 Karin Writzl
Karin Writzl Blaž Mavčič2,3
Blaž Mavčič2,3 Aleš Maver
Aleš Maver Borut Peterlin
Borut Peterlin
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet. , 18 July 2023
Sec. Genetics of Common and Rare Diseases
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1167054
The NONO gene encodes a nuclear protein involved in transcriptional regulation, RNA synthesis and DNA repair. Hemizygous loss-of function, de novo or maternally inherited variants in NONO have been associated with an X-linked syndromic intellectual developmental disorder-34 (OMIM # 300967), characterized by developmental delay, intellectual disability, hypotonia, macrocephaly, elongated face, structural abnormalities of corpus callosum and/or cerebellum, congenital heart defect and left ventricular non-compaction cardiomyopathy. Few patients have been described in the literature and the phenotype data are limited. We report a 17-year-old boy with dolihocephaly, elongated face, strabismus, speech and motor delay, intellectual disability, congenital heart defect (ASD, VSD and Ebstein’s anomaly), left ventricular non-compaction cardiomyopathy, bilateral inguinal hernia and cryptorchidism. Additional features included recurrent fractures due to multiple non-ossifying fibromas, thrombocytopenia, and renal anomalies. Exome sequencing revealed a de novo pathogenic variant (NM_001145408.2: c.348+2_ 348+15del) in intron 5 of the NONO gene. Renal anomalies and thrombocytopenia have been rarely reported in patients with NONO—X-linked intellectual disability syndrome, while recurrent fractures due to multiple non-ossifying fibromas have not previously been associated with this syndrome. The phenotypic spectrum of NONO—X-linked intellectual disability syndrome may be broader than currently known.
The NONO-associated X-linked syndromic intellectual developmental disorder-34 (NONO-XLID) is a rare genetic disorder first described 8 years ago (Mircsof et al., 2015). It is caused by hemizygous loss-of-function variants in the non-POU domain containing, octamer-biding gene, NONO, which encodes an RNA and DNA binding protein involved in RNA synthesis, transcriptional regulation and DNA repair (Mircsof et al., 2015). The disorder is characterized by developmental delay, intellectual disability, macrocephaly and distinctive facial features, structural brain and heart anomalies and left ventricular non-compaction cardiomyopathy. Skeletal abnormalities were rarely described in patients and included kyphoscoliosis and planovalgus but not non-ossifying fibromas (Sewani et al., 2020).
Non-ossifying fibroma (NOF) is the most common bone tumor, thought to affect around 30%–40% of children and adolescents (Choi and Ro, 2021). It is a self-limiting benign tumor, usually diagnosed as an incidental finding on X-rays, that usually regresses after puberty (Choi and Ro, 2021).
Here we report a patient with a diagnosis of NONO-XLID, with characteristic clinical features confirmed by the presence of a germline pathogenic variant in NONO, who also suffered recurrent fractures due to multiple non-ossifying fibromas not previously reported in these patients, and thrombocytopenia and renal anomalies rarely reported patients with NONO-XLID. This has implications for clinical evaluation and management of patients with NONO-XLID.
The patient was a 17-year-old boy of Caucasian European origin, the second child of healthy non-consanguineous parents, who had an older healthy sister and a younger healthy brother. Family history is unremarkable. During pregnancy, hydronephrosis was noted. He was born at 38 weeks of gestation, after a normal vaginal delivery. Birth weight was 3.0 kg (−0.78 z), length 50 cm (−0.64 z), and head circumference 36 cm (0.98 z). Apgar 9/9. Shortly after birth, abdominal ultrasound revealed bilateral kidney dysplasia with bilateral pyeloureteral stenosis and vesicoureteral reflux. Echocardiogram revealed atrial septal defect (ASD), ventricular septal defect (VSD), Ebstein anomaly and spongoformic cardiomyopathy. Brain ultrasound revealed hypoplastic corpus callosum. He was noted to have thrombocytopenia.
His development was delayed; he started to walk at 4 years and spoke his first words at 6 years. He was toilet trained at 6.5 years. The patient’s history also included hearing loss, strabismus, and surgically-corrected bilateral inguinal hernia and cryptorchidism at the age of 2 years.
