
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet., 09 May 2023
Sec. Epigenomics and Epigenetics
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1166831
 Lei Xia1,2,3†
Lei Xia1,2,3† Fan Zhang1,2,3,4,5†‡Yulu Li1,2,3Yuemi Mo1,2,3Lingqiu Zhang1,2,3Qianhua Li1,2,3Minghuang Luo1,2,3Xiaotao Hou1,2,3,5‡
Fan Zhang1,2,3,4,5†‡Yulu Li1,2,3Yuemi Mo1,2,3Lingqiu Zhang1,2,3Qianhua Li1,2,3Minghuang Luo1,2,3Xiaotao Hou1,2,3,5‡ Zhengcai Du1,2,3,5*‡Jiagang Deng1,2,3,5*‡Erwei Hao1,2,3,5*‡
Zhengcai Du1,2,3,5*‡Jiagang Deng1,2,3,5*‡Erwei Hao1,2,3,5*‡As a neurodegenerative disease, Alzheimer’s disease (AD) is characterized by synaptic loss, extracellular plaques of amyloid accumulation, hyperphosphorylation of tau, and neuroinflammation. Various biological processes are affected by epitranscriptomic modifications, which regulate the metabolism of mRNA in cells and regulate the expression of genes. In response to changes in m6A modification levels, the nervous system becomes dysfunctional and plays a significant role in the development of Alzheimer’s disease. As a result of recent research, this paper reviews advances in the understanding of the regulatory mechanisms of m6A modification in the occurrence and development of AD. In addition, the article discusses recent research techniques related to animal models of m6A and AD. Furthermore, it discusses the possibility of studying the pathogenesis of AD at the level of the epitranscriptome, identifying early diagnostic markers, and screening for effective treatment options.
AD is one of the leading causes of dementia and a growing global health problem. Moreover, the vast majority of AD patients are elderly. With the aging of the population, the increasing prevalence of AD is becoming one of the major challenges for the social public health system. Although the understanding of the pathogenesis of AD has made great progress in the past few decades, there is still no cure for AD. The key to initiating treatment is an early and accurate diagnosis. However, preclinical AD is difficult to detect, and patients do not have functional impairments in daily activities, although the cerebral cortex and hippocampus have pathological changes (Dubois et al., 2016). Presently, AD is primarily diagnosed through positron emission tomography (PET) and analysis of cerebrospinal fluid (CSF) proteins. Even though it has a high level of accuracy, AD has already progressed to a chronic stage when pathological symptoms are observed. In light of this, biomarkers may be able to assist in the early detection of Alzheimer’s disease.
m6A is a ubiquitous internal chemical modification in mRNA, which regulates mRNA expression, splicing, decay and translation, and is involved in cell development, embryonic development and stress response (Zhao et al., 2017). The distribution and motif of m6A during human development vary across tissues and developmental processes (Zhang et al., 2020). m6A modification sites are biased to the Coding sequence (CDS) and 3′Untranslated Region (3′UTR) regions, and even a single modification site has a large effect on function (Zhang et al., 2020). A number of recent studies have shown that m6A is abnormally expressed in the brain of AD patients. It may be possible to study the pathogenesis of AD and find new biomarkers from the perspective of m6A modification. Therefore, this article discusses the role of m6A modifying enzymes in the pathological process of AD, and summarizes the animal models used for AD research and the new research techniques related to m6A.
AD is a chronic neurodegenerative disease. As is evident from clinical observations, AD usually typically manifests itself as a progressive decline in higher cognitive functions as well as memory loss. The preclinical stage of AD can be sustained for a long period of time. An initial pathological examination of the cerebral cortex and hippocampus revealed that the patient was experiencing mild memory loss. This is followed by memory loss, emotion changes, dyslexia and, in later stages, abnormal neuropsychiatric symptoms (Knopman et al., 2021; Yun-Ling Zhang et al., 2021). As a pathological condition, AD is characterized by widespread loss of neurons in the brain and the formation of two characteristic proteins, amyloid plaques and neurofibrillary tangles (NFTs), which are produced by hyperphosphorylation of tau. As a result, there is a loss of synaptic plasticity, demyelination, neuroinflammation, and neurotoxicity in the AD brain. In terms of the pathogenesis of AD, two major theories have been proposed: a cascade involving amyloid ß (Aβ) and hyperphosphorylation of tau protein. Aβ deposits as neuroinflammatory plaques and induces AD through its destruction of neuronal cells. Tau hyperphosphorylation then leads to tau pathology leading to NFTs and subsequently to the harmful cascade of neurodegeneration (Busche and Hyman, 2020; Zhang et al., 2021). At the same time, Aβ is considered to be an important factor for neuronal and synaptic dysfunction in the progression of AD. This substance disrupts synaptic plasticity, inhibits long-term potentiation (LTP) in the hippocampus, and causes oxidative stress in the brain through the production of reactive oxygen species (Lustbader et al., 2004). Increasing oxidative stress can lead to oligodendrocyte death and dysfunction, resulting in the failure of Oligodendrocyte progenitor cells (OPCs) to differentiate into oligodendrocytes, which eventually results in abnormalities in the white matter and demyelination of nerves (Nasrabady et al., 2018). There are microglia which are macrophages in the brain, and microglia have a role to play in the removal of amyloid beta. However, with the massive accumulation of Aβ precipitates, in turn, Aβ activated microglia produce inflammatory factors, which in turn cause neuronal cell damage and eventually lead to neuroinflammation (Hickman et al., 2008; Babcock et al., 2015). There was an immune response observed in mice transgenic for human APP (hAPP) at hippocampal and cortical synapses that were engulfed by microglia prior to plaque deposition (Shi Q. et al., 2017) (Figure 1).
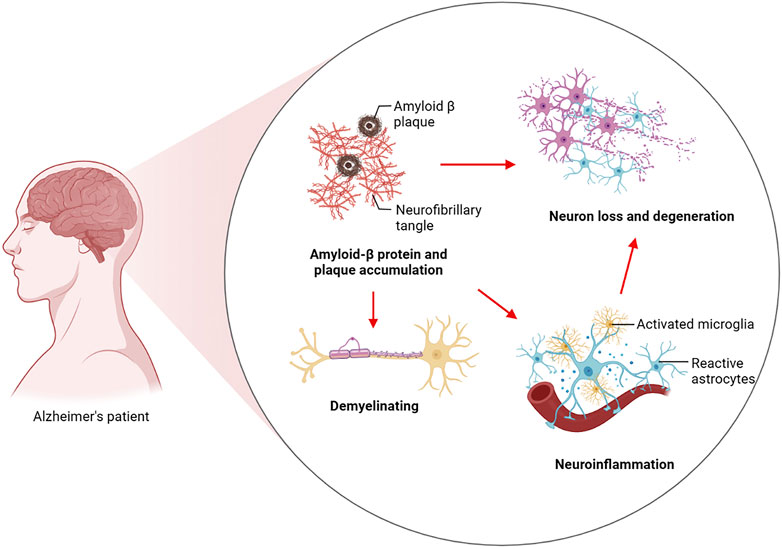
FIGURE 1. Pathogenesis of Alzheimer’s disease. Aβ induces AD by destroying neuronal cells and induces tau hyperphosphorylation to drive the development of tau pathology to form NFTs and then lead to the harmful cascade of neurodegeneration. These include Aβ-induced failure of OPCs to differentiate into oligodendrocytes, resulting in white matter abnormalities and ultimately demyelination of nerve cells. Aβ disrupts synaptic plasticity and activates microglia to promote inflammation, ultimately leading to synaptic loss and neuroinflammation.
The long latency period in the early stages of AD and the lack of obvious clinical manifestations lead to difficulties in the diagnosis of AD. Amyloid accumulation (A), hyperphosphorylation of tau (T) and neurodegeneration (N) are considered to be biomarkers of AD (Jack et al., 2018). Nevertheless, there are operational limitations associated with the use of ATN for the diagnosis of AD in clinical settings. Clinicians still lack effective and efficient biomarkers and detection methods. In terms of drug treatment, only five drugs approved by the FDA for the treatment of AD are on the market, and these drugs are only effective for the treatment of AD symptoms (Briggs et al., 2016). A number of new drugs have been developed for AD in recent years, but none have reached the market. Because AD pathology is diverse and the brain is complex, treating AD is challenging.
In the prevention, diagnosis and treatment of disease, biomarkers play a crucial role. Detecting effective and characteristic markers of AD is important for the prevention and diagnosis of the disease, especially at an early stage. Transcriptomics of epitranscriptomic changes is an emerging field of biological research. Unlike epitranscriptomic modifications of DNA and histones, which affect the expression of genes, chemical modifications of RNA are thought to play a significant role in the occurrence and development of diseases (Fu et al., 2014). Methylation is one of the most prominent forms of RNA modification. Currently, m6A (N6-methyladenosine), which has the highest abundance of internal modifications in messenger RNA (mRNA), is the subject of the most intensive investigation (Zhao et al., 2017). The methylation modifications of specific genes by m6A can serve as potential biomarkers and therapeutic targets in oncology and cardiovascular disease research (Sun et al., 2019; Wu et al., 2020). α-ketoglutarate-dependent dioxygenase alkB homologue 5 (ALKBH5) can reduce the m6A modification of NANOG mRNA, which plays an imperative role in the maintenance of cancer stem cells, thereby enhancing its stability in breast cancer (Zhang et al., 2016a; Zhang et al., 2016b). There is a significant regulatory role played by fat mass and obesity-associated protein (FTO) in the development of heart failure. By selectively demethylating myocardial contractile transcripts, FTO can increase the expression of these transcripts and prevent their degradation, thus protecting the contractile function of cardiomyocytes (Mathiyalagan et al., 2019). The role of m6A modification in the nervous system has attracted considerable attention in recent years. The level of m6A modification in the brain increases with brain development and exhibits tissue specificity, strongly suggesting that m6A is closely related to brain function (Meyer et al., 2012). Meanwhile, the level of m6A in the brains of AD patients differs from that of normal brains, attracting the attention of researchers (Shafik et al., 2021). The discovery of m6A may prove to be a new breakthrough in the search for biomarkers of AD.
