (Borisovna et al., 2019). Examination of blood amino acids (aa) and urine organic acids revealed no significant abnormalities. Optic examination revealed extreme myopia.
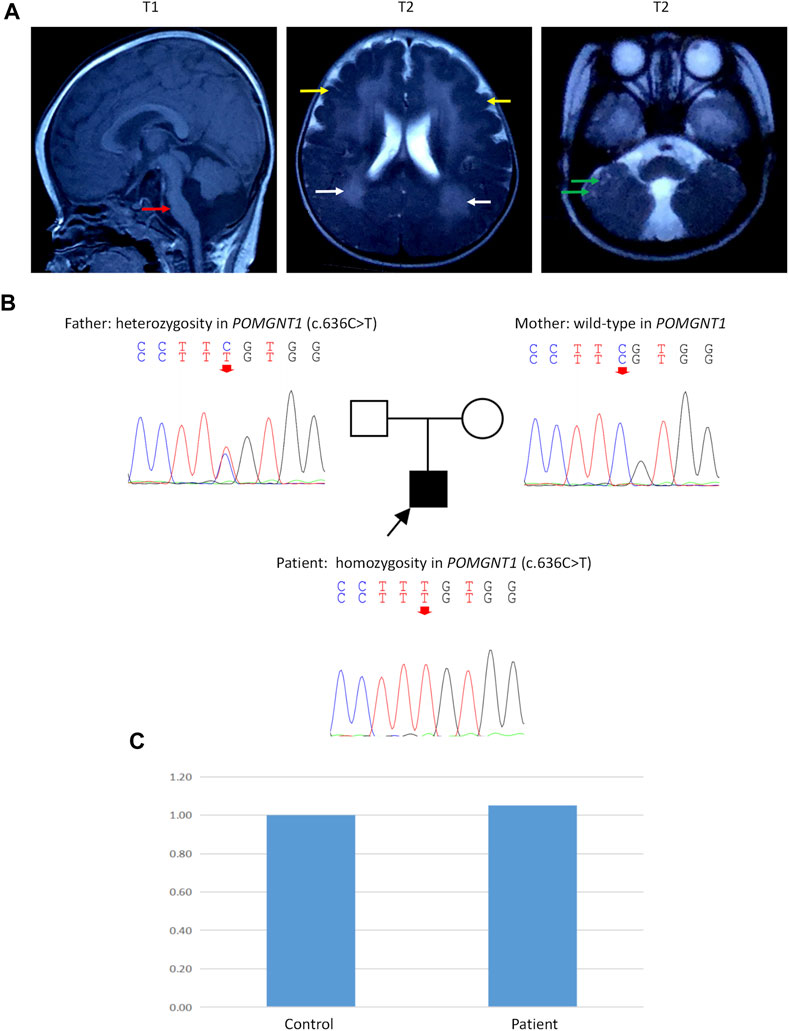
FIGURE 1. Brain magnetic resonance imaging (MRI), Sanger sequencing, and quantitative polymerase chain reaction (q-PCR). (A) Brain MRI of the patient at 8 months of age. T1: Cerebellar and brain stem hypoplasia with a characteristic flattening (red arrow). T2: Pachygyria-polymicrogyria of frontotemporal lobes (type “cobblestone pavement”) (yellow arrows) and white matter lesions (white arrows). T2: Multiple subcortical cerebellar cysts (green arrows). (B) Sanger sequencing showed a homozygous c.636C>T variant in exon 7 of the POMGNT1 gene (NM_001243766) in the patient. His father had a heterozygous c.636C>T variant, and his mother had no variant in this site. (C) Q-PCR of DNA showed a normal copy number in exon 7 of POMGNT1 in the patient.
He achieved rolling over, sitting without support, and standing with support at approximately 1 year of age. He was able to crawl at the age of 2. When he was 3 years old, he could walk several steps with support. After 3 years of age, there was no significant improvement in motor development. At the last follow-up, the boy was 6 years old and was unable to walk independently. He could say “baba” and “mama” but no other words. He wore corrective glasses for extreme myopia (the uncorrected visual acuity was between 0.01 and 0.05).