At the age of 14, he fell from a standing height and was diagnosed with a fracture of the distal diaphysis of the radius and ulna of the left upper limb. The fractures were treated conservatively. X-ray showed osteolytic changes in the distal part of the left radius (Figure 1). After 18 months, he suffered a second fracture in the same area. This time, X-ray imaging of the entire skeleton was performed, which, in addition to osteolytic changes in the area of the left radius, also showed osteolytic changes in the proximal part of the left fibula with fibrous changes after previously unnoted fracture and osteolytic changes in the distal part of the right and left femur (Figure 2). At the age of 16, he underwent MRI of the left lower extremity, which showed a benign lesion on the proximal fibula with a non-aggressive appearance, defined as a non-ossifying fibroma (NOF) or fibrous dysplasia. A biopsy was not performed. Laboratory tests showed the serum levels of calcium, inorganic phosphate, alkaline phosphatase, free T4 and T3, TSH, and PTH, were all normal. Bone mineral density was in the lower range of normal values.

FIGURE 1. (A) Trauma anteroposterior radiograph of the right upper extremity showing a fracture through lesion in the distal radius and a fracture of ulna. (B) Trauma anteroposterior radiograph showing second fracture in the same area 18 months later. (C,D) Anteroposterior radiograph of the upper extremity 25 months (C) and 31 months (D) after the first injury showing united fracture. Fractures are indicated by arrows and non-ossifying fibromas by asterisks.
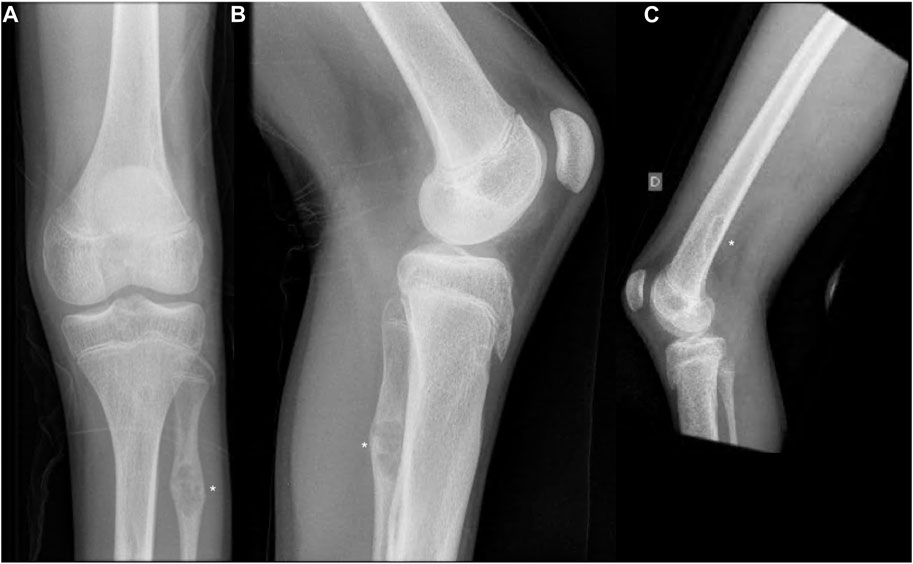
FIGURE 2. (A,B) Anteroposterior and lateral radiographs of the left knee showing non-ossifying fibroma in the distal femur and proximal fibula. (C) Lateral radiographs of the right knee showing non-ossifying fibroma in the distal femur. Non-ossifying fibromas are indicated by asterisks.
At the last examination, at the age of 16 years, his height was 178.5 cm (0.25 z), weight was 43 kg (−4.37 z), head circumference was 58 cm (1.14 z). His physical exam demonstrated dolichocephaly, long face, strabismus, prominent nose, retrognathia, long, thin fingers with partial skin syndactyly of the second, third and fourth fingers and proximal interphalangeal flexion contractions, long toes, planus valgus, and thoracic scoliosis.
He had a moderate intellectual disability. He had regular medical check-ups at cardiology, nephrology, hematology and orthopaedic clinics.