Approximately 80% of RNA methylation modifications in eukaryotes occur through m6A methylation. A m6A modification enzyme system consists of a methylation transferase system (Writers), a demethylase system (Erasers), and a binding protein system (Readers). As a result of their ability to add, remove or identify m6A modification sites, these regulators are able to alter critical biological processes such as splicing, processing, RNA stability, translation, and cellular metabolism. M6A modification plays a significant role in brain development and function as a post-transcriptional regulator. As neurons mature, m6A’s expression levels increases, and m6A-seq functional classification analysis reveals that most of m6A’s target genes are involved in neural development, synaptic transmission, and postsynaptic function (Chang et al., 2017; Huang et al., 2020). The methyltransferase-like protein 3 (METTL3), also known as Writer, is a binding protein for S-adenosylmethionine. METTL3 is a binding protein of S-adenosyl methionine (SAM) that acts as the core of the methyltransferase complex and is involved in methylating mRNA (Zeng et al., 2020). METTL14, another component of the methyltransferase complex, co-localizes with METTL3 in the nucleus and forms a stable heterodimer that enhances METTL3’s catalytic activity (Wang et al., 2016). Wilms tumor 1-associated protein (WTAP) regulates the recruitment of m6A methyltransferase complexes to mRNA targets (Ping et al., 2014). It was discovered that m6A modification is dynamic and reversible when the demethylase fat mass and FTO was discovered (Jia et al., 2011). ALKBH5, which is a demethylase like FTO, belongs to the α-ketoglutarate-dependent dioxygenase protein family and relies on α-ketoglutarate/Fe2+ to perform demethylation function (Zheng et al., 2013). m6A-binding proteins are mainly involved in recognizing m6A-modified mRNA chains and are involved in the splicing, folding, transport, and translation of mRNA. The role of m6A methylation in the development of diseases, including neurological disorders, has attracted increasing attention in recent years. According to the brain synaptic tagging hypothesis, ALKBH5-mediated m6A demethylation can alter RNA methylation and result in synaptic dysfunction as well as reversible disease conditions (Martinez De La Cruz et al., 2021) (Figure 2; Table 1).

FIGURE 2. m6A methylation modification process.
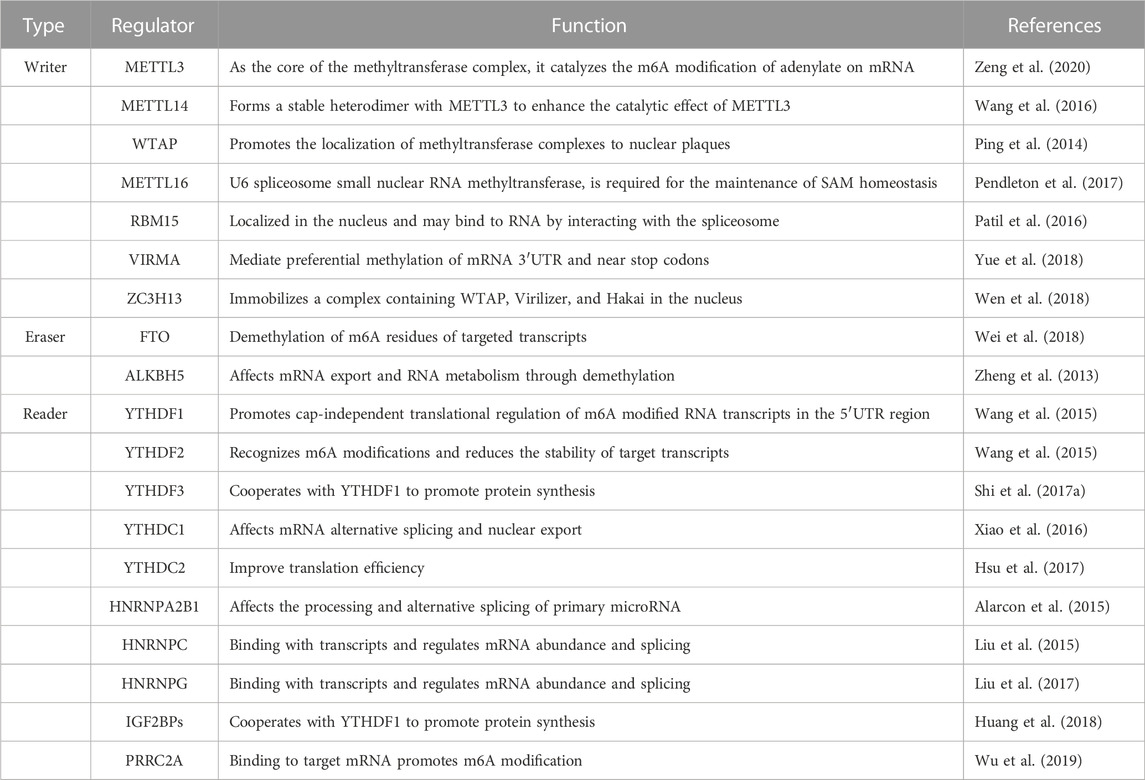
TABLE 1. m6A Modifying Enzymes.
AD is a neurodegenerative disease associated with aging and is the leading cause and major risk factor for dementia in the elderly (Hou et al., 2019). There is an association between m6A modification and neural development (Nussbaum and Ellis, 2003). There was a decrease in m6A methylation levels in the brain of AD patients compared to cognitively intact older adults (Castro-Hernandez et al., 2023). The m6A methylation is often enhanced near the stop codon and in the 3′UTR to affect mRNA expression. A 5XFAD mouse model exhibits lower levels of m6A methylation than a wild type mouse, and the 3′UTR of wild type mice is significantly enriched in m6A modification (Shafik et al., 2021). It is possible that the abnormal levels of m6A in the brains of AD patients and AD model mice are related to the pathological changes that occur in AD brains.
During the development of the human brain, the degree of methylation of m6A changes dynamically, and its expression is spatially specific at specific times (Shafik et al., 2021). There is a reduction in RNA m6A modification in large pyramidal neurons of AD patients compared to that of normal elderly individuals. This is associated with pathological findings including reduced synapses, neuronal atrophy, and increased gliosis. A significant proportion of neurons in the brains of Alzheimer’s patients have been lost. It has been demonstrated that the brain’s hippocampal neurons are closely related to memory, and that damage to these neurons will lead to memory impairment and the manifestations of dementia (Ginsberg et al., 2012). Generally speaking, mild cognitive impairment (MCI) is considered a stage in the progression of more severe dementia and a precursor to AD.
Aβ is considered to be an important factor in neuronal and synaptic dysfunction in the progression of AD, which can disrupt synaptic plasticity, LTP in the hippocampus, and induce the production of reactive oxygen species (Lustbader et al., 2004). Synaptic loss causes cognitive decline, and this alteration in synaptic function is thought to be the primary mechanism underlying AD disorders (Henstridge et al., 2016). There is evidence that m6A is involved in memory formation and contributes to synaptic plasticity (Zhao et al., 2021). In AD patients, the hippocampus expressed a decreased level of the m6A methylases METTL3 and MELLT14 as well as WTAP. Several important synaptic genes, including α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA) and N-methyl-d-aspartic acid receptor (NMDA), were hypermethylated and their expression was significantly reduced in AD mice’s brains compared with control mice’s brains (Han et al., 2020). The middle temporal gyrus is the first area of the brain to show symptoms of AD. The expression of METTL3 is significantly decreased in the middle temporal gyrus of MCI patients, while the expression of METTL14 and FTO is unaffected (Zhao et al., 2021)c. METTL3 has an important in maintaining neuronal survival and synaptic structure, it has a structural and functional role in modifying neural circuits at the synaptic level. Aβ oligomers inhibited METTL3 expression and promoted the reduction of postsynaptic density protein 95 (PSD95) (Zhao et al., 2021). On the contrary, overexpression of METTL3 in vivo rescues synaptic damage and cognitive deficits induced by AβO (Zhao et al., 2021). Accordingly, Mettl3 knockout mice in the hippocampus displayed memory loss and increased oxidative stress, resulting in DNA damage and activation of Caspase 3, a critical factor in neuronal programmed cell death. In addition, postsynaptic deficits and neural mutations result from the knockdown of Mettl3 (Zhao et al., 2021). In other words, METTL3 contributes to the consolidation of hippocampal-dependent memory by promoting the translation of neuronal early response genes, thereby promoting the consolidation of long-term memory (Zhang et al., 2018). It can be seen that METTL3 is an important target of Aβ-induced synaptic loss in the hippocampus of AD brain, and the down-regulation of METTL3 causes a series of reactions that still need to be further studied. This also provides the possibility of METTL3 as a potential biomarker for AD.
In addition to METTL3, other m6A modifying enzymes also play a role in brain memory formation. In the case of Mettl3 and Mettl14 deficient mice, synaptic plasticity, memory, and learning are impaired (Koranda et al., 2018; Zhang et al., 2018). By contrast, Fto knockout mice increased brain m6A abundance and improved memory consolidation. The expression of Fto is reduced and the methylation of mRNA increases in response to fear conditions. Specifically, Fto may be responsible for repressing pre-memory transcript translation as well as promoting the translation of pre-memory transcripts. In response to neuronal stimulation, YTHDF1 promotes the protein synthesis of target transcripts in the hippocampus of adult mice, thereby facilitating learning and memory. It has been established that Ythdf1 knockdown causes hippocampal degeneration as well as deficits in memory and learning, whereas supplementation with Ythdf1 resolves behavioral and synaptic deficits (Shi et al., 2018). Thus, Ythdf1 promotes long-term changes in synaptic plasticity, which facilitate memory formation (Shi et al., 2018).
In the central nervous system, oligodendrocytes, a subclass of glial cells, play an important role in myelination, which constitutes approximately 50% of brain cells (Kuhn et al., 2019). Aβ-induced oxidative stress can lead to oligodendrocyte death and dysfunction, resulting in white matter abnormalities (Nasrabady et al., 2018). Studies of neuroimaging in AD have demonstrated that significant white matter damage and myelin loss occur during the early stages of the disease, often before Aβ plaques and NETs become more apparent (Nasrabady et al., 2018). Olig2 and NG2 are often expressed in oligodendrocytes as Aβ plaque-associated proteins. Aβ plaque-associated protein induces OPCs senescence, turning them into senescent cells with a pro-inflammatory phenotype. In this condition, oligodendrocytes may not differentiate into myelin oligodendrocytes, which may contribute to cognitive deficits and neuroinflammation in AD (Zhang et al., 2019). Recent studies have indicated that Proline rich coiled-coil 2 A (PRRC2A), a novel m6A reader, is highly expressed in oligodendrocytes and regulates the expression of Olig2 mRNA by interacting with PRRC2A, the loss of PRRC2A results in demyelination (Wu et al., 2019). As opposed to this, Fto-mediated m6A demethylation promotes the degradation of Olig2 mRNA and contributes to the reduction of myelin in mice. Interestingly, PRRC2A is characterized by a novel domain that binds methylated RNA specifically and competes with YTHDF2 for RNA binding (Wu et al., 2019). The pathophysiology of AD is largely influenced by myelination (Mathys et al., 2019). As well, METTL14 is essential for the maturation of oligodendrocytes and the formation of myelin in the central nervous system, and the splicing of neural bundle protein mRNA in oligodendrocytes is affected by m6A methylation modifications (Xu et al., 2020).
