 Zhen Li
Zhen Li Wanyu Cheng
Wanyu Cheng Feiyin Zi1
Feiyin Zi1 Xiaoyu Huang
Xiaoyu Huang Xunlun Sheng
Xunlun Sheng Weining Rong
Weining Rong- 1Ningxia Eye Hospital, People’s Hospital of Ningxia Hui Autonomous Region, Third Clinical Medical College of Ningxia Medical University, Yinchuan, China
- 2Department of Ophthalmology, Qingdao West Coast New District Central Hospital, Qingdao, China
- 3Gansu Aier Optometry Hospital, Lanzhou, China
Purpose: To investigate pathogenic variants in six families with cone–rod dystrophy (CORD) presenting various inheritance patterns by using whole-exome sequencing (WES) and analyzing phenotypic features.
Methods: A total of six families with CORD were enrolled in Ningxia Eye Hospital for this study. The probands and their family members received comprehensive ophthalmic examinations, and DNA was abstracted from patients and family members. Whole-exome sequencing was performed on probands to screen the causative variants, and all suspected pathogenic variants were determined via Sanger sequencing. Furthermore, co-segregation analysis was performed on available family members. The pathogenicity of novel variants was predicted using in silico analysis and evaluated according to the American College of Medical Genetics and Genomics (ACMG) guidelines.
Results: Of the six families, two families were assigned as X-linked recessive (XL), two families were assigned as autosomal recessive (AR), and two families were assigned as autosomal dominant (AD). Pathogenic variants were detected in CACNA1F in two X-linked recessive probands, among which family 1 had a hemizygous frameshift variant c.2201del (p.Val734Glyfs*17) and family 2 had a hemizygous missense variant c.245G>A (p.Arg82Gln). Both probands had high myopia, with fundus tessellation accompanied by abnormalities in the outer structure of the macular area. The homozygous splice variant c.2373 + 5G>T in PROM1 and the homozygous nonsense variant c.604C>T (p.Arg202Ter) in ADAM9 were detected in two autosomal recessive families of the probands. Both probands showed different degrees of atrophy in the macular area, and the lesions showed hypofluorescence changes in autofluorescence. The heterozygous variation in CRX c.682C>T (p.Gln228Ter) was detected in two autosomal dominant families. The onset age of the two probands was late, with better vision and severe macular atrophy. According to ACMG guidelines and the analysis of online in silico tools, all variations were labeled as potentially harmful or pathogenic.
Conclusion: Pathogenic variants in CACNA1F, PROM1, ADAM9, and CRX genes were identified in six families affected by the diverse inheritance patterns of CORD. Furthermore, the potential impact of the nonsense-mediated decay (NMD) mechanism on the manifestation of CORD phenotypes was examined and addressed. Simultaneously, the spectrum of pathogenic variants and clinical phenotypes associated with the CORD gene was extended.
1 Introduction
Cone–rod dystrophy (CORD) is a prevalent type of monogenic retinal disease observed in clinical settings. The condition is primarily characterized by atypical or predominantly atypical cone cell functionality, accompanied by various levels of atypical rod cell functionality. Its occurrence is estimated to be approximately 1 in 40,000 individuals (Thiadens et al., 2012). It usually presents as a progressive vision loss starting in adolescence or early adulthood, which may be accompanied by photophobia and varying degrees of color vision abnormalities (Boulanger-Scemama et al., 2019). In the advanced phases, certain individuals may exhibit nystagmus and experience a gradual decline in visual acuity, which may be accompanied by nyctalopia (Boulanger-Scemama et al., 2019).
The inheritance patterns of CORD include autosomal dominant (AD), autosomal recessive (AR), and X-linked recessive (XL) (Wawrocka et al., 2018). So far, 37 genes have been reported in the RetNet database associated with CORD (RetNet: https://sph.uth.edu/Retnet/sum-dis.htm), of which 10 genes were related to autosomal dominant [AIPL1 (Sohocki et al., 1998), CRX (Sohocki et al., 1998), GUCA1A (Downes et al., 2001), GUCY2D (Kelsell et al., 1998), PITPNM3 (Balciuniene et al., 1995), PROM1 (Michaelides et al., 2003), PRPH2 (Arikawa et al., 1992), RIMS1 (Johnson et al., 2003), SEMA4A (Abid et al., 2006), and UNC119 (Kobayashi et al., 2000)], 25 genes were related to autosomal recessive [ABCA4 (Allikmets, 2000), ADAM9 (Danciger et al., 2001), ATF6 (Kohl et al., 2015), C21orf2 (Abu-Safieh et al., 2013), C8orf37 (van Huet et al., 2013), CACNA2D4 (Wycisk et al., 2006), CDHR1 (Ostergaard et al., 2010), CEP78 (Nikopoulos et al., 2016), CERKL (Aleman et al., 2009), CNGA3 (Wissinger et al., 1998), CNGB3 (Michaelides et al., 2004), CNNM4 (Downey et al., 2002), DYNC2I2 (Solaguren-Beascoa et al., 2021), GNAT2 (Aligianis et al., 2002), IFT81 (Dharmat et al., 2017), KCNV2 (Wu et al., 2006), PDE6C (Thiadens et al., 2009), PDE6H (Piri et al., 2005), POC1B (Durlu et al., 2014), RAB28 (Roosing et al., 2013), RAX2 (Wang et al., 2004), RDH5 (Nakamura et al., 2000), RPGRIP1 (Hameed et al., 2003), SLC4A7 (Ahn et al., 2020), and TTLL5 (Sergouniotis et al., 2014)], and two genes were related to X-linked patterns [CACNA1F (Jalkanen et al., 2003a) and RPGR (Yang et al., 2002)].
The earlier diagnosis of CORD mostly relied on clinical and familial history queries, multimodal imaging assessment, and electrophysiological testing (den Hollander et al., 2010). Due to its genetic complexity and clinical variability, CORD exhibits diverse clinical symptoms and signs that may vary throughout different phases. Furthermore, there exists phenotypic similarity between CORD and other inherited retinal diseases (Hamel, 2007). Therefore, accurately diagnosing a condition purely based on clinical presentations or genetic testing outcomes poses a challenge. The integration of genetic screening and clinical phenotypic analysis has the potential to enhance the efficacy and precision of clinical diagnosis in individuals affected by CORD (Birtel et al., 2018a). In this study, whole-exome sequencing (WES) was applied to detect variants in six CORD pedigrees with different inheritance modes, genotypes, and phenotypes; meanwhile, the results are presented.
2 Materials and methods
2.1 Ethical approval
The Ningxia Hui Autonomous Region People’s Hospital Ethics Committee granted approval for the study (approval number 20190909). All participants signed an informed consent form. All experiments were conducted in accordance with the Declaration of Helsinki.
2.2 Clinical data collection
Six CORD pedigrees were recruited from Ningxia Eye Hospital, People’s Hospital of Ningxia Hui Autonomous Region in 2021. The probands and members of their families underwent the required ophthalmologic examinations, including best-corrected visual acuity (BCVA), slit-lamp microscopy, chromoptometry (fifth version color imaginative and prescient examination plates, Ziping YU), dilated fundus examination with photographs (TRC-50DX, Topcon Inc.), optical coherence tomography (OCT, HDOCT4000, Carl Zeiss Meditec, United States), electroretinogram (ERG), and fundus autofluorescence (FAF).
2.3 Whole-exome sequencing
Peripheral venous blood (5 mL) samples were obtained from all participants for genomic DNA extraction using a QIAmp DNA Mini Blood Kit (Qiagen, Hilden, Germany). Whole-exome sequencing was performed on probands. The Agilent SureSelect exon capture kit was previously used to capture the exome. A high-throughput sequencer was once utilized to provide sequencing services (Illumina, HiSeq X Ten). Illumina base-calling software 1.7 was used to evaluate the raw sequencing data, which were then compared to the NCBI human genomic DNA reference sequence (NCBI construct 37.1). SOAP (http://soap.genomics.org.cn) and BWA software applications (http://bio-bwa.sourceforge.net/) were used to examine single-nucleotide variants (SNVs) and insertion and deletion editions (Indel). To reflect every variation present in the samples’ DNA sequences, co-segregation of the genotype and phenotype was previously established in ordinary household members, and Sanger validation was originally employed to remove false positives for potentially harmful variants. According to the genetic patterns of AD, AR, and XL, the family history was analyzed to establish their inheritance pattern.