The ASD and VSD spontaneously closed. He had Ebstein’s anomaly with moderate tricuspid valve regurgitation and left ventricular noncompaction cardiomyopathy with a normal-sized left ventricle with slightly impaired function.
He had bilateral renal dysplasia, with the right kidney being markedly more affected than the left. At the age of 6 years, the right kidney was already virtually non-functional and contributed about 5% of the total function. There was pyeloureteral stenosis on the right side and grade 2 vesicoureteral reflux, while the reflux on the left side had resolved spontaneously. Abdominal ultrasound examination at the age of 16 years revealed that the right kidney was smaller, measuring about 9 cm, with a uniformly thinned parenchyma measuring about 7 mm. The left kidney measured about 10, 5 cm, with reasonably wide parenchyma (about 15 mm).
He was followed up at the haematology clinic for thrombocytopenia, which was classified as mild [Platelets = 104 10^9/L (150–410^9/L)] at the last follow-up at the age of 16 years. The complete blood count with differential did not reveal any additional abnormalities.
He unexpectedly died during sleep at the age of 17.
Trio Exome sequencing (ES) was performed on genomic DNA of the patient and his parents using the Illumina Nextera Rapid Capture Expanded exome kit for exome enrichment. Sequencing was performed on the Illumina NextSeq 550 sequencer and the data analysed as previously described (Bergant et al., 2018). A median coverage of 211x was attained across exonic regions and 99.9% of exonic regions were covered with a minimum 20x coverage. Briefly, the data was analysed using a pipeline based on bwa alignment and variant calling in accordance with GATK best practice guidelines. The functional consequences of the resulting variants were predicted using the snpEff v.5.1 software and variants were annotated with bcftools 1.9 using the frequency data from the gnomAD exomes project v.2.1.1 and dbNSFP v3.0 resource (DePristo et al., 2011). We identified a novel de novo hemizygous splice-site variant NM_001145408.2: c.348+2_ 348+15del in NONO (PS2_MOD). The genome reference nomenclature of the variant was NC_000023.10:g.70511824_70511837del. The variant was absent from databases (gnomAD and the in-house population database) (PM2_SUP), was predicted to alter or cause deletion of the canonical sequence of the donor site adjacent to exon 5, possibly leading to the loss of functional protein product [NC_000023.10(NM_001145408.2):r.spl, NC_000023.10(NP_001138880.1):p.?, PVS1] and was classified as a pathogenic variant in accordance with the ACMG criteria (PVS1, PS2_MOD, PM2_SUP, Richards et al. (2015). In the classification process, the strength of PVS1 evidence was rated as very strong based on the published recommendations (Abou Tayoun et al., 2018), as the variant impacts a canonical splice site adjacent to a non-symmetric exon 5. Furthermore, the PS2 was weighted as moderate evidence in support of pathogenicity based on the ClinGen’s recommendations, due to a consistent albeit non-specific clinical presentation in the patient (ClinGen SVI recommendations for De novo criteria, version 1.1, available from https://clinicalgenome.org/working-groups/sequence-variant-interpretation/). Furthermore, the weight of PM2 was downgraded to PM2_SUP in line with the recent recommendations of the ClinGen SVI working group (Recommendation for Absence/Rarity Criterion PM2, version 1.0, available from https://clinicalgenome.org/working-groups/sequence-variant-interpretation/).
Twenty-five patients with pathogenic variants of the NONO gene have been reported so far, of which 7 were prenatal cases (Mircsof et al., 2015; Reinstein et al., 2016; Scott et al., 2017; Carlston et al., 2019; Sun et al., 2020a; Sun et al., 2020b; Sewani et al., 2020; Coetzer and Moosa, 2022; Safi et al., 2022; Roessler et al., 2023). While in the prenatal period, the main clinical features are congenital heart defects and/or cardiomyopathy (7/7; 100%), the core phenotype in the postnatal period is developmental delay/intellectual disability (13/13; 100%), macrocephaly, corpus callosum anomalies (10/11; 91%), facial dysmorphic features, congenital heart defect (10/11; 91%) and left ventricular non-compaction cardiomyopathy (8/11; 73%). The patient presented here had all of these core phenotype clinical features. In addition, he had congenital thrombocytopenia, previously reported in three patient and renal abnormalities, previously described in a fetus that miscarried at 16 weeks and was found at autopsy to have renal agenesis Sewani et al., 2020. Although previously rarely reported, both congenital thrombocytopenia and renal anomalies are likely to be part of the clinical picture of patients with NONO-XLID and should be checked for.