Inflammatory factors can be a double-edged sword. In the pathogenesis of AD, inflammatory factors can remove the deposited Aβ, but Aβ activates inflammatory factors produced by microglia and astrocytes, which guide more inflammatory reactions at the site of injury and further produce neuroinflammation (Dhapola et al., 2021). As part of the pathological development of AD, neuroinflammation can also occur, including the activation of inflammatory factors and the activation of astrocytes and microglia to perform immune functions. Numerous neurodegenerative diseases are characterized by microglia-mediated cellular inflammation. Among the many functions that microglia play in neural development, these include forming synapse connections, eliminating synapse connections, and phagocytosing cells. Microglia that have been activated can become M1 or M2 phenotypes. As M1 is primarily involved in inflammatory factors and neurotoxicity, and M2 is primarily involved in tissue repair and restoration of homeostasis, microglia in AD can be continuously activated to the M1 phenotype, resulting in inflammatory responses that ultimately result in irreversible neuronal damage (Tang and Le, 2016). Inducing different phenotypes of microglia allowed us to examine m6A modifications on mRNA and IncRNA, which indicated that RNA m6A methylation differs greatly among different microglia phenotypes. A significant increase in m6A modified genes was observed when microglia switched from M0 to M1 phenotype, and these genes may be involved in the inflammatory response (Li et al., 2021).
The activation of microglia in the brain of AD patients was correlated with neuroinflammation and increased levels of MHC-II and CD68 positive cells (Hopperton et al., 2018). Microglial cells that are unable to clear Aβ deposits begin to fail, with lipofuscin deposition and a “dystrophic” state of cell fragmentation from multiple cytoplasmic protrusions to spherical swelling, cytoplasmic fragmentation, and vesicle formation (Streit et al., 2020). A significant change in the methylation levels of multiple inflammation-related factors occurs during the activation of microglia into pro-inflammatory microglia (Li et al., 2021). Microglial inflammation induced by lipopolysaccharide was shown to be accompanied by increased METTL3 expression, and METTL3 overexpression was found to activate TRAF6/NF-κB to promote microglial inflammatory responses in an in vitro model (Wen et al., 2022). Furthermore, IGF2BP1 could regulate the activation of microglia in this model, and its expression levels increased significantly based on the LPS concentration (Ding et al., 2022).
The NETs is composed of filamentous aggregates of the microtubule-associated protein tau. During neurodegeneration, Tau protein undergoes phosphorylation modifications and conformation changes, which lead to its separation from microtubules and binding to aggregates of varying sizes. One of the causes of AD is believed to be these aggregates that are capable of producing neurotoxicity (Niewiadomska et al., 2021).
FTO expression was increased in the brain tissue of 3×Tg mice. As Fto is knocked down in neurons, tau protein is phosphorylated at a lower level, whereas FTO overexpression in neurons increases tau protein phosphorylation. By activating mTOR and its downstream signaling pathways, FTO induces Tau phosphorylation and the mTORC1 inhibitor rapamycin blocks this action, indicating that FTO induces Tau phosphorylation (p-Tau) based on mTOR signaling (Li et al., 2018). It has been demonstrated that HNRNPA2B1 functions as a m6A reader in the nucleus and that Tau oligomerization induces its translocation to the cytoplasm. It is interesting to note that HNRNPA2B1 promotes tau oligomerization but not pTau oligomerization. HNRNPA2B1 and pTau were able to interact more effectively with one another due to tau oligomerization facilitated by pTau. For tau oligomers to bind to m6A, HNRNPA2B1 is required. In the context of Tau oligomerization, METTL3 knockdown decreased m6A methylation and prevented HNRNPA2B1 accumulation in the cytoplasm. Knocking down HNRNPA2B1 may partially reverse neurotoxicity (Jiang et al., 2021). NOP2/Sun RNA methyltransferase 2 (NSun2) is well known as an m5C RNA methylase (Hussain et al., 2013). However, NSun2 also appears to function as an m6A RNA methylase for m6A modification of microRNA. NSun2 is located in the nucleus of neurons in the hippocampus and prefrontal cortex of the brain, and its expression is reduced in AD patients. Its downregulation promotes Tau phosphorylation (Kim et al., 2022). In contrast, NSun2 overexpression resulted in a significant reduction in phosphorylated tau levels, restored neuronal cell viability and reduced AβO-induced toxicity, indicating a neuroprotective role of NSun2. It is noteworthy that regions of the brain that were not affected by AD pathology, such as the cerebellum, had NSun2 levels comparable to those in the control cerebellum, indicating that NSun2 deficiency is limited to those brain regions most vulnerable to AD (Kim et al., 2022). Further, METTL3, also a ‘Writer’, can improve AD through the enhancement of autophagy clearance of p-Tau through a m6A-dependent STUB1 pathway (Tang et al., 2023).
Most of the components of the brain are composed of lipids, and lipid metabolism is often associated with pathological conditions of the brain. APOE is a class of glycoproteins that are widely expressed in astrocytes, microglia, and vascular wall cells in the central nervous system (CNS). In addition to being involved in lipid transport and metabolism, APOE4 is one of the most prevalent genetic risk factors for late-onset Alzheimer’s disease (Chew et al., 2020). It has been shown in AD that APOE4 enhances microglia-induced inflammation, promotes neurodegeneration, alters lipid raft structure within neuroglia membranes, and accelerates the destruction of Aβ and tau proteins. According to the bioinformatics data, APOEɛ4 may have a close relationship to the m6A methylation regulators (METTL3, METTL16, YTHDC2, LRPPRC) in brain tissue from AD patients, but the exact mechanism needs to be determined by further experimental studies (Du et al., 2021).
In summary, cognitive impairment, white matter demyelination, neuroinflammation, and neurotoxicity are all associated with the presence of Aβ-insoluble starch and hyperphosphorylated Tau. Under stimulation of Aβ, the expression level of METTL3 in neurons is down-regulated, oxidative stress occurs, the apoptosis signaling pathway is activated, and PSD95 protein is reduced, which eventually leads to synaptic loss (Zhao et al., 2021). However, METTL3 expression is up-regulated in microglia, which activates the TRAF6/NF-κB signaling pathway and contributes to neuroinflammation (Wen et al., 2022). In addition, m6A modifying enzymes participate in the processes of Tau oligomerization, phosphorylation, and myelination. Multiple factors contribute to the pathogenesis of Alzheimer’s disease. Previously, it was mentioned that Aβ and Tau proteins promote each other. Down-regulation of METTL3 by Aβ stimulating in neurons leads to the translocation of HNRNPA2B1 originally located in the nucleus to the cytoplasm to promote tau oligomerization (Jiang et al., 2021). A notable observation was that METTL3 is accumulated in insoluble plaques in the postmortem brains of AD patients, and its expression level is positively correlated with Tau levels. In contrast, METTL3 expression was reduced in the hippocampal region (Huang et al., 2020). Aside from the modifying enzymes mentioned above, RBM15B, ALKBH5 and ELVAL3 showed significant changes in the m6A gene in patients with AD (Huang et al., 2020). Currently, there are limited studies on the signaling pathways involved in AD that are mediated by m6A-modifying enzymes. The study of m6A-modifying enzymes may provide insight into the occurrence and development of AD, as well as identify potential biomarkers and therapeutic targets for early detection of the disease (Figure 3).
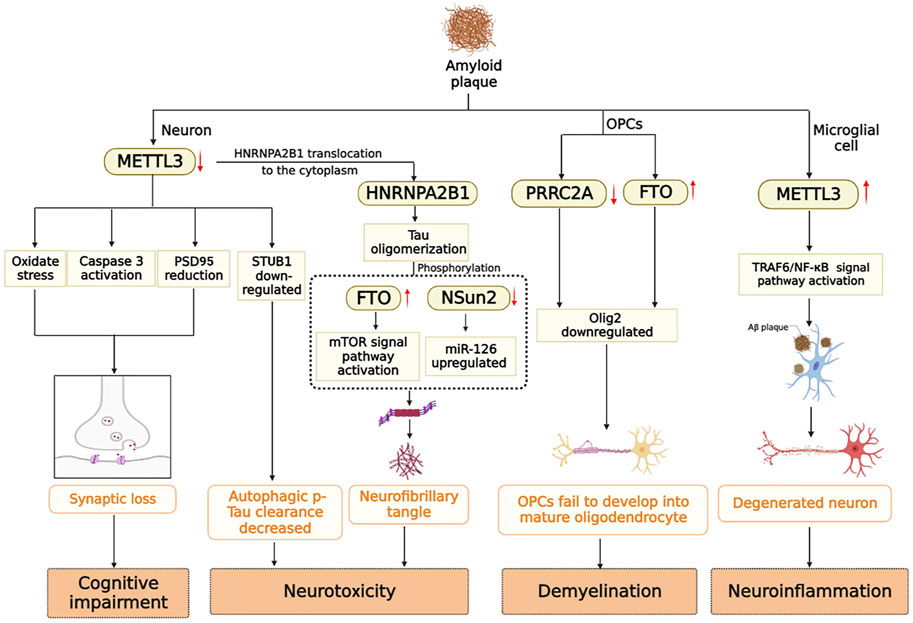
FIGURE 3. m6A modifying enzymes are involved in the pathogenesis induced by Aβ and Tau protein in AD brain.
Since the 21st century, the incidence of AD has increased year by year while it has also been extensively studied. To explore the pathogenesis of AD, researchers have not only studied brain samples, brain cell lines and genomes from postmortem AD patients and normal individuals. The studies of AD pathogenesis and the development of targeted drugs have also been significantly aided by a variety of animal disease models (Figure 4; Table 2).