Genetic findings
Peripheral blood samples were collected from the patient and his parents after obtaining informed consent. A panel testing of genetic myopathy-related genes was first performed, and the candidate variants were verified using Sanger sequencing. The gene panel testing identified a homozygous c.636C>T (p.Phe212Phe) variant in exon 7 of the POMGNT1 gene (NM_001243766) in the patient, and the pathogenicity of this variant has been reported previously (Bouchet et al., 2007). Sanger sequencing revealed that the father had a heterozygous c.636C>T variant, but the mother had no variant at this site (Figure 1B).
Then, quantitative polymerase chain reaction (q-PCR) of DNA was used to identify whether there was an abnormal copy number in exon 7 of POMGNT1 in the patient. However, no abnormalities were found (Figure 1C), suggesting that there may be another cause of the homozygous variant. Next, trio-based whole-exome sequencing (trio-WES) was performed to identify other variants and UPD. As a result, a possible paternal UPD on chromosome 1 of the patient was identified using trio-WES data analysis (Figure 2), which revealed the homozygous variant, and no other pathogenic variants were detected. Finally, chromosomal microarray analysis (CMA) of the patient revealed a 120,451 kb loss of heterozygosity (LOH) on 1p36.33-p11.2 (888,658_121,339,317), encompassing POMGNT1, and a 99,319 kb LOH on 1q21.2-q44 (149,879,544_249,198,164) (Figure 3A). Thus, UPD was ascertained.

FIGURE 2. Analysis of uniparental disomy (UPD) using trio-based whole-exome sequencing (trio-WES). It showed that paternal UPD existed on chromosome 1. UI_P: Uniparental Paternal Isodisomy. UA_P: Uniparental Paternal Heterdisomy. UI_M: Uniparental Maternal Isodisomy. UA_M: Uniparental Maternal Heterdisomy. BPI: Bi-Parental Inheritance. Het: Heterozygous. Hom: Homozygous.
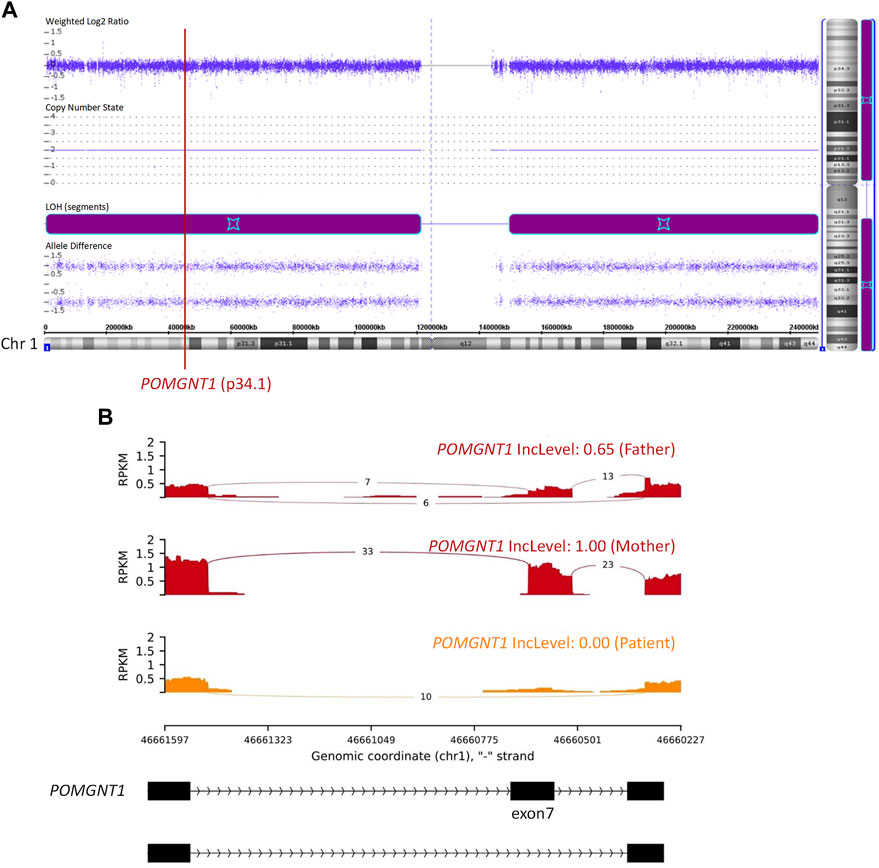
FIGURE 3. Chromosomal microarray analysis (CMA) and RNA sequencing (RNA-seq). (A) CMA showed a 120,451 kb loss of heterozygosity (LOH) on 1p36.33-p11.2 (888,658_121,339,317) and a 99,319 kb LOH on 1q21.2-q44 (149,879,544_249,198,164). The former encompassed the POMGNT1 gene. (B) RNA-seq showed skipped exon 7 in the POMGNT1 mRNA of the patient and the father. RPKM: Reads Per Kilobase per Million mapped reads. IncLevel: Inclusion Level.