Non-ossifying fibromas (NOFs) have not been previously reported in patients with NONO-XLID. The presented patient suffered recurrent fractures due to multiple NOFs at atypical locations. NOFs are benign tumours that typically appear in adolescence and then resolve. In less than 5%, NOFs are multifocal. The principal locations of NOF are metaphysis of the distal femur, proximal and distal tibia, but can also be found in the proximal humerus, fibula and distal radius (Mankin et al., 2009; Sakamoto et al., 2017). Pathologic fractures may occur with non-physiological loading or low-impact injuries and have been reported to occur in up to 20% of NOFs (Friedrich et al., 2021; Emori et al., 2022).
Multiple NOFs, with atypical location and fracture propensity, have been previously described in patients with neurofibromatosis type 1, and multifocal NOF have also been reported in patients with Jaffe-Campanacci syndrome and Oculoectodermal Syndrome (Peacock et al., 2015; Vannelli et al., 2020; Friedrich et al., 2021). All these disorders are known as RASopathies and are caused by germline or post-zygotic mosaic mutations in genes encoding RAS/MAPK signaling pathways components. Recently, NOF has been defined as a genetically driven benign neoplasm, caused by activation of Ras-MAPK signaling by somatic mutation in KRAS, FGFR1 and NF1 genes (Baumhoer et al., 2019), which also provides a causal genetic explanation for the occurrence of NOFs in RASopathies.
NONO-XLID is caused by hemizygeous pathogenic loss of function variants in the NONO gene and functional studies in patients showed loss of the NONO protein in patients’ cells (Mircsof et al., 2015; Roessler et al., 2023). NONO protein plays an important role in human tumorigenesis as either oncogene or tumour suppressor and is involved in many biological processes including cell proliferation, apoptosis, migration, and DNA damage repair (Feng et al., 2020). It has been suggested to play an important role in esophageal squamous cell carcinoma tumorigenesis by activation of the Erk1/2/MAPK pathway (Cheng et al., 2018) and is overexpressed in most cancers, while also lower NONO levels promote tumorigenesis in certain cancers (Pavao et al., 2001).
In summary, we report a patient with a diagnosis of NONO-XLID who suffered recurrent fractures due to multiple non-ossifying fibromas. The occurrence of multiple NOFs in unusual locations and recurrent fracturing, which is very rare in the general population, and the association of the NONO gene with tumourigenesis support the possibility that NOFs could be part of the clinical picture of NONO-XLID. In any case, the description of NOFs in a larger number of patients with NONO-XLID will be needed to confirm this. We suggest that patients with a NONO pathogenic variant who suffer low-force fractures should undergo skeletal X-ray imaging and a targeted search for lytic areas in the skeleton.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/clinvar/variation/2429456/, submission name: SUB12837649; submission number: SCV003799180.
Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Patient workup: KW and BM. Genetic analysis: AH and AM. Drafted the manuscript: KW, BM, AM, and BP. Final approval of the version to be published: KW, BM, AM, AH, and BP. Agreement to be accountable for all aspects of the work: KW, BM, AM, AH, and BP. All authors contributed to the article and approved the submitted version.
This work was funded by the ARRS programme: P3-0326.
The authors wish to thank the family for their collaboration.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
ASD, atrial septal defect; NOF, non-ossifying fibroma; PTH, parathyroid hormone; RAS/MAPK pathway, RAS/Mitogen Activated Protein Kinase pathway; T3, triiodothyronine; T4, thyroxine; TSH, thyroid stimulating hormone; VSD, ventricular septal defect; XLID, X-linked intellectual developmental disorder.