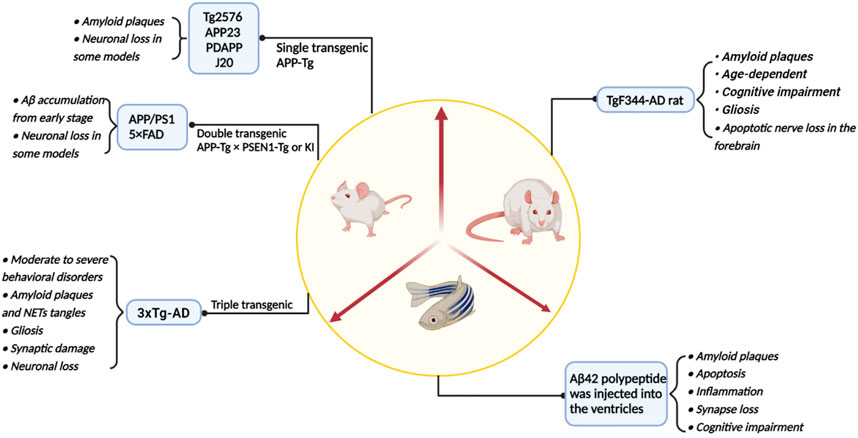
FIGURE 4. A summary of the main model animals used to construct AD models and their pathological representations.

TABLE 2. Role of m6A modifying enzymes in model animals and AD pathology.
Mice have been a major tool for AD research because they share most of the major brain regions and neurotransmitter systems with humans. During aging, mouse brains show the same elevated levels of m6A methylation as human brains, as well as a high degree of overlap between m6A sites in mouse brains and human brains (Shafik et al., 2021). A majority of AD mouse models are transgenic models. Edited amyloid precursor protein (APP) is closely associated with AD, and APP is broken down by ß-protease and γ-protease to produce toxic Aβ plaques. APP single transgenic mice were first applied to the study of AD based on the hypothesis of insoluble amyloid in the brain of AD patients (Games et al., 1995). APP single transgenic mice exhibited Aβ pathological plaques but did not exhibit neurofibrillary tangles or significant neuronal loss. In preclinical studies for AD drugs, APP transgenic mice are used as preclinical study models to evaluate drugs such as BACE1 and aducanumab monoclonal antibodies, etc (Kastanenka et al., 2016; Neumann et al., 2018).
PS19 transgenic mice express mutant human microtubule-associated protein tau (MART) driven by the mouse prion protein (Prnp) promoter. At 8 months of age, PS19 mice with mutant human tau expression were found to have neuronal loss and brain atrophy in the hippocampus. In other brain regions, extensive neuronal fibrillary tangles were formed, accompanied by microgliosis and astrocytosis (Yoshiyama et al., 2007). RNA transcription is regulated by HNRNPA2B1, a reading protein for m6A in the nucleus. Moreover, HNRNPA2B1, along with other reader proteins, has been shown to promote the interaction between oTau and m6A transcripts in the cytoplasms (Jiang et al., 2021). It was found that HNRNPA2B1 expression increased in the cytoplasm of brain neurons of three-month-old P301S tau mice and was stably co-localized with m6A by 9 months of age. Conversely, HNRNPA2B1 expression was limited to the nucleus of WT mice aged 3–6 months (Jiang et al., 2021). In addition to the classical single transgenic mouse models described above, knockout or overexpression of m6A modifying enzymes in mice also exhibited similar AD phenotypes. The brains of Mettl3−/− mice and Ythdf1−/− mice showed cognitive impairment similar to AD, in which the brains of Mettl3−/− mice showed microglia activation, and METTL3 overexpression mice prevented AβO-induced synaptic loss and cognitive impairment (Shi et al., 2018; Zhao et al., 2021). Mettl14−/− mice exhibited white matter abnormalities (Koranda et al., 2018). Similarly, the brains of Prrc2a−/− mice also showed hypomyelination and behavioral and cognitive impairments, as did Fto overexpression mice (Wu et al., 2019). The brain of NSun2 −/−mice showed significantly increased levels of phosphorylated Tau (Kim et al., 2022).
In APP/PS1 transgenic mice, pathological changes in the brain are similar to those observed in AD patients, as well as behavioral and cognitive dysfunction. The levels of m6A methylation were elevated in the cortex and hippocampus of APP/PS1 transgenic mice, along with increased expression of METTL3 and decreased expression of FTO (Han et al., 2020). As compared with the APP mouse model, the APP/PS1 mouse brain showed Aβ deposition at an early stage. Neither model was capable of generating NET tangles and complete neuronal loss. Due to this, APP/Tau double transgenic mice develop both Aβ plaque deposition and neurofibrillary tangles. 5×FAD mice exhibit neuronal loss and memory deficits associated with amyloid pathology (Oakley et al., 2006). As an important animal model to ensure clinical marketing of novel AD drugs, the AD transgenic mouse model can predict almost all of the side effects observed in human trials. The triple transgenic 3×Tg-AD mice behaviorally exhibit moderate to severe behavioral impairment of AD, downregulation of FTO expression, formation of Aβ plaques and NETs, as well as gliosis, synaptic damage, and neuronal loss in neuropathology (Wang et al., 2021). TYRO protein kinase-binding protein (TYROBP) knockout mice develop learning and memory deficits as well as increased levels of phosphorylated Tau and Aβ amyloid in the brain. The severity of these symptoms and pathological features increases with age. The deficiency of TYROBP appears to be associated with the regulation of m6A RNA methylation. A decrease in the expression levels of m6A methyltransferases METTL3, METTL14, and WTAP was observed in the brains of Tyrobp−/− mice compared to wild type mice, which is consistent with this trend found in the brains of AD patients (Zhao et al., 2021; Lv et al., 2022).
Rat tau protein more closely resembles human tau protein, and human amyloid precursor protein (APPsw) and progerin 1 (PS1ΔE9) mutant TgF344-AD rats phenotypically exhibit amyloid plaques, age-dependent cognitive deficits, amyloid-β oligomers, glial cell proliferation, and forebrain apoptotic neural loss typical of AD (Cohen et al., 2013). The accumulation of tau protein in the blue spot region and noradrenergic dysfunction may play a critical role in AD disease progression clinically (Braak et al., 2011). Cognitive performance in old age is significantly influenced by the integrity of the nucleus ceruleus (LC)-norepinephrine system (NE) (Mather and Harley, 2016). A preclinical model of this pathological feature may be provided by TgF344-AD rats. TgF344-AD rats have hyperphosphorylation of tau in the locus ceruleus, which is associated with a deficiency in norepinephrine in the hippocampal area (Rorabaugh et al., 2017).
In terms of biomedical research, zebrafish is an important model vertebrate and is widely used in the study of human disease and development. This species is characterized by a fast reproduction rate, a low cost, and ease of observation. According to genetic analysis, zebrafish and humans share an average genetic similarity of 87%. A zebrafish has the neural structure of vertebrates, and its brain has a similar structure to that of a mammal (Kalueff et al., 2014). A Furthermore, zebrafish have a functional blood-brain barrier (BBB), which does not develop during embryonic life, allowing drugs to enter the brain and may thus be used to screen drugs for neurodegenerative disorders (Kim et al., 2017). Zebrafish have many genes and neural systems that are involved in AD (Santana et al., 2012). In current pharmacological research on brain dysfunction and related drugs, zebrafish, both juvenile and adult, are used (Kalueff et al., 2014). As a translucent vertebrate, zebrafish are physiologically homologous to Homo sapiens, making them an ideal model for studying pathways and mechanisms relevant to human disease prognosis and clinical treatment. The AD model with Aβ starch deposition can be constructed by injecting Aβ42 polypeptide into the ventricles of zebrafish. The zebrafish model of AD also exhibits a number of pathological characteristics of the disease, including apoptosis, inflammation, reduced synapses, and cognitive impairment (Bhattarai et al., 2016).
The brain tissue of zebrafish contains a significant amount of m6A modifications (Li et al., 2022). There is evidence that the m6A reader Ythdf in zebrafish is capable of deadenylating maternal mRNA and causing attenuation of maternal zebrafish transcripts, and the sequence of Ythdf in zebrafish is similar to the sequence of Human YTHDFs (Kontur et al., 2020). Similarly, loss of YTHDF2 in mammals affects early zygote development in mammalian oocytes (Ivanova et al., 2017). METTL3 and WTAP knockouts resulted in severe embryonic defects in zebrafish embryos. The knockout of WTAP caused zebrafish to have developmental defects, such as smaller eyes and ventricles, whereas the knockout of Mettl13 had only a minor effect on the embryos. In situ hybridization of zebrafish whole tissues revealed that WTAP and METTL3 were commonly expressed early in embryonic development and enriched in brain regions at 36 hpf (Ping et al., 2014). Thus, the development of zebrafish is dependent on m6A methyltransferases. In both rodent and human brains, the methyltransferase family protein METTL5 has been found to be enriched in cellular elements and synapses of hippocampal neurons, and knockdown of the METTL5 gene in zebrafish results in the same phenotype as in humans (Richard et al., 2019).
Zebrafish spinal cord contains different functional OPC subtypes (Marisca et al., 2020). The rapid culture cycle of zebrafish allows the molecular characteristics of oligodendrocyte differentiation at each time period and the changes of m6A modification in oligodendrocyte differentiation by single-cell RNA sequencing. In zebrafish, the heterogeneity of microglia associated with neuroinflammation is almost preserved into adulthood, whereas in other rodents the opposite is true (Li et al., 2019; Silva et al., 2021). The findings of this study will help us establish a zebrafish model of AD to study the role of microglia and m6A modification in the development of AD. The mammalian neural stem/progenitor cells (NSPCs) has a limited ability to regenerate neurons, and the neurons in the brains of people with Alzheimer’s disease cannot grow new neurons once they have lost them (Tanaka and Ferretti, 2009). In contrast to mammals, zebrafish neurons are capable of regeneration (Kyritsis et al., 2012). Thus, the zebrafish model of AD can be designed to study the molecular differences between NSPCs and mammalian NSPCS, and to then utilize them as a form of neuronal regeneration therapy (Cosacak et al., 2015). A variety of new gene editing technologies have recently made it possible to develop zebrafish models of neurodegenerative diseases with greater feasibility and selectivity. A technology for zebrafish that is designed to knock out specific tissues and cell types through a conditional knockout switch method, zCKOIS (zebrafish Conditional Knockout-knock In Switch). By inserting donor plasmids containing reverse knockout markers indicated by red fluorescent protein and forward knockout markers indicated by green fluorescent protein into the gene through Cas9 site-directed operation in the gene intron, it utilizes the efficiency of non-homologous recombination. Combined with tissue-specific Cre expression, it achieves visual gene knockout in specific tissues. Using this method, gene-targeted knockouts in zebrafish can be efficiently performed, which fills a technical gap for studying gene function in zebrafish (Li et al., 2020b).