2.4 In silico analysis
Standards and Guidelines for the Interpretation of Sequence Variants established in 2015 by the American College of Medical Genetics and Genomics (ACMG) were used to evaluate the pathogenicity of novel variations for genetic variation. The variant sites were filtered and screened by inserting the sites into the normal human databases, including the 1,000 genomes with normal population gene frequency, the Exome Aggregation Consortium (ExAC), and ExAC-EAS (approximately 4,000 East Asian data under ExAC). MAF <0.005 was used as a criterion to exclude benign variants. The gnomAD (all_gnomAD and eas_gnomAD) was used to analyze the frequency of variants in the normal population (and the normal East Asian population) of the gnomAD. Tools such as MutationTaster (http://mutationtaster.org/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.jcvi.Org/www/SIFT.chr.coords.submit.html), CADD (https://cadd.gs.washington.edu/score), and REVEL (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5065685/) were used to predict the effects of variant sites on protein function. The measurements of the conservation of gene sequences across species in evolution were made using websites like GERP++ (https://bio.tools/gerp). If the projected score (HDIV/HVAR) of PolyPhen-2 is close to 1, then it is likely to be harmful (D); otherwise, it is either possibly damaging (P) or benign (B). The sequences underwent SIFT analysis, and amino acids that have probabilities below 0.05 are considered (D). The disease probability increases as the MutationTaster analysis score approaches 1, whose scores range from 0 to 1. REVEL is an ensemble score based on 13 individual scores for predicting the pathogenicity of missense variants, and its scores range from 0 to 1. The larger the score, the more likely the SNP to be damaging. The CADD raw score was used for the functional prediction of SNP. The larger the score, the more likely the SNP to be damaging. Its scores range from−6.458163 to 18.301497 in dbNSFP. The CADD PHRED-like score is a PHRED-like rank score based on whole-genome CADD raw scores. The larger the score, the more likely the SNP to be damaging. When considering the anticipated values for the conservativeness of the amino acid sequence, GERP++ values larger than 2 imply that the sequence is relatively conservative. The sequence is assumed to be highly conserved and deleterious if it has a high GERP score. When all predictions turned out to be accurate, variations were categorized as potentially pathogenic when used in conjunction with further data. Pathogenic variants were defined as frameshift, nonsense, and variants with the experimental proof of causing the loss of protein function. For the conservativeness study of variant loci, the online analysis tool MultAlin (http://sacs.ucsf.edu/cgi-bin/multalin.py) was employed. The spatial structures of these proteins were modeled using the freeware programs AlphaFold2 and Missense3D and then aligned using PyMOL 2.3.
3 Results
3.1 Family 1
The proband of family 1 was a 34-year-old man who complained of reduced vision in both eyes with photophobia for more than 10 years. He denied a consanguineous marriage history (Figure 1 F1-A). He had dyserythrochloropsia and BCVA of 0.4 in both eyes (Table 1). The anterior segment was normal, fundus tessellation with no reflection was observed in the macular fovea, and the peripheral retina did not show obvious bone-spicule pigmentation. OCT showed the disappearance of the light reflection signal in the chimeric zone of fovea centralis, and autofluorescence showed hypofluorescence in the macula of both eyes (Figure 2-F1). ERG recording revealed a mild decline in scotopic and severe or moderately reduced photopic responses (Table 1). The condition was diagnosed as CORD according to the clinical phenotype. The proband and his affected cousin carried a hemizygous frameshift variant c.2201del (p.Val734Glyfs*17) in CACNA1F, and Sanger sequencing showed that the unaffected mother and aunt carried the same heterozygous variant in CACNA1F (Table 2) (PP1_Supporting), suggesting the co-segregation of a genotype and clinical phenotype (Figure 1 F1-A), consistent with the mode of the X-linked recessive inheritance. The East Asian population (ExAC_EAS) and gnomAD databases and prior reports of the frameshift variant were negative (PM2_Moderate). The c.2201del variant led to a premature stop codon at position 17 of the new reading frame and caused a frameshift beginning with codon valine 734, changing this amino acid to a glycine residue (p.Val734Glyfs*17). Most of the proteins produced as a consequence of the truncated variants that were inactive or lost their normal function, which resulted in the premature termination of polypeptide chain synthesis (PVS1_Very Strong). It is anticipated that CACNA1F protein production (including truncated polypeptides) will be eliminated from mutant alleles by nonsense-mediated mRNA decay (NMD).Various bioinformatics computing software programs indicated the deleterious impact of this variant (PP3_Supporting). Moreover, proteome conservation analysis revealed that the amino acid at position 734 was substantially conserved across several taxa (Figure 1 F1-C), indicating that the variation at this location is more likely to have an impact on the CACNA1F protein’s structure and function. According to ACMG guidelines, we therefore, believed that the hemizygous frameshift variation c.2201del (p.Val734Glyfs*17) in CACNA1F was more likely the pathogenic variant in family 1 with X-linked recessive CORD (PVS1 + PM2 + PP1 + PP3).
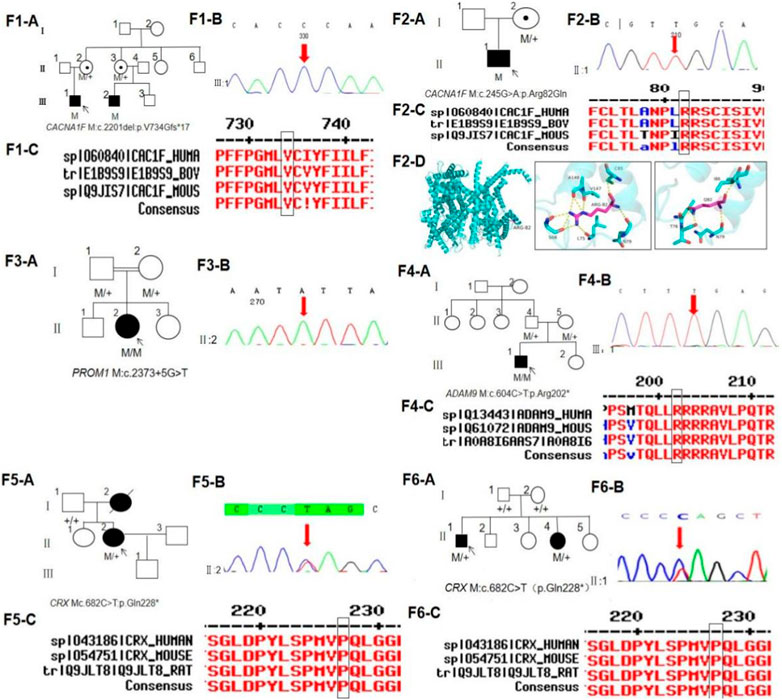
FIGURE 1. Pedigree, sequence analysis of six families with CORD. (F1-A) Pedigree of family 1. The filled black symbols represent the affected members, and the arrow denotes the proband. (F1-B) Sequence chromatograms of identified mutations in family 1. (F1-C) Homology of amino acid sequences between human CACNA1F and other species. The amino acid at position 734 is highly conserved among species. Mutated residue 734 is boxed and indicated. (F2-A) Pedigree of family 2. (F2-B) Sequence chromatograms of identified mutations in family 2. (F2-C) Homology of amino acid sequences between human CACNA1F and other species. The amino acid at position 82 is highly conserved among species. Mutated residue 82 is boxed and indicated. (F2-D) Homology model of the CACNA1F homeodomain (green) of family 2. Purple indicates the location of p.Arg82Gln in the protein structure. (F3-A) Pedigree of family 3. (F3-B) Sequence chromatograms of identified mutations in family 3. (F4-A) Pedigree of family 4. (F4-B) Sequence chromatograms of identified mutations in family 4. (F4-C) Homology of amino acid sequences between human ADAM9 and other species. The amino acid at position 202 is highly conserved among species. Mutated residue 202 is boxed and indicated. (F5-A) Pedigree of family 5. (F5-B) Sequence chromatograms of identified mutations in family 5. (F5-C) Homology of amino acid sequences between human CRX and other species. The amino acid at position 228 is highly conserved among species. Mutated residue 202 is boxed and indicated. (F6-A) Pedigree of family 6. (F6-B) Sequence chromatograms of identified mutations in family 6. (F6-C) Homology of amino acid sequences between human CRX and other species.