Abou Tayoun, A. N., Pesaran, T., DiStefano, M. T., Oza, A., Rehm, H. L., Biesecker, L. G., et al. (2018). Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 39, 1517–1524. doi:10.1002/humu.23626
Baumhoer, D., Kovac, M., Sperveslage, J., Ameline, B., Strobl, A., Krause, A., et al. (2019). Activating mutations in the MAP-kinase pathway define non-ossifying fibroma of bone. J. Pathol. 248, 116–122. doi:10.1002/path.5216
Bergant, G., Maver, A., Lovrecic, L., Čuturilo, G., Hodzic, A., and Peterlin, B. (2018). Comprehensive use of extended exome analysis improves diagnostic yield in rare disease: A retrospective survey in 1,059 cases. Genet. Med. 20, 303–312. doi:10.1038/gim.2017.142
Carlston, C. M., Bleyl, S. B., Andrews, A., Meyers, L., Brown, S., Bayrak-Toydemir, P., et al. (2019). Expanding the genetic and clinical spectrum of the NONO-associated X-linked intellectual disability syndrome. Am. J. Med. Genet. 179, 792–796. doi:10.1002/ajmg.a.61091
Cheng, R., Zhu, S., Guo, S., Min, L., Xing, J., Guo, Q., et al. (2018). Downregulation of NONO induces apoptosis, suppressing growth and invasion in esophageal squamous cell carcinoma. Oncol. Rep. 39, 2575–2583. doi:10.3892/or.2018.6334
Choi, J. H., and Ro, J. Y. (2021). The 2020 WHO classification of tumors of bone: An updated review. Adv. Anatomic Pathology 28, 119–138. doi:10.1097/PAP.0000000000000293
Coetzer, K. C., and Moosa, S. (2022). Novel hemizygous loss-of-function variant in NONO identified in a South African boy. Am. J. Med. Genet. Part A 188, 373–376. doi:10.1002/ajmg.a.62509
DePristo, M. A., Banks, E., Poplin, R., Garimella, K. V., Maguire, J. R., Hartl, C., et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498. doi:10.1038/ng.806
Emori, M., Tsuchie, H., Teramoto, A., Shimizu, J., Mizushima, E., Murahashi, Y., et al. (2022). Non-ossifying fibromas and fibrous cortical defects around the knee - an epidemiologic survey in a Japanese pediatric population. BMC Musculoskelet. Disord. 23, 378. doi:10.1186/s12891-022-05330-9
Feng, P., Li, L., Deng, T., Liu, Y., Ling, N., Qiu, S., et al. (2020). NONO and tumorigenesis: More than splicing. J. Cell. Mol. Med. 24, 4368–4376. doi:10.1111/jcmm.15141
Friedrich, R. E., Zustin, J., Luebke, A. M., Rosenbaum, T., Gosau, M., Hagel, C., et al. (2021). Neurofibromatosis type 1 with cherubism-like phenotype, multiple osteolytic bone lesions of lower extremities, and alagille-syndrome: Case report with literature. Surv. Vivo 35, 1711–1736. doi:10.21873/invivo.12431
Mankin, H. J., Trahan, C. A., Fondren, G., and Mankin, C. J. (2009). Non-ossifying fibroma, fibrous cortical defect and jaffe–campanacci syndrome: A biologic and clinical review. Musculoskelet. Surg. 93, 1–7. doi:10.1007/s12306-009-0016-4
Mircsof, D., Langouët, M., Rio, M., Moutton, S., Siquier-Pernet, K., Bole-Feysot, C., et al. (2015). Mutations in NONO lead to syndromic intellectual disability and inhibitory synaptic defects. Nat. Neurosci. 18, 1731–1736. doi:10.1038/nn.4169
Pavao, M., Huang, Y.-H., Hafer, L. J., Moreland, R. B., and Traish, A. M. (2001). Immunodetection of nmt55/p54nrbisoforms in human breast cancer. BMC Cancer 1, 15. doi:10.1186/1471-2407-1-15
Peacock, J. D., Dykema, K. J., Toriello, H. V., Mooney, M. R., Scholten, D. J., Winn, M. E., et al. (2015). Oculoectodermal syndrome is a mosaic RASopathy associated with KRAS alterations. Am. J. Med. Genet. 167, 1429–1435. doi:10.1002/ajmg.a.37048