Human-specific pathologies are often not replicated by animal models, and the complex interactions in the in vivo environment are not properly reflected by cell model cultures. Organoids are 3D cell cultures that are derived from adult stem cells in vitro and have similar histological characteristics to human organs. Organoids can also partially reproduce the physiological functions of human organs. Organoid culture, however, requires a relatively long culture period. In 16–20 days, human fibroblasts can be transformed into specific types of neurons by sequentially adding small molecule inhibitors to the cell culture medium every other day (Hu et al., 2015). A 3D brain organoid can be used as a tool to study brain development, differentiation of brain cells, aging, metabolic processes, drug screening, and disease modeling. Organoids are induced by region-specific growth factors, differentiation factors, and cell inhibitors, which can facilitate the study of the expression level of m6A in different populations of the AD brain, as well as the pathogenic mechanism that causes the disease.
Currently, human pluripotent stem cells (hPSCs) can be used to generate AD organoid models in three different ways. The first method involves the use of Aβ42 oligomers or inducers to induce the generation of AD models (Pavoni et al., 2018). Alternatively, iPSCs can be generated from AD-mutant cells and induced to differentiate into various types of neural cells (Gonzalez et al., 2018). CRISPR/Cas9 gene editing can be used to generate AD organoids by causing mutations in APP, PS1, and APOE4 genes (Lin et al., 2018). Organoids derived from hPSCs have been successfully used to model various neurological disorders, and AD organoids can be created by overexpressing mutant PS1 and APP proteins (Choi et al., 2014). Organoid brain models of AD derived from iPSCs of familial AD patients exhibit progressive accumulation of Aβ protein accompanied by the development of NFTs associated with amyloid plaques (Gonzalez et al., 2018). The construction of AD models using microfluidics by neurons, astrocytes, and microglia can provide an insight into key features of AD pathology, such as Aβ amyloid deposition, tau hyperphosphorylation, neuroinflammation, and neurotoxicity (Park et al., 2018). In addition to being more accurate than other models, the model can be used to develop and screen drugs for interaction with neuroglial cells. Organoids can offer the same advantages as cell cultures as well as the ability to model the complex interactions between cells. It is important to note, however, that studying AD on the basis of AD organoids still have some limitations. As AD is a disease associated with aging, genetic changes as well as changes in the global transcriptional profile of cells accompany the aging process. In spite of this, it is difficult to mimic aging-related phenotypes in iPSCs-induced organoids (Camp et al., 2015; Gerakis and Hetz, 2019). Additionally, AD organoids fail to consider the effects of oxygen and nutrients transported from blood vessels on neuronal cells and their connections.
As the study of m6A modifications has intensified, it has become urgent to develop detection techniques and research methods for m6A modifications. There have been several advances in the detection of m6A modifications in recent years in order to meet the needs of most researchers.
As the first developed method, methylated RNA immunoprecipitation (MeRIP) utilizes m6A specific antibodies to recognize methylated RNA on mRNA and RNA immunoprecipitation to enrich methylated fragments (Dominissini et al., 2012). As part of this method, high-throughput sequencing and quantitative PCR were used to analyze RNA regions with m6A methylation modification. However, this method is limited by the lack of specific antibodies or compounds, and by the inability to identify individual modifications of RNA molecules. On the basis of differential cleavage of ribonucleases, the MAZTER-seq system was able to quantify single nucleotides at 16%–25% of the m6A expression sites (Garcia-Campos et al., 2019). Nevertheless, this approach is only able to quantify a subset of m6A modification sites that are located at ACA sites and are situated at an appropriate distance from adjacent ACA sites. The m6Anet is a MIL-based neural network model that identifies potential m6A sites directly from RNA-Seq data and can be applied across multiple cell lines and species without reducing its precision (Hendra et al., 2022). As a result of m6Anet, the stoichiometry of modification is quantified at the site level without the use of control samples, providing further insight into the relationship between RNA modifications and m6A methylation (Hendra et al., 2022). With APEX seq, adjacent RNAs are labeled using the peroxidase enzyme APEX2, which allows for high-resolution RNA mapping of diverse subcellular sites at the nanoscale. It may be applied to any subcellular region, and it allows for the comparison of RNA variants and isoforms (Fazal et al., 2019). The method of CAP-seq involves photooxidation of RNA bases prior to photoactivation, followed by affinity purification and enrichment for sequencing (Wang et al., 2019).
As of now, the most common method for manipulating RNA methylation modification is to regulate the expression of RNA methyltransferases and demethylases. Techniques to target RNA methylation have been less widely reported. A metabolite of riboflavin kinase, flavin mononucleotide (FMN), mediates substantial demethylation of RNA m6A residues in living cells under blue light irradiation (Xie et al., 2019). However, FMN is unable to modify RNA m6A in a targeted manner. By targeting specific genes, the CRISPR/Cas system is a simple and stable gene editing technology. A mutation in the structural domain of the Cas13 nuclease protein can lead to the generation of a catalytic death enzyme that binds with affinity to RNA (Abudayyeh et al., 2016). Accordingly, recent studies have demonstrated the effectiveness of targeting specific mRNAs for demethylation via CRISPR-Cas13b (Li et al., 2020a). Compared to Cas9, the gRNA of the Cas13 system has no PAM targeting sequence restrictions or motif preference around the target site and has a higher mismatch tolerance (Cox et al., 2017). There is a novel nuclease, MTR1, that can transfer a methyl group to a nitrogen atom of a target RNA, thus adding methyl groups to both synthetic and natural RNA strands (Scheitl et al., 2020). This novel nuclease may be useful in understanding the interplay between RNA methylation, structure, and function. Furthermore, it can be used as an RNA probe to dynamically observe changes in m6A methylation levels over during the course of the disease.
This review summarizes the key role of m6A modification in AD, and we additionally summarize the research models of Alzheimer’s disease and the studies related to m6A among them, as well as the current m6A research technologies in recent years. There is an emphasis on the potential role of m6A modification in the development of AD. So far, several RNA m6A modifying enzymes and expression changes of m6A modifying enzymes have been discovered during the development of AD. Nevertheless, many knowledge gaps remain to be filled. The brain environment is complex, and the substances transported by blood vessels, the information transmission between synapses, and the interconnection between cell populations can affect the expression level of key proteins. Proteases involved in the modification of m6A have only recently been discovered. It remains unclear where they are localized in different tissues, what the preferential transcript modification sites are, and what is involved in disease pathogenesis. In AD brain, different modifying enzymes are differentially expressed in different cell populations and involve different signaling pathways. The expression of METTL3 is decreased in the hippocampus of AD brain, leading to cognitive impairment, but its expression is increased in microglia, which has the role of promoting neuroinflammation (Zhao et al., 2021; Wen et al., 2022). The direction of its effect on the pathological development of AD was also opposite in these two regions. Interestingly, METTL3 was highly expressed in insoluble deposits in the brain (Huang et al., 2020). This may be related to the role of Aβ in promoting the activation of microglia, and the specific mechanism needs to be further studied. Through gene targeted knockout, the role of m6A modifying enzymes in different cell populations should be considered to screen out the key factors that can prevent or reverse the disease. Whether m6A modifying enzymes can be used as AD biomarkers in plasma needs further study.
AD presents several diagnostic and therapeutic challenges. First, AD has the characteristic features of long latency and inconspicuous clinical manifestations in its early stages. This is the reason why patients often come to the hospital for tests after the MCI stage. The growing incidence of AD has also placed a significant burden on nursing care. Second, based on the current theoretical conception of AD, it is not yet possible to identify the key pathogenesis of AD and the limitations of current drugs for its treatment. A new perspective on the pathogenesis of AD is needed, as is an exploration of its characteristic biomarkers. In AD brains, m6A methylation levels are abnormal and its modification enzymes have been linked to pathological processes such as synapse loss, oligodendrocyte differentiation, and neuroinflammation. Several new detection methods and research techniques have been reported in recent years to address the limitations of m6A studies. However, research on the pathogenesis of AD at the level of m6A methylation remains shallow. Consequently, this provides a greater opportunity for further analysis of the potential role of m6A methylation modification in AD.
LX: Writing—original draft, including substantive translation; FZ: Writing—review and editing; YL: Visualization; YM: tables sorting LZ: Literature curation; QL: Literature search; ML: Literature search; XH: Funding acquisition; ZD: Conceptualization; JD: Project administration; EH: Conceptualization. All authors read and approved the final manuscript. All authors contributed to the article and approved the submitted version.