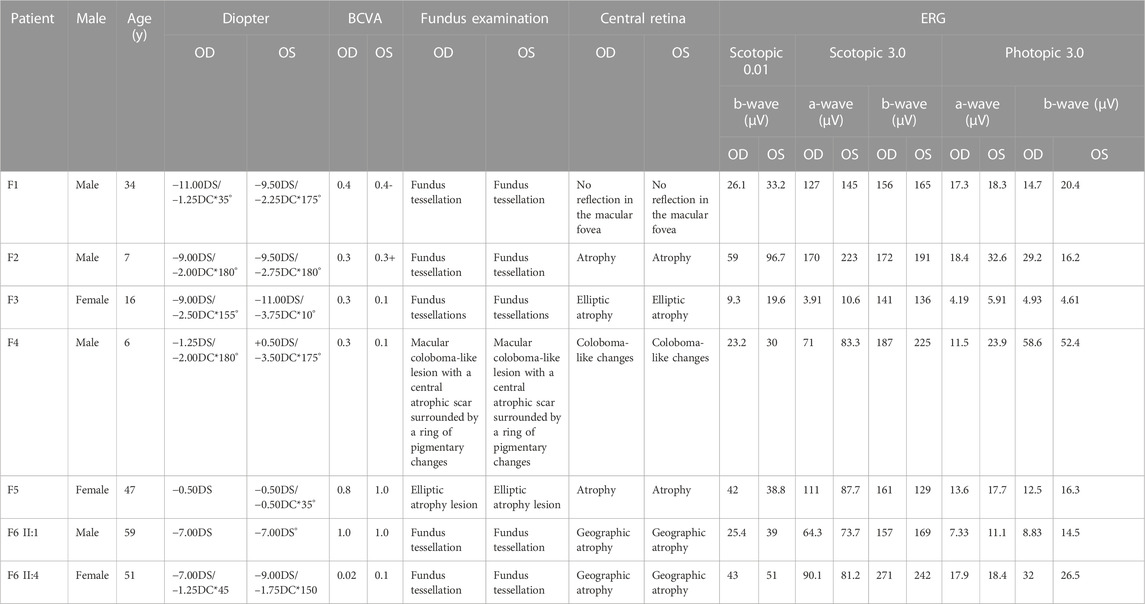
TABLE 1. Clinical phenotype and ERG data in six Chinese families with CORD.
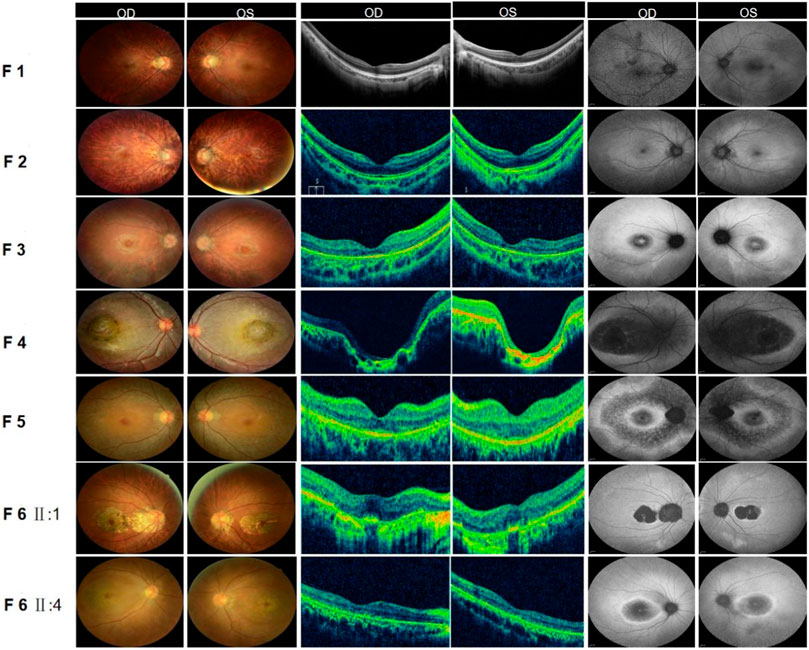
FIGURE 2. Fundus examination of six families with CORD. (F1) Fundus of both eyes in family 1: Fundus tessellation with no reflection was observed in the macular fovea; the peripheral retina did not show obvious bone-spicule pigmentation; OCT of the macula in both eyes suggested the disappearance of the light reflection signal in the chimeric zone of the fovea centralis; autofluorescence in both eyes suggested hypofluorescence in the macula. (F2) Fundus of both eyes in family 2: Fundus tessellation with macular atrophy was observed; the peripheral retina did not show obvious bone-spicule pigmentation; OCT of the macula in both eyes showed the thinning of the outer layer of the macula and the disappearance of the light reflection signal in the ellipsoid zone and chimeric zone of the fovea centralis; autofluorescence in both eyes suggested hypofluorescence in the macula. (F3) Fundus of both eyes in family 3: Fundus tessellation with an elliptic atrophy lesion was observed in the macular area and the peripheral retina did not show obvious bone-spicule pigmentation; OCT of the macula in both eyes showed significant atrophy and the thinning of the outer retina in the macula; autofluorescence in both eyes suggested that hyperfluorescence lesions were surrounded by low fluorescence in the macula of both eyes. (F4) Fundus of both eyes in family 4: Central retina with a macular coloboma-like lesion with a central atrophic scar surrounded by a ring of pigmentary changes, including a yellowish pigmentation of both eyes, was observed; OCT of the macula in both eyes showed the atrophic and ill-defined retinal layers, alterations and disruptions of the retinal pigment epithelium, and macular posterior staphyloma; autofluorescence showed hypofluorescence in the macula of both eyes. (F5) Fundus of both eyes in family 5: Elliptic atrophy lesion with no foveal reflection was observed in the macular area; OCT of the macula in both eyes showed atrophy of the outer retina and RPE layer in the macula; autofluorescence results of both eyes suggested that hyperfluorescence lesions were surrounded by low fluorescence in the macula of both eyes. (F6) Fundus of both eyes in family 6: Fundus photograph: The proband (II:1) and his sister (II: 4) all showed fundus tessellation with geographic atrophy in the macula; OCT: The proband (II:1) showed obvious thinning of central retinal thickness, and the outer nuclear layer and the ellipsoid zone disappeared in both eyes; OCT of his sister (II: 4) showed atrophy of the outer retina and RPE layer in the macula; FAF: The proband (II:1) and his sister (II: 4) all showed that hyperfluorescence lesions were surrounded by low fluorescence in the macula of both eyes.