Reinstein, E., Tzur, S., Cohen, R., Bormans, C., and Behar, D. M. (2016). Intellectual disability and non-compaction cardiomyopathy with a de novo NONO mutation identified by exome sequencing. Eur. J. Hum. Genet. 24, 1635–1638. doi:10.1038/ejhg.2016.72
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Roessler, F., Beck, A. E., Susie, B., Tobias, B., Begtrup, A., Biskup, S., et al. (2023). Genetic and phenotypic spectrum in the NONO-associated syndromic disorder. Am. J. Med. Genet. Part A 191A, 469–478. doi:10.1002/ajmg.a.63044
Safi, S., Sanders, S. P., Zhao, M., and Carreon, C. K. (2022). Biventricular non-compaction cardiomyopathy and tricuspid hypoplasia in a novel non-POU domain-containing octamer-binding gene variant. Cardiol. Young 32, 1–5. doi:10.1017/S1047951121004923
Sakamoto, A., Arai, R., Okamoto, T., and Matsuda, S. (2017). Non-ossifying fibromas: Case series, including in uncommon upper extremity sites. World J. Orthop. 8, 561. doi:10.5312/wjo.v8.i7.561
Scott, D. A., Hernandez-Garcia, A., Azamian, M. S., Jordan, V. K., Kim, B. J., Starkovich, M., et al. (2017). Congenital heart defects and left ventricular non-compaction in males with loss-of-function variants in NONO. J. Med. Genet. 54, 47–53. doi:10.1136/jmedgenet-2016-104039
Sewani, M., Nugent, K., Blackburn, P. R., Tarnowski, J. M., Hernandez-Garcia, A., Amiel, J., et al. (2020). Further delineation of the phenotypic spectrum associated with hemizygous loss-of-function variants in NONO. Am. J. Med. Genet. 182, 652–658. doi:10.1002/ajmg.a.61466
Sun, H., Han, L., Zhang, X., Hao, X., Zhou, X., Pan, R., et al. (2020a). Case report: Characterization of a novel NONO intronic mutation in a fetus with X-linked syndromic mental retardation-34. Front. Genet. 11, 593688. doi:10.3389/fgene.2020.593688
Sun, H., Zhou, X., Hao, X., Zhang, Y., Zhang, H., and He, Y. (2020b). Characteristics of cardiac phenotype in prenatal familial cases with NONO mutations. Circ Genomic Precis. Med. 13, e002847. doi:10.1161/CIRCGEN.119.002847
Vannelli, S., Buganza, R., Runfola, F., Mussinatto, I., Andreacchio, A., and de Sanctis, L. (2020). Jaffe-campanacci syndrome or neurofibromatosis type 1: A case report of phenotypic overlap with detection of NF1 gene mutation in non-ossifying fibroma. Ital. J. Pediatr. 46, 58. doi:10.1186/s13052-020-0813-9
Keywords: NONO, non-ossifying fibromas, left ventricular non-compaction cardiomyopathy, X-linked intellectual disability syndrome, thrombocytopenia, recurrent fractures
Citation: Writzl K, Mavčič B, Maver A, Hodžić A and Peterlin B (2023) Case Report: Non-ossifying fibromas with pathologic fractures in a patient with NONO-associated X-linked syndromic intellectual developmental disorder. Front. Genet. 14:1167054. doi: 10.3389/fgene.2023.1167054
Received: 15 February 2023; Accepted: 24 May 2023;
Published: 18 July 2023.
Edited by:
Mahmood Rasool, King Abdulaziz University, Saudi ArabiaReviewed by:
Muhammad Asif, Balochistan University of Information Technology, Engineering and Management Sciences, PakistanCopyright © 2023 Writzl, Mavčič, Maver, Hodžić and Peterlin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karin Writzl, a2FyaW53cml0emxAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.