This review was supported by The Guangxi Youth Science Foundation (2020GXNSFBA159020); National Natural Science Foundation of China (82060742).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abudayyeh, O. O., Gootenberg, J. S., Konermann, S., Joung, J., Slaymaker, I. M., Cox, D. B., et al. (2016). C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 353 (6299), aaf5573. doi:10.1126/science.aaf5573
Alarcon, C. R., Goodarzi, H., Lee, H., Liu, X., Tavazoie, S., and Tavazoie, S. F. (2015). HNRNPA2B1 is a mediator of m(6)a-dependent nuclear RNA processing events. Cell 162 (6), 1299–1308. doi:10.1016/j.cell.2015.08.011
Babcock, A. A., Ilkjaer, L., Clausen, B. H., Villadsen, B., Dissing-Olesen, L., Bendixen, A. T., et al. (2015). Cytokine-producing microglia have an altered beta-amyloid load in aged APP/PS1 Tg mice. Brain Behav. Immun. 48, 86–101. doi:10.1016/j.bbi.2015.03.006
Bhattarai, P., Thomas, A. K., Cosacak, M. I., Papadimitriou, C., Mashkaryan, V., Froc, C., et al. (2016). IL4/STAT6 signaling activates neural stem cell proliferation and neurogenesis upon amyloid-β42 aggregation in adult zebrafish brain. Cell Rep. 17 (4), 941–948. doi:10.1016/j.celrep.2016.09.075
Braak, H., Thal Dr Fau - Ghebremedhin, E., Ghebremedhin E Fau - Del Tredici, K., and Del Tredici, K. (2011). Stages of the pathologic process in alzheimer disease: Age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 70, 960–969. doi:10.1097/NEN.0b013e318232a379
Briggs, R., Kennelly, S. P., and O'Neill, D. (2016). Drug treatments in Alzheimer's disease. Clin. Med. (Lond) 16, 247–253. doi:10.7861/clinmedicine.16-3-247
Busche, M. A., and Hyman, B. T. (2020). Synergy between amyloid-beta and tau in Alzheimer's disease. Nat. Neurosci. 23 (10), 1183–1193. doi:10.1038/s41593-020-0687-6
Camp, J. G., Badsha, F., Florio, M., Kanton, S., Gerber, T., Wilsch-Brauninger, M., et al. (2015). Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc. Natl. Acad. Sci. U. S. A. 112 (51), 15672–15677. doi:10.1073/pnas.1520760112
Castro-Hernandez, R., Berulava, T., Metelova, M., Epple, R., Pena Centeno, T., Richter, J., et al. (2023). Conserved reduction of m(6)A RNA modifications during aging and neurodegeneration is linked to changes in synaptic transcripts. Proc. Natl. Acad. Sci. U. S. A. 120 (9), e2204933120. doi:10.1073/pnas.2204933120
Chang, M., Lv, H., Zhang, W., Ma, C., He, X., Zhao, S., et al. (2017). Region-specific RNA m(6)A methylation represents a new layer of control in the gene regulatory network in the mouse brain. Open Biol. 7 (9), 170166. doi:10.1098/rsob.170166
Chew, H., Solomon, V. A., and Fonteh, A. N. (2020). Involvement of lipids in Alzheimer's disease pathology and potential therapies. Front. Physiol. 11, 598. doi:10.3389/fphys.2020.00598
Choi, S. H., Kim, Y. H., Hebisch, M., Sliwinski, C., Lee, S., D'Avanzo, C., et al. (2014). A three-dimensional human neural cell culture model of Alzheimer's disease. Nature 515 (7526), 274–278. doi:10.1038/nature13800
Cohen, R. M., Rezai-Zadeh, K., Weitz, T. M., Rentsendorj, A., Gate, D., Spivak, I., et al. (2013). A transgenic alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric A, and frank neuronal loss. J. Neurosci. 33 (15), 6245–6256. doi:10.1523/jneurosci.3672-12.2013
Cosacak, M. I., Papadimitriou, C., and Kizil, C. (2015). Regeneration, plasticity, and induced molecular programs in adult zebrafish brain. Biomed. Res. Int. 2015, 769763. doi:10.1155/2015/769763
Cox, D. B. T., Gootenberg, J. S., Abudayyeh, O. O., Franklin, B., Kellner, M. J., Joung, J., et al. (2017). RNA editing with CRISPR-Cas13. Science 358 (6366), 1019–1027. doi:10.1126/science.aaq0180
Dhapola, R., Hota, S. S., Sarma, P., Bhattacharyya, A., Medhi, B., and Reddy, D. H. (2021). Recent advances in molecular pathways and therapeutic implications targeting neuroinflammation for Alzheimer's disease. Inflammopharmacology 29 (6), 1669–1681. doi:10.1007/s10787-021-00889-6
Ding, L., Wu, H., Wang, Y., Li, Y., Liang, Z., Xia, X., et al. (2022). m6A reader Igf2bp1 regulates the inflammatory responses of microglia by stabilizing Gbp11 and cp mRNAs. Front. Immunol. 13, 872252. doi:10.3389/fimmu.2022.872252
Dominissini, D., Moshitch-Moshkovitz, S., Schwartz, S., Salmon-Divon, M., Ungar, L., Osenberg, S., et al. (2012). Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485 (7397), 201–206. doi:10.1038/nature11112
Du, B., Zhang, Y., Liang, M., Du, Z., Li, H., Fan, C., et al. (2021). N6-methyladenosine (m6A) modification and its clinical relevance in cognitive dysfunctions. Aging (Albany NY) 13, 20716–20737. doi:10.18632/aging.203457
Dubois, B., Hampel, H., Feldman, H. H., Scheltens, P., Aisen, P., Andrieu, S., et al. (2016). Preclinical Alzheimer's disease: Definition, natural history, and diagnostic criteria. Alzheimers Dement. 12 (3), 292–323. doi:10.1016/j.jalz.2016.02.002
Fazal, F. M., Han, S., Parker, K. R., Kaewsapsak, P., Xu, J., Boettiger, A. N., et al. (2019). Atlas of subcellular RNA localization revealed by APEX-seq. Cell 178 (2), 473–490. doi:10.1016/j.cell.2019.05.027
Fu, Y., Dominissini, D., Rechavi, G., and He, C. (2014). Gene expression regulation mediated through reversible m⁶A RNA methylation. Nat. Rev. Genet. 15 (5), 293–306. doi:10.1038/nrg3724
Games, D., Adams D Fau - Alessandrini, R., Alessandrini R Fau - Barbour, R., Barbour R Fau - Berthelette, P., Berthelette P Fau - Blackwell, C., Blackwell C Fau - Carr, T., et al. (1995). Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 373, 523–527. doi:10.1038/373523a0
Garcia-Campos, M. A., Edelheit, S., Toth, U., Safra, M., Shachar, R., Viukov, S., et al. (2019). Deciphering the "m(6)A code" via antibody-independent quantitative profiling. Cell 178 (3), 731–747. doi:10.1016/j.cell.2019.06.013
Gerakis, Y., and Hetz, C. (2019). Brain organoids: A next step for humanized Alzheimer's disease models? Mol. Psychiatry 24 (4), 474–478. doi:10.1038/s41380-018-0343-7
Ginsberg, S. D., Alldred, M. J., and Che, S. (2012). Gene expression levels assessed by CA1 pyramidal neuron and regional hippocampal dissections in Alzheimer's disease. Neurobiol. Dis. 45 (1), 99–107. doi:10.1016/j.nbd.2011.07.013
Gonzalez, C., Armijo, E., Bravo-Alegria, J., Becerra-Calixto, A., Mays, C. E., and Soto, C. (2018). Modeling amyloid beta and tau pathology in human cerebral organoids. Mol. Psychiatry 23 (12), 2363–2374. doi:10.1038/s41380-018-0229-8
Han, M., Liu, Z., Xu, Y., Liu, X., Wang, D., Li, F., et al. (2020). Abnormality of m6A mRNA methylation is involved in Alzheimer's disease. Front. Neurosci. 14, 98. doi:10.3389/fnins.2020.00098
Hendra, C., Pratanwanich, P. N., Wan, Y. K., Goh, W. S. S., Thiery, A., and Goke, J. (2022). Detection of m6A from direct RNA sequencing using a multiple instance learning framework. Nat. Methods 19 (12), 1590–1598. doi:10.1038/s41592-022-01666-1
Henstridge, C. M., Pickett, E., and Spires-Jones, T. L. (2016). Synaptic pathology: A shared mechanism in neurological disease. Ageing Res. Rev. 28, 72–84. doi:10.1016/j.arr.2016.04.005
Hickman, S. E., Allison, E. K., and El Khoury, J. (2008). Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice. J. Neurosci. 28 (33), 8354–8360. doi:10.1523/JNEUROSCI.0616-08.2008
Hopperton, K. E., Mohammad, D., Trepanier, M. O., Giuliano, V., and Bazinet, R. P. (2018). Markers of microglia in post-mortem brain samples from patients with Alzheimer's disease: A systematic review. Mol. Psychiatry 23 (2), 177–198. doi:10.1038/mp.2017.246
Hou, Y., Dan, X., Babbar, M., Wei, Y., Hasselbalch, S. G., Croteau, D. L., et al. (2019). Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 15 (10), 565–581. doi:10.1038/s41582-019-0244-7
Hsu, P. J., Zhu, Y., Ma, H., Guo, Y., Shi, X., Liu, Y., et al. (2017). Ythdc2 is an N(6)-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 27 (9), 1115–1127. doi:10.1038/cr.2017.99
Hu, W., Qiu, B., Guan, W., Wang, Q., Wang, M., Li, W., et al. (2015). Direct conversion of normal and Alzheimer's disease human fibroblasts into neuronal cells by small molecules. Cell Stem Cell 17, 204–212. doi:10.1016/j.stem.2015.07.006
Huang, H., Camats-Perna, J., Medeiros, R., Anggono, V., and Widagdo, J. (2020). Altered expression of the m6A methyltransferase METTL3 in Alzheimer's disease. eNeuro 7, ENEURO.0125. doi:10.1523/ENEURO.0125-20.2020
Huang, H., Weng, H., Sun, W., Qin, X., Shi, H., Wu, H., et al. (2018). Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 20 (3), 285–295. doi:10.1038/s41556-018-0045-z
Hussain, S., Sajini, A. A., Blanco, S., Dietmann, S., Lombard, P., Sugimoto, Y., et al. (2013). NSun2-mediated cytosine-5 methylation of vault noncoding RNA determines its processing into regulatory small RNAs. Cell Rep. 4 (2), 255–261. doi:10.1016/j.celrep.2013.06.029
Ivanova, I., Much, C., Di Giacomo, M., Azzi, C., Morgan, M., Moreira, P. N., et al. (2017). The RNA m(6)A reader YTHDF2 is essential for the post-transcriptional regulation of the maternal transcriptome and oocyte competence. Mol. Cell 67 (6), 1059–1067. doi:10.1016/j.molcel.2017.08.003
Jack, C. R., Bennett, D. A., Blennow, K., Carrillo, M. C., Dunn, B., Haeberlein, S. B., et al. (2018). NIA-AA Research Framework: Toward a biological definition of Alzheimer's disease. Alzheimers Dement. 14 (4), 535–562. doi:10.1016/j.jalz.2018.02.018