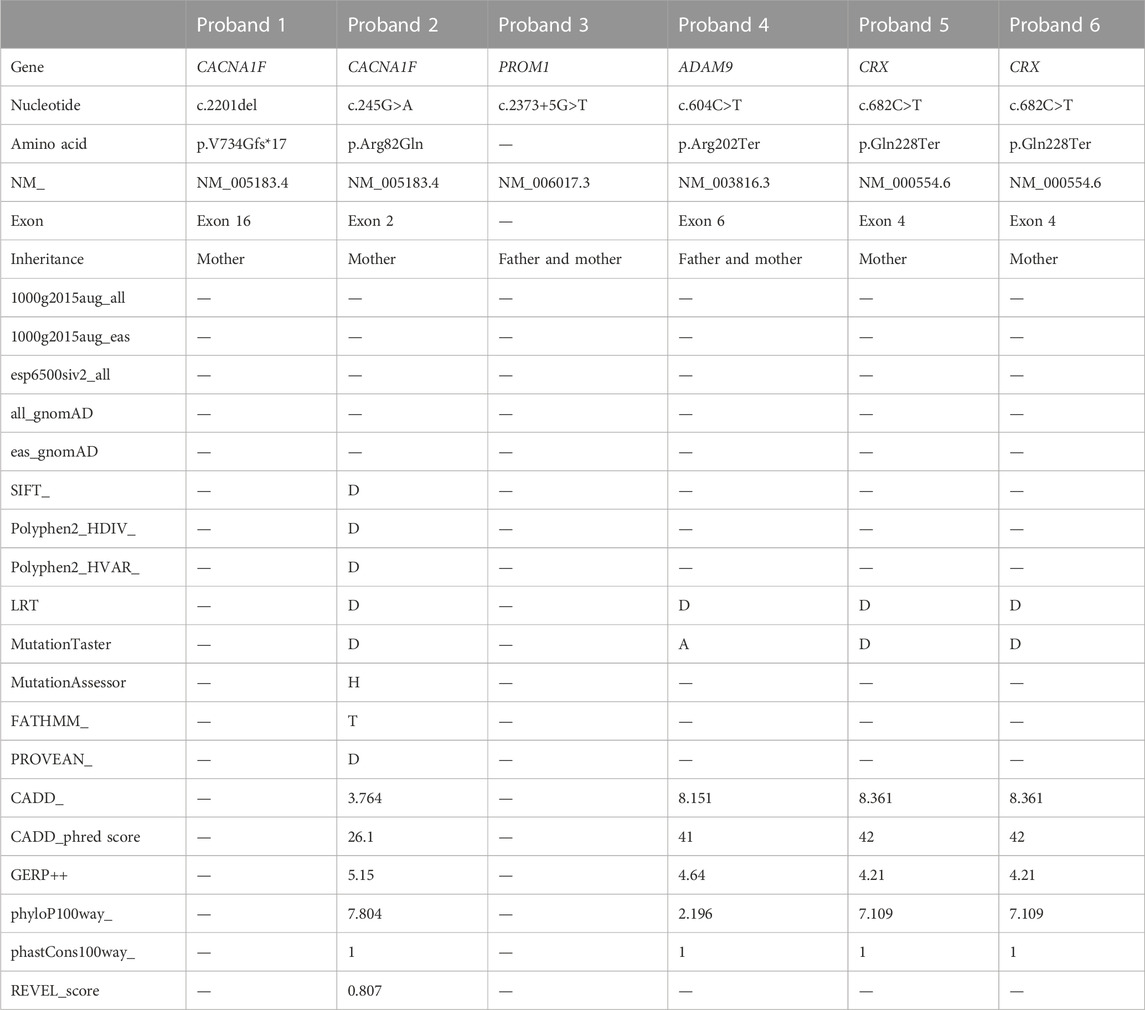
TABLE 2. Bioinformatics and pathogenicity analyses of variants in six Chinese families with CORD.
3.2 Family 2
The proband of family 2 was a 7-year-old boy who complained of progressively reduced vision in both eyes with photophobia for 1 year. He denied a family history of hereditary disease and consanguineous marriage (Figure 1 F2-A). He had dyserythrochloropsia and BCVA of 0.3 in both eyes (Table 1). The anterior segment was normal, fundus tessellation with macular atrophy was observed, and the peripheral retina did not show obvious bone-spicule pigmentation. OCT showed thinning of the outer layer of the macula and the disappearance of the light reflection signal in the ellipsoid zone and chimeric zone of fovea centralis, and autofluorescence in both eyes suggested hypofluorescence in the macula (Figure 2-F2). ERG recording showed slightly reduced scotopic and significantly diminished photopic responses (Table 1). The condition was diagnosed as CORD according to the clinical phenotype. The proband carried a hemizygous missense variant c.245G>A (p.Arg82Gln) in CACNA1F, and Sanger sequencing showed that the proband’s unaffected mother carried the same heterozygous variant in CACNA1F (Table 2), consistent with the mode of X-linked recessive inheritance. The East Asian population (ExAC_EAS) and the gnomAD databases and prior reports of the variant were negative (PM2_Moderate). This variant has been reported in the literature in congenital stationary night blindness and early detection of high myopia (Zeitz et al., 2015; Sun et al., 2015; Zeitz et al., 2019) (PS1_Strong). The substitution, c.245G>A, caused the amino acid change from arginine to glutamine at residue 82 (p.Arg82Gln), and four software predictions of SIFT, PolyPhen-2, CADD, and REVEL_score indicated the deleterious impact of this variant (PP3_Supporting). Moreover, proteome conservation analysis revealed that the amino acid at position 82 was substantially conserved across several taxa (Figure 1 F2-C), indicating that the variation at this location is more likely to have an impact on the CACNA1F protein’s structure and function. The codon arginine 82 was located in the topological domain. The missense variant c.245G>A (p.Arg82Gln) would lead to the conversion of charged residues to uncharged residues, and the interacting amino acid residues would change from S68, L75, N79, C85, V147, and A148 to T76, N79, and I86, resulting in topological changes that affect protein function (Figure 1 F2-D). According to ACMG guidelines, we therefore believed that the hemizygous missense variant c.245G>A (p.Arg82Gln) in the CACNA1F gene was more likely the pathogenic variant in family 2 with X-linked recessive CORD (PS1 + PM2 + PP3).
3.3 Family 3
The proband of family 3 was a 16-year-old girl who complained of reduced vision in both eyes for 3 years, and she had a history of consanguineous marriage of her parents (Figure 1 F3-A). She had dyserythrochloropsia in both eyes. BCVA was 0.3 in the right eye and 0.1 in the left eye (Table 1). The anterior segment was normal, fundus tessellation with elliptic atrophy lesion was observed in the macular area, and the peripheral retina did not show obvious bone-spicule pigmentation. OCT showed significant atrophy and the thinning of the outer retina in the macula, and autofluorescence suggested that hyperfluorescence lesions were surrounded by low fluorescence in the macula of both eyes (Figure 2-F3). ERG suggested a significant decrease in response amplitude for both scotopic adaptation and photopic adaptation (Table 1). The condition was diagnosed as CORD according to the clinical phenotype. The proband carried a homozygous splice variant c.2373+5G>T in PROM1, and both the phenotypically normal father and mother carried the same heterozygous variant at this locus as validated by Sanger sequencing (Table 2), suggesting the co-segregation of the genotype and clinical phenotype, consistent with the mode of an autosomal recessive inheritance. The variant has not been detected in the East Asian population (ExAC_EAS) and gnomAD databases. This variant has been identified in the literature in a consanguineous family member with retinitis pigmentosa (Chen et al., 2013). The proband was clinically consistent with the disease caused by this gene, and various bioinformatics computing software programs predicted that the variant affected mRNA splicing. We therefore believed that the homozygous splice variant c.2373+5G>T in PROM1 was more likely the pathogenic variant present in family 3 with CORD.