The c.636C>T (p.Phe212Phe) variant is synonymous. RNA sequencing (RNA-seq) of peripheral blood samples was performed to determine whether splicing was affected. It was confirmed to cause the skipping of exon 7 in the POMGNT1 mRNA (Figure 3B), resulting in a frameshift change (p.Asp179Valfs*23). Based on the ACMG guidelines (Richards et al., 2015), c.636C>T is considered a pathogenic variant.
Discussion
MEB is a phenotype of DGP, which is characterized by hypotonia at birth, brain structural abnormalities, and ocular malformations (Jiao et al., 2013). Considering the typical symptoms in the muscles, eyes, and brain of the patient, making a diagnosis of MEB is clear.
POMGNT1 is the most common gene responsible for MEB (Song et al., 2021). A homozygous c.636C>T (p.Phe212Phe) variant in exon 7 of POMGNT1 was found in the patient. However, only his father had a heterozygous c.636C>T variant and his mother was the wild type. Initially, copy number variation (CNV), a de novo variant, or UPD were considered. Then, the result of q-PCR excluded CNV of exon 7 on DNA level. Finally, paternal UPD on chromosome 1 was confirmed by using trio-WES and CMA. RNA-seq revealed skipped exon 7 in the patient and the father on mRNA level. RNA-seq also showed a lower RPKM (Reads Per Kilobase per Million mapped reads) value of the father and the patient than the mother. For the three samples of this family were sequenced at the same time and the sequencing depth was standardized in calculation, the lower RPKM value of the father and the patient may be caused by nonsense-mediated mRNA decay.
The POMGNT1 protein contains 660 aa and has four domains: an N-terminal cytoplasmic tail (aa 1–37), a transmembrane domain (aa 38–58), a stem domain (aa 59–300), and a catalytic domain (aa 301–660) (Akasaka-Manya et al., 2004). The c.636C>T (p.Asp179Valfs*23) variant found in the present study leads to the loss of part of the stem domain and all of the catalytic domain. In previous studies, the c.636C>T variant was reported in fetal forms of type II lissencephaly, a severe clinical presentation of CMD and in an 8-year-old girl presenting CMD with clinical features compatible with MEB or severe FCMD (Bouchet et al., 2007; Oliveira et al., 2008). Different from the patient in the present study, the 8-year-old girl with a homozygous c.636C>T variant in the literature had epilepsy in addition to hypotonia, visual impairment, and brain structure abnormalities (Oliveira et al., 2008).