Jia, G., Fu, Y., Zhao, X., Dai, Q., Zheng, G., Yang, Y., et al. (2011). N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 7 (12), 885–887. doi:10.1038/nchembio.687
Jiang, L., Lin, W., Zhang, C., Ash, P. E. A., Verma, M., Kwan, J., et al. (2021). Interaction of tau with HNRNPA2B1 and N(6)-methyladenosine RNA mediates the progression of tauopathy. Mol. Cell 81 (20), 4209–4227.e12. doi:10.1016/j.molcel.2021.07.038
Kalueff, A. V., Stewart, A. M., and Gerlai, R. (2014). Zebrafish as an emerging model for studying complex brain disorders. Trends Pharmacol. Sci. 35 (2), 63–75. doi:10.1016/j.tips.2013.12.002
Kastanenka, K. V., Bussiere, T., Shakerdge, N., Qian, F., Weinreb, P. H., Rhodes, K., et al. (2016). Immunotherapy with aducanumab restores calcium homeostasis in Tg2576 mice. J. Neurosci. 36 (50), 12549–12558. doi:10.1523/JNEUROSCI.2080-16.2016
Kim, S. S., Im, S. H., Yang, J. Y., Lee, Y.-R., Kim, G. R., Chae, J. S., et al. (2017). Zebrafish as a screening model for testing the permeability of blood–brain barrier to small molecules. Zebrafish 14 (4), 322–330. doi:10.1089/zeb.2016.1392
Kim, Y. A., Siddiqui, T., Blaze, J., Cosacak, M. I., Winters, T., Kumar, A., et al. (2022). RNA methyltransferase NSun2 deficiency promotes neurodegeneration through epitranscriptomic regulation of tau phosphorylation. Acta Neuropathol. 145 (1), 29–48. doi:10.1007/s00401-022-02511-7
Knopman, D. S., Amieva, H., Petersen, R. C., Chetelat, G., Holtzman, D. M., Hyman, B. T., et al. (2021). Alzheimer disease. Nat. Rev. Dis. Prim. 7 (1), 33. doi:10.1038/s41572-021-00269-y
Kontur, C., Jeong, M., Cifuentes, D., and Giraldez, A. J. (2020). Ythdf m(6)A readers function redundantly during zebrafish development. Cell Rep. 33 (13), 108598. doi:10.1016/j.celrep.2020.108598
Koranda, J. L., Dore, L., Shi, H., Patel, M. J., Vaasjo, L. O., Rao, M. N., et al. (2018). Mettl14 is essential for epitranscriptomic regulation of striatal function and learning. Neuron 99 (2), 283–292. doi:10.1016/j.neuron.2018.06.007
Kuhn, S., Gritti, L., Crooks, D., and Dombrowski, Y. (2019). Oligodendrocytes in development, myelin generation and beyond. Cells 8 (11), 1424. doi:10.3390/cells8111424
Kyritsis, N., Kizil C Fau - Zocher, S., Zocher S Fau - Kroehne, V., Kroehne, V., Fau - Kaslin, J., Kaslin J Fau - Freudenreich, D., et al. (2012). Acute inflammation initiates the regenerative response in the adult zebrafish brain. Science 338, 1353–1356. (Electronic)). doi:10.1126/science.1228773
Li, H., Ren, Y., Mao, K., Hua, F., Yang, Y., Wei, N., et al. (2018). FTO is involved in Alzheimer's disease by targeting TSC1-mTOR-Tau signaling. Biochem. Biophys. Res. Commun. 498 (1), 234–239. doi:10.1016/j.bbrc.2018.02.201
Li, J., Chen, Z., Chen, F., Xie, G., Ling, Y., Peng, Y., et al. (2020a). Targeted mRNA demethylation using an engineered dCas13b-ALKBH5 fusion protein. Nucleic Acids Res. 48 (10), 5684–5694. doi:10.1093/nar/gkaa269
Li, J., Li, H. Y., Gu, S. Y., Zi, H. X., Jiang, L., and Du, J. L. (2020b). One-step generation of zebrafish carrying a conditional knockout-knockin visible switch via CRISPR/Cas9-mediated intron targeting. Sci. China Life Sci. 63 (1), 59–67. doi:10.1007/s11427-019-1607-9
Li, Q., Cheng, Z., Zhou, L., Darmanis, S., Neff, N. F., Okamoto, J., et al. (2019). Developmental heterogeneity of microglia and brain myeloid cells revealed by deep single-cell RNA sequencing. Neuron 101 (2), 207–223. doi:10.1016/j.neuron.2018.12.006
Li, Q., Wen, S., Ye, W., Zhao, S., and Liu, X. (2021). The potential roles of m(6)A modification in regulating the inflammatory response in microglia. J. Neuroinflammation 18 (1), 149. doi:10.1186/s12974-021-02205-z
Li, W., Li, X., Ma, X., Xiao, W., and Zhang, J. (2022). Mapping the m1A, m5C, m6A and m7G methylation atlas in zebrafish brain under hypoxic conditions by MeRIP-seq. BMC Genomics 23 (1), 105. doi:10.1186/s12864-022-08350-w
Lin, Y. T., Seo, J., Gao, F., Feldman, H. M., Wen, H. L., Penney, J., et al. (2018). APOE4 causes widespread molecular and cellular alterations associated with Alzheimer's disease phenotypes in human iPSC-derived brain cell types. Neuron 98 (6), 1141–1154. doi:10.1016/j.neuron.2018.05.008
Liu, N., Dai, Q., Zheng, G., He, C., Parisien, M., and Pan, T. (2015). N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 518 (7540), 560–564. doi:10.1038/nature14234
Liu, N., Zhou, K. I., Parisien, M., Dai, Q., Diatchenko, L., and Pan, T. (2017). N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 45 (10), 6051–6063. doi:10.1093/nar/gkx141
Lustbader, J. W., Cirilli, M., Lin, C., Xu, H. W., Takuma, K., Wang, N., et al. (2004). ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science 304 (5669), 448–452. doi:10.1126/science.1091230
Lv, Z., Xu, T., Li, R., Zheng, D., Li, Y., Li, W., et al. (2022). Downregulation of m6A methyltransferase in the Hippocampus of Tyrobp (-/-) mice and implications for learning and memory deficits. Front. Neurosci. 16, 739201. doi:10.3389/fnins.2022.739201
Marisca, R., Hoche, T., Agirre, E., Hoodless, L. J., Barkey, W., Auer, F., et al. (2020). Functionally distinct subgroups of oligodendrocyte precursor cells integrate neural activity and execute myelin formation. Nat. Neurosci. 23 (3), 363–374. doi:10.1038/s41593-019-0581-2
Martinez De La Cruz, B., Markus, R., Malla, S., Haig, M. I., Gell, C., Sang, F., et al. (2021). Modifying the m(6)A brain methylome by ALKBH5-mediated demethylation: A new contender for synaptic tagging. Mol. Psychiatry 26 (12), 7141–7153. doi:10.1038/s41380-021-01282-z
Mather, M., and Harley, C. W. (2016). The locus coeruleus: Essential for maintaining cognitive function and the aging brain. Trends Cogn. Sci. 20 (3), 214–226. doi:10.1016/j.tics.2016.01.001
Mathiyalagan, P., Adamiak, M., Mayourian, J., Sassi, Y., Liang, Y., Agarwal, N., et al. (2019). FTO-dependent N(6)-methyladenosine regulates cardiac function during remodeling and repair. Circulation 139 (4), 518–532. doi:10.1161/CIRCULATIONAHA.118.033794
Mathys, H., Davila-Velderrain, J., Peng, Z., Gao, F., Mohammadi, S., Young, J. Z., et al. (2019). Single-cell transcriptomic analysis of Alzheimer's disease. Nature 570 (7761), 332–337. doi:10.1038/s41586-019-1195-2
Meyer, K. D., Saletore, Y., Zumbo, P., Elemento, O., Mason, C. E., and Jaffrey, S. R. (2012). Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRs and near stop codons. Cell 149 (7), 1635–1646. doi:10.1016/j.cell.2012.05.003
Nasrabady, S. E., Rizvi, B., Goldman, J. E., and Brickman, A. M. (2018). White matter changes in Alzheimer's disease: A focus on myelin and oligodendrocytes. Acta Neuropathol. Commun. 6 (1), 22. doi:10.1186/s40478-018-0515-3
Neumann, U., Ufer, M., Jacobson, L. H., Rouzade-Dominguez, M. L., Huledal, G., Kolly, C., et al. (2018). The BACE-1 inhibitor CNP520 for prevention trials in Alzheimer's disease. EMBO Mol. Med. 10 (11), e9316. doi:10.15252/emmm.201809316
Niewiadomska, G., Niewiadomski, W., Steczkowska, M., and Gasiorowska, A. (2021). Tau oligomers neurotoxicity. Life (Basel) 11 (1), 28. doi:10.3390/life11010028
Nussbaum, R. L., and Ellis, C. E. (2003). Alzheimer's disease and Parkinson's disease. N. Engl. J. Med. 348 (14), 1356–1364. doi:10.1056/NEJM2003ra020003
Oakley, H., Cole, S. L., Logan, S., Maus, E., Shao, P., Craft, J., et al. (2006). Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 26 (40), 10129–10140. doi:10.1523/JNEUROSCI.1202-06.2006
Park, J., Wetzel, I., Marriott, I., Dreau, D., D'Avanzo, C., Kim, D. Y., et al. (2018). A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer's disease. Nat. Neurosci. 21 (7), 941–951. doi:10.1038/s41593-018-0175-4
Patil, D. P., Chen, C. K., Pickering, B. F., Chow, A., Jackson, C., Guttman, M., et al. (2016). m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 537 (7620), 369–373. doi:10.1038/nature19342
Pavoni, S., Jarray, R., Nassor, F., Guyot, A. C., Cottin, S., Rontard, J., et al. (2018). Small-molecule induction of Aβ-42 peptide production in human cerebral organoids to model Alzheimer's disease associated phenotypes. PLoS One 13 (12), e0209150. doi:10.1371/journal.pone.0209150
Pendleton, K. E., Chen, B., Liu, K., Hunter, O. V., Xie, Y., Tu, B. P., et al. (2017). The U6 snRNA m(6)A methyltransferase METTL16 regulates SAM synthetase intron retention. Cell 169 (5), 824–835. doi:10.1016/j.cell.2017.05.003
Ping, X. L., Sun, B. F., Wang, L., Xiao, W., Yang, X., Wang, W. J., et al. (2014). Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 24 (2), 177–189. doi:10.1038/cr.2014.3
Richard, E. M., Polla, D. L., Assir, M. Z., Contreras, M., Shahzad, M., Khan, A. A., et al. (2019). Bi-Allelic variants in METTL5 cause autosomal-recessive intellectual disability and microcephaly. Am. J. Hum. Genet. 105 (4), 869–878. doi:10.1016/j.ajhg.2019.09.007
Rorabaugh, J. M., Chalermpalanupap, T., Botz-Zapp, C. A., Fu, V. M., Lembeck, N. A., Cohen, R. M., et al. (2017). Chemogenetic locus coeruleus activation restores reversal learning in a rat model of Alzheimer's disease. Brain 140 (11), 3023–3038. doi:10.1093/brain/awx232
Santana, S., Rico Ep Fau - Burgos, J. S., and Burgos, J. S. (2012). Can zebrafish be used as animal model to study Alzheimer's disease? Am. J. Neurodegener. Dis. 1, 32–48.