3.4 Family 4
The proband of family 4 was a 6-year-old boy who complained of reduced vision in both eyes for 3 years. He denied a family history of hereditary disease and consanguineous marriage (Figure 1 F4-A). He had dyserythrochloropsia in both eyes. BCVA was 0.3 in the right eye and 0.1 in the left eye (Table 1). The anterior segment was normal and the central retina had a macular coloboma-like lesion with a central atrophic scar surrounded by a ring of pigmentary changes, including a yellowish pigmentation of both eyes. OCT showed the atrophic and ill-defined retinal layers, alterations, and disruptions of the retinal pigment epithelium and macular posterior staphyloma. Autofluorescence showed hypofluorescence in the macula of both eyes (Figure 2-F4). ERG results suggested a mild reduction in the b-wave amplitude of the photopic cone response and a severe reduction in the b-wave amplitude of the scotopic rod response in both eyes (Table 1). The condition was diagnosed as CORD according to the clinical phenotype. The proband carried a homozygous nonsense variant c.604C>T (p.Arg202Ter) in ADAM9, and the phenotypically normal father and mother carried the same heterozygous variant (Table 2). The variant has not been detected in the East Asian population (ExAC_ EAS) and gnomAD databases, and it has not been reported previously (PM2_Moderate). The nonsense variant, c.604C>T, was anticipated to create an early termination codon at residue 202, causing the early termination of polypeptide chain synthesis. As a result, the majority of the proteins generated were inactive or lost their usual function (PVS1_Very Strong), and various bioinformatics computing software programs indicated the deleterious impact of this variant (PP3_Supporting). It is anticipated that mutant alleles’ expression of the ADAM9 protein (including shortened polypeptides) will be eliminated via NMD. Moreover, proteome conservation analysis revealed that the amino acid at position 202 was substantially conserved across several taxa (Figure 1 F4-C), indicating that the variation at this location is more likely to have an impact on the ADAM9 protein’s structure and function. According to ACMG guidelines, we therefore believed that the homozygous nonsense variant c.604C>T (p.Arg202Ter) in ADAM9 was more likely the pathogenic variant present in family 4 with CORD (PVS1 + PM2 + PP3).
3.5 Family 5
The proband of family 5 was a 47-year-old woman who complained of poor night vision in both eyes for 1 year. She denied a consanguineous marriage history, and her mother had impaired vision and night blindness for many years (Figure 1 F5-A). The proband had normal color vision, and BCVA was 0.8 in the right eye and 1.0 in the left eye (Table 1). Elliptic atrophy lesion was observed in the retinal region with no foveal reflection, and the anterior segment was normal. OCT showed atrophy of the outer retina and RPE layer in the macula, and autofluorescence suggested that hyperfluorescence lesions were surrounded by low fluorescence in the macula of both eyes (Figure 2-F5). ERG recording revealed a moderate decline in scotopic and severe or moderately reduced photopic responses (Table 1). The condition was diagnosed as CORD according to the clinical phenotype. The proband carried the heterozygous nonsense variant c.682C>T (p.Gln228Ter) in CRX (Figure 1 F5-B Table 2). This variant has not been previously reported and was also not detected in the East Asian population (ExAC_EAS) and gnomAD databases (PM2_Moderate). This variant has been reported in the literature in a retinitis pimentosa patient (Ge et al., 2015) (PP5_Supporting). The nonsense variant, c.682C>T, was predicted to generate a premature termination codon at residue 228, which would result in a premature termination of polypeptide chain synthesis, and most of the proteins produced were inactive or lost their normal function (PVS1_Very Strong), and various bioinformatics computing software programs indicated the deleterious impact of this variant (PP3_Supporting). The nonsense variation was predicted to cause protein truncation length >10% but not nonsense-mediated mRNA degradation because it was positioned in the final exon. Moreover, proteome conservation analysis revealed that the amino acid at position 228 was substantially conserved across several taxa (Figure 1 F5-C), indicating that the variation at this location is more likely to have an impact on the CRX protein’s structure and function. According to ACMG guidelines, we therefore believed that the heterozygous nonsense variant c.682C>T (p.Gln228Ter) in CRX was more likely the pathogenic variant observed in family 4 with CORD (PVS1 + PM2 + PP3 + PP5).
3.6 Family 6
The proband of family 6 was a 59-year-old man who complained of reduced vision in both eyes for 3 years. He denied a consanguineous marriage history, and his younger sister had severe vision loss (Figure 1 F6-A). His color vision was normal, and both of his eyes had a BCVA of 1.0 (Table 1). The anterior segment was normal, and fundus tessellation with geographic atrophy was observed in the macula. OCT showed obvious thinning of central retinal thickness, and the outer nuclear layer and the ellipsoid zone disappeared in both eyes. Autofluorescence showed hypofluorescence lesions in the macula of both eyes (Figure 2-F6 Ⅱ:1). A slight fall in scotopic and a sharp reduction in photopic responses were detected in ERG recordings (Table 1). His younger sister had a normal color vision, and BCVA was 0.02 in the right eye and 0.1 in the left eye (Table 1). The anterior segment was normal, and fundus tessellation with macular atrophy was observed. OCT showed atrophy of the outer retina and RPE layer in the macula, and autofluorescence suggested that hyperfluorescence lesions were surrounded by low fluorescence in the macula of both eyes (Figure 2-F6 II:4). ERG recording revealed a moderate decline in scotopic and severe or moderately reduced photopic responses (Table 1). The condition was diagnosed as CORD in both patients according to the clinical phenotype. The proband as well as his younger sister carried the heterozygous nonsense variant c.682C>T (p.Gln228Ter) in CRX (Table 2). Moreover, proteome conservation analysis revealed that the amino acid at position 228 was substantially conserved across several taxa (Figure 1 F6-C), indicating that the variation at this location is more likely to have an impact on the CRX protein’s structure and function.
4 Discussion
X-linked cone–rod dystrophy (CORDX) is characterized by genetic variations found in three specific loci on the X chromosome. Particularly, CORDX1 is caused by variants in RPGR (Xp21.1), CORDX2 is located at Xq27.2−28, and CORDX3 is caused by variants in CACNA1F (Xp11.23) (Jalkanen et al., 2003b; Zeitz et al., 2015). The voltage-dependent calcium channel α1F subunit (CACNA1F) gene, located on human chromosome Xp11.23, contains 48 exons and encodes a retina-specific expression of the Cav1.4 ion channel α1F subunit consisting of 1,977 amino acids (McRory et al., 2004; McRory et al., 2004). Cav1.4 is typically observed in the outer plexiform, inner nuclear, inner plexiform, and nerve fiber layers of the retina (McRory et al., 2004). Furthermore, it is also localized at the distal regions of optic rods and cone cells and facilitates the exocytosis of neurotransmitters through the ribbon synapse of retinal photoreceptors, hence exerting a crucial function in the transmission of signals from photoreceptors to secondary retinal neurons (Morgans, 2001). CACNA1F is the first gene found on the X chromosome to cause congenital stationary night blindness (CSNB) (Bech-Hansen et al., 1998). The identification of a splicing variant in the CACNA1F gene was initially reported in a family affected with CSNB in Finland in the year 2006 (Jalkanen et al., 2006), confirming the association of such gene with the occurrence of CORDX. Subsequent reports have successively confirmed the association of CACNA1F with the occurrence of CORDX (Hauke et al., 2013). In the present investigation, it was shown that individuals harboring the CACNA1F variation exhibited fundus tessellation, concomitant with irregularities in the outer macular layer. Nevertheless, it is worth noting that the patient harboring the nonsensical variant exhibited an earlier onset of symptoms and a decline in visual acuity, potentially indicating an association with genetic variability. The variant locus c.245G>A (p.Arg82Gln) in CACNA1F has been previously reported in families with CSNB (Zeitz et al., 2015), but the clinical phenotype was not described in detail, whereas the patient carrying the same variant in this study had progressive vision loss as the chief complaint, and the patient did not complain of night blindness, and as questioned repeatedly, we diagnosed the patient with CORDX in combination with the mild decrease in the amplitudes of b-wave of scotopic 0.01 ERG and a- and b-waves of 3.0 ERG and the severe decrease in the amplitudes of a- and b-wave of photopic 3.0 ERG. Additionally, some studies found that CACNA1F is closely associated with the onset of Aland Island eye disease (Jalkanen et al., 2007), characterized by fundus pigmentation, reduced visual acuity, nystagmus, astigmatism, color vision defects, and dark maladjustment. Thus, it is evident that there is a significant overlap in the clinical phenotypes of the three diseases caused by CACNA1F. Hence, conducting a thorough and extensive clinical examination and analysis can serve to elucidate the diagnosis and mitigate the occurrence of misinterpretation and overlooked diagnoses in clinical settings, alongside the utilization of genetic testing.