UPD occurs with an estimated overall prevalence of 1 in 2,000 births (Nakka et al., 2019), not all of which causes phenotypic consequences. UPD can lead to clinical presentation when it involves disrupting imprinting (chromosomes 6, 7, 11, 14, 15, and 20) and uncovering recessive alleles in blocks of isodisomy (Nakka et al., 2019; Benn, 2021). UPD for any chromosome is associated with an increased risk for a recessive disorder since it can result in an affected child when only one parent is a carrier of a pathogenic variant (Del Gaudio et al., 2020). LOH is one mechanism that leads to UPD. Somatic cell recombination between chromatids results in two populations of cells with reciprocal segments with LOH (Benn, 2021). In clinical practice, UPD can typically be ascertained using CMA testing (Del Gaudio et al., 2020). Computational algorithms can also be used to detect UPD greater than 10 Mb from trio exome or genome sequencing data (King et al., 2014; Magi et al., 2014; Bis et al., 2017; Del Gaudio et al., 2020). In the present study, both CMA and trio-WES were employed to identify UPD.
Among DGP that has 18 responsible genes, only two cases associated with UPD have been reported previously. A case of LGMD with a variant in POMT2 and a case of a milder phenotype with a variant in B3GALNT2 caused by UPD was reported in 2018 and 2022, respectively (Brun et al., 2018; D'Haenens et al., 2022).
In conclusion, to the best of our knowledge, we present the first case of MEB caused by UPD, providing valuable insights into the genetic mechanisms underlying this condition.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The study was approved by the Ethics Committee of Peking University First. Hospital, and the patient gave written informed consent before the data were obtained. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
Y-DL, D-DT, and D-YS collected the data. HX, Y-BF, X-NF, and LG participated in the diagnosis of the patient. WW provided technical support. Y-DL wrote the paper. HX designed the present study and revised the paper. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the Natural Science Foundation of Beijing Municipality (No. 7212116), National Natural Science Foundation of China (No. 82171393), National High Level Hospital Clinical Research Funding (High Quality Clinical Research Project of Peking University First Hospital, no. 2022CR69), National Key Research and Development Program of China (No. 2016YFC0901505) and Beijing Key Laboratory of Molecular Diagnosis and Study on Pediatric Genetic Diseases (No. BZ0317).
Conflict of interest
WW was empolyed by Beijing Kangso Medical Inspection Co., Ltd.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Akasaka-Manya, K., Manya, H., Kobayashi, K., Toda, T., and Endo, T. (2004). Structure-function analysis of human protein O-linked mannose beta1,2-N-acetylglucosaminyltransferase 1, POMGnT1. Biochem. Biophys. Res. Commun. 320, 39–44. doi:10.1016/j.bbrc.2004.05.129
Benn, P. (2021). Uniparental disomy: Origin, frequency, and clinical significance. Prenat. Diagn. 41, 564–572. doi:10.1002/pd.5837
Bis, D. M., Schule, R., Reichbauer, J., Synofzik, M., Rattay, T. W., Soehn, A., et al. (2017). Uniparental disomy determined by whole-exome sequencing in a spectrum of rare motoneuron diseases and ataxias. Mol. Genet. Genomic Med. 5, 280–286. doi:10.1002/mgg3.285
Borisovna, K. O., Yurievna, K. A., Yurievich, T. K., Igorevna, K. O., Olegovich, K. D., Igorevna, D. A., et al. (2019). Compound heterozygous POMGNT1 mutations leading to muscular dystrophy-dystroglycanopathy type A3: A case report. BMC Pediatr. 19, 98. doi:10.1186/s12887-019-1470-2