Scheitl, C. P. M., Ghaem Maghami, M., Lenz, A. K., and Hobartner, C. (2020). Site-specific RNA methylation by a methyltransferase ribozyme. Nature 587 (7835), 663–667. doi:10.1038/s41586-020-2854-z
Shafik, A. M., Zhang, F., Guo, Z., Dai, Q., Pajdzik, K., Li, Y., et al. (2021). N6-methyladenosine dynamics in neurodevelopment and aging, and its potential role in Alzheimer's disease. Genome Biol. 22 (1), 17. doi:10.1186/s13059-020-02249-z
Shi, H., Wang, X., Lu, Z., Zhao, B. S., Ma, H., Hsu, P. J., et al. (2017a). YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res. 27 (3), 315–328. doi:10.1038/cr.2017.15
Shi, H., Zhang, X., Weng, Y. L., Lu, Z., Liu, Y., Lu, Z., et al. (2018). m(6)A facilitates hippocampus-dependent learning and memory through YTHDF1. Nature 563 (7730), 249–253. doi:10.1038/s41586-018-0666-1
Shi, Q., Chowdhury, S., Ma, R., Le, K. X., Hong, S., Caldarone, B. J., et al. (2017b). Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med. 9 (392), eaaf6295. doi:10.1126/scitranslmed.aaf6295
Silva, N. J., Dorman, L. C., Vainchtein, I. D., Horneck, N. C., and Molofsky, A. V. (2021). In situ and transcriptomic identification of microglia in synapse-rich regions of the developing zebrafish brain. Nat. Commun. 12 (1), 5916. doi:10.1038/s41467-021-26206-x
Streit, W. J., Khoshbouei, H., and Bechmann, I. (2020). Dystrophic microglia in late-onset Alzheimer's disease. Glia 68 (4), 845–854. doi:10.1002/glia.23782
Sun, T., Wu, R., and Ming, L. (2019). The role of m6A RNA methylation in cancer. Biomed. Pharmacother. 112, 108613. doi:10.1016/j.biopha.2019.108613
Tanaka, E. M., and Ferretti, P. (2009). Considering the evolution of regeneration in the central nervous system. Nat. Rev. Neurosci. 10 (10), 713–723. doi:10.1038/nrn2707
Tang, Y., and Le, W. (2016). Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. 53 (2), 1181–1194. doi:10.1007/s12035-014-9070-5
Tang, Z., Cao, J., Yao, J., Fan, X., Zhao, J., Zhao, M., et al. (2023). KDM1A-mediated upregulation of METTL3 ameliorates Alzheimer's disease via enhancing autophagic clearance of p-Tau through m6A-dependent regulation of STUB1. Free Radic. Biol. Med. 195, 343–358. doi:10.1016/j.freeradbiomed.2022.12.099
Walters, B. J., Mercaldo, V., Gillon, C. J., Yip, M., Neve, R. L., Boyce, F. M., et al. (2017). The role of the RNA demethylase FTO (fat mass and obesity-associated) and mRNA methylation in hippocampal memory formation. Neuropsychopharmacology 42 (7), 1502–1510. doi:10.1038/npp.2017.31
Wang, P., Doxtader, K. A., and Nam, Y. (2016). Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol. Cell 63 (2), 306–317. doi:10.1016/j.molcel.2016.05.041
Wang, P., Tang, W., Li, Z., Zou, Z., Zhou, Y., Li, R., et al. (2019). Mapping spatial transcriptome with light-activated proximity-dependent RNA labeling. Nat. Chem. Biol. 15 (11), 1110–1119. doi:10.1038/s41589-019-0368-5
Wang, W. Z., Li, M. W., Chen, Y., Liu, L. Y., Xu, Y., Xia, Z. H., et al. (2021). 3×Tg-AD mice overexpressing phospholipid transfer protein improves cognition through decreasing amyloid-β production and tau hyperphosphorylation. J. Alzheimers Dis. 82 (4), 1635–1649. doi:10.3233/JAD-210463
Wang, X., Zhao, B. S., Roundtree, I. A., Lu, Z., Han, D., Ma, H., et al. (2015). N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell 161 (6), 1388–1399. doi:10.1016/j.cell.2015.05.014
Wei, J., Liu, F., Lu, Z., Fei, Q., Ai, Y., He, P. C., et al. (2018). Differential m(6)A, m(6)A(m), and m(1)A demethylation mediated by FTO in the cell nucleus and cytoplasm. Mol. Cell 71 (6), 973–985. doi:10.1016/j.molcel.2018.08.011
Wen, J., Lv, R., Ma, H., Shen, H., He, C., Wang, J., et al. (2018). Zc3h13 regulates nuclear RNA m(6)A methylation and mouse embryonic stem cell self-renewal. Mol. Cell 69 (6), 1028–1038. doi:10.1016/j.molcel.2018.02.015
Wen, L., Sun, W., Xia, D., Wang, Y., Li, J., and Yang, S. (2022). The m6A methyltransferase METTL3 promotes LPS-induced microglia inflammation through TRAF6/NF-κB pathway. Neuroreport 33 (6), 243–251. doi:10.1097/WNR.0000000000001550
Wu, R., Li, A., Sun, B., Sun, J. G., Zhang, J., Zhang, T., et al. (2019). A novel m(6)A reader Prrc2a controls oligodendroglial specification and myelination. Cell Res. 29 (1), 23–41. doi:10.1038/s41422-018-0113-8
Wu, S., Zhang, S., Wu, X., and Zhou, X. (2020). m(6)A RNA methylation in cardiovascular diseases. Mol. Ther. 28 (10), 2111–2119. doi:10.1016/j.ymthe.2020.08.010
Xiao, W., Adhikari, S., Dahal, U., Chen, Y. S., Hao, Y. J., Sun, B. F., et al. (2016). Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol. Cell 61 (4), 507–519. doi:10.1016/j.molcel.2016.01.012
Xie, L. J., Yang, X. T., Wang, R. L., Cheng, H. P., Li, Z. Y., Liu, L., et al. (2019). Identification of flavin mononucleotide as a cell-active artificial N(6) -methyladenosine RNA demethylase. Angew. Chem. Int. Ed. Engl. 58 (15), 5028–5032. doi:10.1002/anie.201900901
Xu, H., Dzhashiashvili, Y., Shah, A., Kunjamma, R. B., Weng, Y. L., Elbaz, B., et al. (2020). m(6)A mRNA methylation is essential for oligodendrocyte maturation and CNS myelination. Neuron 105 (2), 293–309. doi:10.1016/j.neuron.2019.12.013
Yoshiyama, Y., Higuchi, M., Zhang, B., Huang, S. M., Iwata, N., Saido, T. C., et al. (2007). Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53 (3), 337–351. doi:10.1016/j.neuron.2007.01.010
Yue, Y., Liu, J., Cui, X., Cao, J., Luo, G., Zhang, Z., et al. (2018). VIRMA mediates preferential m(6)A mRNA methylation in 3'UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 4, 10. doi:10.1038/s41421-018-0019-0
Yun-Ling Zhang, X.-L. H., Li, Y-H., Zhao, C-X., Abudushalamu, X-L., Pang, H-Q., Sun, L., et al. (2021). International clinical practice guideline of Chinese medicine alzheimer. World J. Traditional Chin. Med. 7 (2), 265–275. doi:10.4103/wjtcm.wjtcm_28_21
Zeng, C., Huang, W., Li, Y., and Weng, H. (2020). Roles of METTL3 in cancer: Mechanisms and therapeutic targeting. J. Hematol. Oncol. 13 (1), 117. doi:10.1186/s13045-020-00951-w
Zhang, C., Samanta, D., Lu, H., Bullen, J. W., Zhang, H., Chen, I., et al. (2016a). Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m⁶A-demethylation of NANOG mRNA. Proc. Natl. Acad. Sci. U. S. A. 113 (14), E2047–E2056. doi:10.1073/pnas.1602883113
Zhang, C., Zhi, W. I., Lu, H., Samanta, D., Chen, I., Gabrielson, E., et al. (2016b). Hypoxia-inducible factors regulate pluripotency factor expression by ZNF217- and ALKBH5-mediated modulation of RNA methylation in breast cancer cells. Oncotarget 4, 64527–64542. doi:10.18632/oncotarget.11743
Zhang, H., Shi, X., Huang, T., Zhao, X., Chen, W., Gu, N., et al. (2020). Dynamic landscape and evolution of m6A methylation in human. Nucleic Acids Res. 48 (11), 6251–6264. doi:10.1093/nar/gkaa347
Zhang, H., Wei, W., Zhao, M., Ma, L., Jiang, X., Pei, H., et al. (2021). Interaction between Aβ and tau in the pathogenesis of Alzheimer's disease. Int. J. Biol. Sci. 17 (9), 2181–2192. doi:10.7150/ijbs.57078
Zhang, P., Kishimoto, Y., Grammatikakis, I., Gottimukkala, K., Cutler, R. G., Zhang, S., et al. (2019). Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 22 (5), 719–728. doi:10.1038/s41593-019-0372-9
Zhang, Z., Wang, M., Xie, D., Huang, Z., Zhang, L., Yang, Y., et al. (2018). METTL3-mediated N(6)-methyladenosine mRNA modification enhances long-term memory consolidation. Cell Res. 28 (11), 1050–1061. doi:10.1038/s41422-018-0092-9
Zhao, B. S., Roundtree, I. A., and He, C. (2017). Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 18 (1), 31–42. doi:10.1038/nrm.2016.132
Zhao, F., Xu, Y., Gao, S., Qin, L., Austria, Q., Siedlak, S. L., et al. (2021). METTL3-dependent RNA m(6)A dysregulation contributes to neurodegeneration in Alzheimer's disease through aberrant cell cycle events. Mol. Neurodegener. 16 (1), 70. doi:10.1186/s13024-021-00484-x
Keywords: Alzheimer’s disease, m6A modification, synaptic loss, animal model, techniques
Citation: Xia L, Zhang F, Li Y, Mo Y, Zhang L, Li Q, Luo M, Hou X, Du Z, Deng J and Hao E (2023) A new perspective on Alzheimer’s disease: m6A modification. Front. Genet. 14:1166831. doi: 10.3389/fgene.2023.1166831
Received: 15 February 2023; Accepted: 26 April 2023;
Published: 09 May 2023.
Edited by:
Serge Weis, Kepler University Hospital GmbH, AustriaReviewed by:
Zhenyu Wu, First People’s Hospital of Foshan, ChinaCopyright © 2023 Xia, Zhang, Li, Mo, Zhang, Li, Luo, Hou, Du, Deng and Hao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhengcai Du, ZHV6aGVuZ2NhaThAMTYzLmNvbQ==; Jiagang Deng, ZGVuZ2pnNTNAMTI2LmNvbQ==; Erwei Hao, aGFvZXdAZ3h0Y211LmVkdS5jbg==
†These authors have contributed equally to this work and share first authorship
‡ORCID ID: Fan Zhang, https://orcid.org/0000-0001-7230-7077; Xiaotao Hou, https://orcid.org/0000-0001-8251-220X; Zhengcai Du , https://orcid.org/0000-0002-9239-3336; Jiagang Deng, https://orcid.org/0000-0003-2079-981X; Erwei Hao, https://orcid.org/0000-0001-7394-6022
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.