PROM1 is a transmembrane glycoprotein originally thought to be a biomarker for human hematopoietic stem and progenitor cells (Miraglia et al., 1997) and in the retina. PROM1 is mainly localized in the outer segment of retinal photoreceptors and plays a key role in the membranous disc morphogenesis of the photoreceptor outer segment (Yang et al., 2008). Some studies have shown that in CORD, approximately 1%–9.5% of cases are caused by the PROM1 variant (Littink et al., 2010; Boulanger-Scemama et al., 2015; Birtel et al., 2018b). Overall, the PROM1 variant is associated with multiple diseases with overlapping phenotypes, such as retinitis pigmentosa, cone–rod dystrophy, Stargardt-like macular dystrophy, and bull’s eye macular dystrophy (Littink et al., 2010; Boulanger-Scemama et al., 2015; Birtel et al., 2018b; Michaelides et al., 2010). Depending on the variant, the age of the onset, first symptoms, and the severity of the disease vary among patients. The PROM1 variant can present both recessive and dominant genetic inheritance (Boulanger-Scemama et al., 2015). Compared with retinal dystrophy caused by the dominantly inherited PROM1 variant, patients with retinal dystrophy associated with recessive inherited PROM1 variants, such as CORD and Leber congenital amaurosis, have an early onset and more severe clinical phenotype and can present high myopia and vision loss at the early stage of the disease (Michaelides et al., 2010; Eidinger et al., 2015; Cehajic-Kapetanovic et al., 2019), which is highly consistent with the clinical phenotype of the patient in this study. The majority of frameshift and nonsense variations caused premature termination codons, which hastens the nonsense-mediated decay of shortened modified RNAs before translation (Maw et al., 2000). Missense and splice variants associated with recessive disease may cause null (or near null loss of function) effects, as evident from similar phenotypes of patients with homozygote missense and splice variants and patients with truncating variants of similar age, and these loss-of-function (LOF) variants are associated with optic disc membrane disorders and photoreceptor degeneration (Zacchigna et al., 2009), which can lead to disease. The homozygous variant c.2373+5G>T in PROM1 detected in this study was previously detected in an RP patient with a consanguineous family history, but the clinical phenotype was unknown (Chen et al., 2013). The parents of the patient in this study were also consanguineous, and the patient complained of reduced vision in both eyes. Clinically, CORD patients need to be differentiated from RP when they show significant rod dysfunction in the late stages often accompanied by night blindness symptoms. Typical RP usually starts with night blindness as the first symptom, often not accompanied by photophobia and high myopia. In this study, fundus tessellation with an elliptic atrophy lesion was observed in the macular area of the patient carrying the PROM1 variant. The lesions showed hypofluorescence changes in autofluorescence, and there was no typical RP fundus appearance. Repeatedly questioning the medical history, the patient had no night blindness, and the ERG results suggested severe dysfunction of both cone and rod cells. Based on the patient’s chief complaint, clinical phenotype, and ERG examination, this patient was finally diagnosed with CORD.
ADAM9 is located on chromosome 8 and contains 22 exons, which are involved in a number of biological processes, such as cell division, proteolysis, cell adhesion, cell fusion, and signal transmission, which encode membrane proteins (Mahimkar et al., 2005). The brain, spinal cord, retina, and lens all express ADAM9, which participates in a variety of pathways including extracellular matrix interactions (Schwettmann and Tschesche, 2001). It primarily functions in the link between RPE and photoreceptor outer segments in the human retina (Seals and Courtneidge, 2003). Parry et al. (2009) first reported the ADAM9 variant causing CORD in four consanguineously married families with poor vision before the age of 10 years without nystagmus and photophobia. Atrophy of the outer layer of the retina was observed in the macula area of the fundus. Scattered white patches were observed in the posterior pole and optic disc in most patients, which may be accompanied by pre-equatorial pigmentosa retinopathy in older patients. CORD caused by the ADAM9 variant as reported so far were all from consanguineously married families, and the type of variant was a homozygous splice or nonsense variant, all of which could produce termination codon. It was found from the studies that the pathogenic mechanism of ADAM9 is LOF, and the early occurrence of the termination codon can lead to nonsense-mediated mRNA degradation, which leads to the onset of the disease (Parry et al., 2009). The family of this study denied a consanguineous marriage history, the onset age was young, and the disease was severe, which was consistent with pathogenesis characteristics reported in previous studies. However, there was no significant characteristic of fundus lesions caused by this gene variant (Hull et al., 2015). Since CORD studies related to the ADAM9 variant were all case reports and lacked large sample studies, the relationship between the genotype and clinical phenotype cannot be determined yet and needs further exploration.
CRX, located on chromosome 19q13.33, is a cone–rod isoform gene (OMIM: 602225). It encodes a 299-amino acid isoform transcription factor necessary for photoreceptor development and survival and is highly similar to the OTX family isoform gene (Freund et al., 1997). Animal studies have shown that CRX is most prominently expressed in the vertebrate retina’s photoreceptor cells and pineal cells (Chen et al., 1997) and plays a key role in the differentiation and maintenance of photoreceptor cells by interacting synergistically with other transcription factors, such as the neural retina-specific leucine zipper protein (NRL), retina and anterior neural fold homeobox protein (RAX), and nuclear receptor subfamily 2 group E member 3 (NR2E3) (Peng et al., 2005). CRX variants can lead to not only dominant CORD but also dominant retinitis pigmentosa and Leber congenital amaurosis (Sohocki et al., 2001; Freund et al., 1998). CRX proteins have five conserved functional domains, namely, the homeodomain (HD) (from N-terminal to C-terminal), glutamine-rich (Gln) domain, infrastructure domain, WSP domain, and OTX-tail domain. The homeodomain is associated with DNA binding, while the C-terminal region of the protein is associated with transcriptional regulation (Chau et al., 2000). It was found from the studies that the missense variants in CRX occur in the homeodomain, while frameshift and nonsense variants are confined to the C-terminal region of the protein (Tran and Chen, 2014).
In this study, the nonsense and frameshift variations in four families with CORD generate premature stop codons and C-terminal truncation variations in CACNA1F, ADAM9, and CRX. They might introduce premature translational-termination codons (PTCs) that NMD can detect and degrade. NMD functions as a widespread RNA surveillance system for mRNA quality in eukaryotic cells. By stopping the translation of the aberrant mRNA and changing the phenotype, it can identify mutant mRNAs carrying PTCs and quickly degrade and remove aberrant transcripts to prevent the buildup of shortened and potentially dangerous proteins. PTCs situated upstream of the last exon junction complex (EJC) make NMD effective (Khajavi et al., 2006). Many rules that were consistent with our findings govern this efficiency. The final exon rule and the 50-nt rule are considered to be canonical rules (Nagy and Maquat, 1998; Le Hir et al., 2001; Holbrook et al., 2004). The 50-nt rule states that PTCs that are fewer than 50 nucleotides (nt) upstream of the final exon–exon junction will cause NMD, whereas PTCs that are located in the final exon of a gene or in the final 50 bases of the penultimate exon will not cause NMD (the last exon rule). Family 1 in our study involved a variation, p.Val734Glyfs*17, located on exon 6 in CACNA1. Consistent with the 50-nt rule, the physical sites of this variant were not on the last or the penultimate exon as NMD is believed to play a role in the phenotypes, presenting a mild clinical phenotype. Families 5 and 6 in our study involved the same variation, p.Gln228Ter, located on exon 4 in CRX. Consistent with the last exon rule, the physical site of this variant was on the last exon as NMD is not triggered to play a role in the phenotypes (the last exon rule), presenting severe clinical phenotypes (Conti and Izaurralde, 2005; Inoue et al., 2004; Kerr et al., 2001).