Bouchet, C., Gonzales, M., Vuillaumier-Barrot, S., Devisme, L., Lebizec, C., Alanio, E., et al. (2007). Molecular heterogeneity in fetal forms of type II lissencephaly. Hum. Mutat. 28, 1020–1027. doi:10.1002/humu.20561
Brun, B. N., Willer, T., Darbro, B. W., Gonorazky, H. D., Naumenko, S., Dowling, J. J., et al. (2018). Uniparental disomy unveils a novel recessive mutation in POMT2. Neuromuscul. Disord. 28, 592–596. doi:10.1016/j.nmd.2018.04.003
D'Haenens, E., Vergult, S., Menten, B., Dheedene, A., Kooy, R. F., and Callewaert, B. (2022). Expanding the phenotype of B3GALNT2-related disorders. Genes (Basel) 13. doi:10.3390/genes13040694
Del Gaudio, D., Shinawi, M., Astbury, C., Tayeh, M. K., Deak, K. L., Raca, G., et al. (2020). Diagnostic testing for uniparental disomy: A points to consider statement from the American college of medical genetics and genomics (ACMG). Genet. Med. 22, 1133–1141. doi:10.1038/s41436-020-0782-9
Fu, X., Yang, H., Jiao, H., Wang, S., Liu, A., Li, X., et al. (2017). Novel copy number variation of POMGNT1 associated with muscle-eye-brain disease detected by next-generation sequencing. Sci. Rep. 7, 7056. doi:10.1038/s41598-017-07349-8
Jiao, H., Manya, H., Wang, S., Zhang, Y., Li, X., Xiao, J., et al. (2013). Novel POMGnT1 mutations cause muscle-eye-brain disease in Chinese patients. Mol. Genet. Genomics 288, 297–308. doi:10.1007/s00438-013-0749-5
King, D. A., Fitzgerald, T. W., Miller, R., Canham, N., Clayton-Smith, J., Johnson, D., et al. (2014). A novel method for detecting uniparental disomy from trio genotypes identifies a significant excess in children with developmental disorders. Genome Res. 24, 673–687. doi:10.1101/gr.160465.113
Magi, A., Tattini, L., Palombo, F., Benelli, M., Gialluisi, A., Giusti, B., et al. (2014). H3M2: Detection of runs of homozygosity from whole-exome sequencing data. Bioinformatics 30, 2852–2859. doi:10.1093/bioinformatics/btu401
Nakka, P., Pattillo Smith, S., O'Donnell-Luria, A. H., McManus, K. F., andMe Research, T., Mountain, J. L., et al. (2019). Characterization of prevalence and health consequences of uniparental disomy in four million individuals from the general population. Am. J. Hum. Genet. 105, 921–932. doi:10.1016/j.ajhg.2019.09.016
Oliveira, J., Soares-Silva, I., Fokkema, I., Goncalves, A., Cabral, A., Diogo, L., et al. (2008). Novel synonymous substitution in POMGNT1 promotes exon skipping in a patient with congenital muscular dystrophy. J. Hum. Genet. 53, 565–572. doi:10.1007/s10038-008-0263-5
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Santavuori, P., Leisti, J., and Kruus, S. J. N. (1977). Muscle, eye and brain disease. A New Syndr. 8, 553.
Song, D., Dai, Y., Chen, X., Fu, X., Chang, X., Wang, N., et al. (2021). Genetic variations and clinical spectrum of dystroglycanopathy in a large cohort of Chinese patients. Clin. Genet. 99, 384–395. doi:10.1111/cge.13886
Keywords: uniparental disomy (UPD), muscle-eye-brain disease (MEB), PomGnT1, dystroglycanopathy (DGP), splice-site variant
Citation: Liu Y-D, Tan D-D, Song D-Y, Fan Y-B, Fu X-N, Ge L, Wei W and Xiong H (2023) Uniparental disomy for chromosome 1 with POMGNT1 splice-site variant causes muscle-eye-brain disease. Front. Genet. 14:1170089. doi: 10.3389/fgene.2023.1170089
Received: 20 February 2023; Accepted: 25 May 2023;
Published: 05 June 2023.
Edited by:
Xin-Ming Shen, Mayo Clinic, United StatesReviewed by:
Hoi Shan Sophelia Chan, The University of Hong Kong, Hong Kong SAR, ChinaRuxu Zhang, Central South University, China
Copyright © 2023 Liu, Tan, Song, Fan, Fu, Ge, Wei and Xiong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Xiong, eGhfYmpiakAxNjMuY29t