However, sometimes NMD does not absolutely degrade all transcripts containing PTCs, and some truncated mutant proteins are likely to be expressed, which leads to a more severe clinical phenotype. In this study, family 4 involved a variation, p.Arg202Ter, located on exon 6 in ADAM9. Consistent with the 50-nt rule, the physical sites of this variant were not on the last or the penultimate exon as NMD might be triggered to play a role in the phenotypes, presenting a mild clinical phenotype. However, because NMD fails to completely degrade all transcripts containing PTCs, some truncated mutant proteins are likely to be expressed, resulting in severe visual impairment and macular degeneration of the patient phenotype. In addition, it is likely that NMD is not the only pathway that is actually responsible for mRNA stability/degradation, thus modulating disease severity (Khajavi et al., 2005; Chan et al., 1998).
It is worth noting that the cases of patients in the two CORDX families, one AR CORD family, and one AD CORD family in this study were complicated with high myopia. It was found from the studies that approximately 39% of pathogenic variants in people with high myopia are found in genes associated with retinal inherited disease (Haarman et al., 2022), and many patients with retinal dystrophy often present high myopia or even early-onset high myopia in the early stages, while symptoms associated with retinal dystrophies such as dyschromatopsia and night blindness are not prominent. Therefore, these disorders are easily overlooked and often result in misdiagnosis and missed diagnosis. Two investigations of children with high myopia conducted in the community have further supported this (Logan et al., 2004; Marr et al., 2001). In light of this, we advise clinicians to deeply interrogate patients with high myopia about their medical history and family history in order to identify a particular genetic pattern. This should be followed by a detailed ophthalmologic examination and the necessary systemic examination to look for clinical signs other than myopia, and if the patient is suspected of having a Mendelian disorder, whole-genome exome sequencing can be performed to further clarify the diagnosis.
In conclusion, our study successfully identified three previously unreported genetic variations in CORD genes across six distinct CORD families, each exhibiting unique inheritance patterns. Our study was accomplished through the utilization of WES) and Sanger sequencing techniques. This discovery extends the range of pathogenic variants in CORD genes, hence offering valuable insights into genetic counseling in families.
Data availability statement
The data that support the findings of this study are openly available in the GenBank repository (https://www.ncbi.nlm.nih.gov/genbank/). Accession numbers: 2634202, 2634179, 2634114, 2634096.
Ethics statement
The studies involving humans were approved by the Ningxia Hui Autonomous Region People’s Hospital Ethics Committee. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
ZL and WR wrote the main manuscript. WC, FZ, and XH collected case data and followed up patients. WR and XS refined the article. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Natural Science Foundation of Ningxia Hui Autonomous Region (2021AAC03314), the Key Research and Development Project of Ningxia Hui Autonomous Region (2020BEG03047 and 2021BEG02045), the Training Project of the Scientific Innovation Commanding Talented Person in Ningxia Hui Autonomous Region (KJT2020013), and the National Natural Science Foundation of China (82260206).
Acknowledgments
The authors would like to thank all patients and their family members for their participation.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abid, A., Ismail, M., Mehdi, S. Q., and Khaliq, S. (2006). Identification of novel mutations in the SEMA4A gene associated with retinal degenerative diseases. J. Med. Genet. 43 (4), 378–381. Epub 2005 Sep 30. doi:10.1136/jmg.2005.035055
Abu-Safieh, L., Alrashed, M., Anazi, S., Alkuraya, H., Khan, A. O., Al-Owain, M., et al. (2013). Autozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Res. 23 (2), 236–247. doi:10.1101/gr.144105.112
Ahn, J., Chiang, J., and Gorin, M. B. (2020). Novel mutation in SLC4A7 gene causing autosomal recessive progressive rod-cone dystrophy. Ophthalmic Genet. 41 (4), 386–389. doi:10.1080/13816810.2020.1783691
Aleman, T. S., Soumittra, N., Cideciyan, A. V., Sumaroka, A. M., Ramprasad, V. L., Herrera, W., et al. (2009). CERKL mutations cause an autosomal recessive cone-rod dystrophy with inner retinopathy. Invest. Ophthalmol. Vis. Sci. 50 (12), 5944–5954. doi:10.1167/iovs.09-3982
Aligianis, I. A., Forshew, T., Johnson, S., Michaelides, M., Johnson, C. A., Trembath, R. C., et al. (2002). Mapping of a novel locus for achromatopsia (ACHM4) to 1p and identification of a germline mutation in the alpha subunit of cone transducin (GNAT2). J. Med. Genet. 39 (9), 656–660. doi:10.1136/jmg.39.9.656
Allikmets, R. (2000). Simple and complex ABCR: genetic predisposition to retinal disease. Am. J. Hum. Genet. 67 (4), 793–799. doi:10.1086/303100
Arikawa, K., Molday, L. L., Molday, R. S., and Williams, D. S. (1992). Localization of peripherin/rds in the disk membranes of cone and rod photoreceptors: relationship to disk membrane morphogenesis and retinal degeneration. J. Cell. Biol. 116 (3), 659–667. doi:10.1083/jcb.116.3.659
Balciuniene, J., Johansson, K., Sandgren, O., Wachtmeister, L., Holmgren, G., and Forsman, K. (1995). A gene for autosomal dominant progressive cone dystrophy (CORD5) maps to chromosome 17p12-p13. Genomics 30 (2), 281–286. doi:10.1006/geno.1995.9876
Bech-Hansen, N. T., Naylor, M. J., Maybaum, T. A., Pearce, W. G., Koop, B., Fishman, G. A., et al. (1998). Loss-of-function mutations in a calcium-channel alpha1-subunit gene in Xp11.23 cause incomplete X-linked congenital stationary night blindness. Nat. Genet. 19 (3), 264–267. doi:10.1038/947
Birtel, J., Eisenberger, T., Gliem, M., Muller, P. L., Herrmann, P., Betz, C., et al. (2018a). Clinical and genetic characteristics of 251 consecutive patients with macular and cone/cone-rod dystrophy. Sci. Rep. 8 (1), 4824. doi:10.1038/s41598-018-22096-0
Birtel, J., Eisenberger, T., Gliem, M., Müller, P. L., Herrmann, P., Betz, C., et al. (2018b). Clinical and genetic characteristics of 251 consecutive patients with macular andcone/cone-rod dystrophy. Sci. Rep. 8, 4824. doi:10.1038/s41598-018-22096-0
Boulanger-Scemama, E., El Shamieh, S., Demontant, V., Condroyer, C., Antonio, A., Michiels, C., et al. (2015). Next Generation sequencing applied to a large French cone andcone-rod dystrophy cohort: mutation spectrum and new genotype-phenotype correlation. Orphanet J. Rare Dis. 10, 85. doi:10.1186/s13023-015-0300-3
Boulanger-Scemama, E., Mohand-Said, S., El, S. S., Demontant, V., Condroyer, C., Antonio, A., et al. (2019). Phenotype analysis of retinal dystrophies in light of the underlying genetic defects: application to cone and cone-rod dystrophies. Int. J. Mol. Sci. 20 (19), 4854. doi:10.3390/ijms20194854
Cehajic-Kapetanovic, J., Birtel, J., McClements, M. E., Shanks, M. E., Clouston, P., Downes, S. M., et al. (2019). Clinical and molecular characterization of PROM1-related retinal degeneration. JAMA NetwOpen 2, e195752. doi:10.1001/jamanetworkopen.2019.5752
Chan, D., Weng, Y. M., Graham, H. K., Sillence, D. O., and Bateman, J. F. (1998). A nonsense mutation in the carboxyl-terminal domain of type X collagen causes haploinsufficiency in schmid metaphyseal chondrodysplasia. J. Clin. Invest. 101 (7), 1490–1499. doi:10.1172/JCI1976
Chau, K., Chen, S., Zack, D., and Ono, S. J. (2000). Functional domains of the cone-rod homeobox (CRX) transcription factor. J. Biol. Chem. 275, 37264–37270. doi:10.1074/jbc.M002763200
Chen, S., Wang, Q. L., Nie, Z., Sun, H., Lennon, G., Copeland, N. G., et al. (1997). Crx, a novel Otx-like paired-homeodomain protein, binds to and transactivates photoreceptor cell-specific genes. Neuron 19 (5), 1017–1030. doi:10.1016/s0896-6273(00)80394-3
Chen, X., Zhao, K., Sheng, X., Li, Y., Gao, X., Zhang, X., et al. (2013). Targeted sequencing of 179 genes associated with hereditary retinal dystrophies and 10 candidate genes identifies novel and known mutations in patients with various retinal diseases. Invest. Ophthalmol. Vis. Sci. 54 (3), 2186–2197. Published 2013 Mar 1. doi:10.1167/iovs.12-10967
Conti, E., and Izaurralde, E. (2005). Nonsense-mediated mRNA decay: molecular insights and mechanistic variations across species. Curr. Opin. Cell. Biol. 17, 316–325. doi:10.1016/j.ceb.2005.04.005
Danciger, M., Hendrickson, J., Lyon, J., Toomes, C., McHale, J. C., Fishman, G. A., et al. (2001). CORD9 a new locus for arCRD: mapping to 8p11, estimation of frequency, evaluation of a candidate gene. Invest. Ophthalmol. Vis. Sci. 42 (11), 2458–2465.
den Hollander, A. I., Black, A., Bennett, J., and Cremers, F. P. (2010). Lighting a candle in the dark: advances in genetics and gene therapy of recessive retinal dystrophies. J. Clin. Invest. 120 (9), 3042–3053. doi:10.1172/JCI42258
Dharmat, R., Liu, W., Ge, Z., Sun, Z., Yang, L., Li, Y., et al. (2017). IFT81 as a candidate gene for nonsyndromic retinal degeneration. Invest. Ophthalmol. Vis. Sci. 58 (5), 2483–2490. doi:10.1167/iovs.16-19133
Downes, S. M., Holder, G. E., Fitzke, F. W., Payne, A. M., Warren, M. J., Bhattacharya, S. S., et al. (2001). Autosomal dominant cone and cone-rod dystrophy with mutations in the guanylate cyclase activator 1A gene-encoding guanylate cyclase activating protein-1. Arch. Ophthalmol. 119 (1), 96–105.
Downey, L. M., Keen, T. J., Jalili, I. K., McHale, J., Aldred, M. J., Robertson, S. P., et al. (2002). Identification of a locus on chromosome 2q11 at which recessive amelogenesis imperfecta and cone-rod dystrophy cosegregate. Eur. J. Hum. Genet. 10 (12), 865–869. doi:10.1038/sj.ejhg.5200884
Durlu, Y. K., Köroğlu, Ç., and Tolun, A. (2014). Novel recessive cone-rod dystrophy caused by POC1B mutation. JAMA Ophthalmol. 132 (10), 1185–1191. doi:10.1001/jamaophthalmol.2014.1658
Eidinger, O., Leibu, R., Newman, H., Rizel, L., and PerlmanIBen-Yosef, T. (2015). An intronic deletion in the PROM1gene leads to autosomal recessive cone-rod dystrophy. Mol. Vis. 21, 1295–1306. PMID: 26702251;.
Freund, C. L., Gregory-Evans, C. Y., Furukawa, T., Papaioannou, M., Looser, J., Ploder, L., et al. (1997). Cone-rod dystrophy due to mutations in a novel photoreceptor-specific homeobox gene (CRX) essential for maintenance of the photoreceptor. Cell. 91 (4), 543–553. doi:10.1016/s0092-8674(00)80440-7
Freund, C., Wang, Q., Chen, S., Muskat, B. L., Wiles, C. D., Sheffield, V. C., et al. (1998). De novo mutations in the CRX homeobox gene associated with Leber congenital amaurosis. Nat 18, 311–312. doi:10.1038/ng0498-311
Ge, Z., Kristen, B., Kerry, G., Scholl, H. P. N., Wang, F., Wang, X., et al. (2015). NGS-based Molecular diagnosis of 105 eyeGENE(®) probands with Retinitis Pigmentosa. Sci. Rep. 5, 18287. doi:10.1038/srep18287
Haarman, A. E. G., Thiadens, AAHJ, van Tienhoven, M., Loudon, S. E., de Klein, J. E. M. M. A., Brosens, E., et al. (2022). Whole exome sequencing of known eye genes reveals genetic causes for high myopia. Hum. Mol. Genet. 31 (19), 3290–3298. doi:10.1093/hmg/ddac113
Hameed, A., Abid, A., Aziz, A., Ismail, M., Mehdi, S. Q., and Khaliq, S. (2003). Evidence of RPGRIP1 gene mutations associated with recessive cone-rod dystrophy. J. Med. Genet. 40 (8), 616–619. doi:10.1136/jmg.40.8.616
Hauke, J., Schild, A., Neugebauer, A., Lappa, A., Fricke, J., Fauser, S., et al. (2013). A novel large in-frame deletion within the CACNA1F gene associates with a cone-rod dystrophy 3-like phenotype. PLoS ONE 8 (10), e76414. doi:10.1371/journal.pone.0076414
Holbrook, J. A., Neu-Yilik, G., Hentze, M. W., and Kulozik, A. E. (2004). Nonsense-mediated decay approaches the clinic. Nat. Genet. 36 (8), 801–808. doi:10.1038/ng1403
Hull, S., Arno, G., Plagnol, V., Robson, A., Webster, A. R., and Moore, A. T. (2015). Exome sequencing reveals ADAM9 mutations in a child with cone-rod dystrophy. Acta Ophthalmol. 93 (5), e392–e393. doi:10.1111/aos.12592
Inoue, K., Khajavi, M., Ohyama, T., Hirabayashi, S. i., Wilson, J., Reggin, J. D., et al. (2004). Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat. Genet. 36, 361–369. doi:10.1038/ng1322
Jalkanen, R., Demirci, F. Y., Tyynismaa, H., Bech-Hansen, T., Meindl, A., Peippo, M., et al. (2003a). A new genetic locus for X linked progressive cone-rod dystrophy. J. Med. Genet. 40 (6), 418–423. doi:10.1136/jmg.40.6.418
Jalkanen, R., Demirci, F. Y., Tyynismaa, H., Bech-Hansen, T., Meindl, A., Peippo, M., et al. (2003b). A new genetic locus for X linked progressive cone-rod dystrophy. J. Med. Genet. 40, 418–423. doi:10.1136/jmg.40.6.418
Jalkanen, R., Mäntyjärvi, M., Tobias, R., Isosomppi, J., Sankila, E. M., Alitalo, T., et al. (2006). X linked cone-rod dystrophy, CORDX3, is caused by a mutation in the CACNA1F gene. J. Med. Genet. 43 (8), 699–704. doi:10.1136/jmg.2006.040741
Jalkanen, R., Bech-Hansen, N. T., Tobias, R., Sankila, E. M., Mäntyjärvi, M., Forsius, H., et al. (2007). A novel CACNA1F gene mutation causes Aland Island eye disease. Invest. Ophthalmol. Vis. Sci. 48 (6), 2498–2502. doi:10.1167/iovs.06-1103
Johnson, S., Halford, S., Morris, A. G., Patel, R. J., Wilkie, S. E., Hardcastle, A. J., et al. (2003). Genomic organisation and alternative splicing of human RIM1, a gene implicated in autosomal dominant cone-rod dystrophy (CORD7). Genomics 81 (3), 304–314. doi:10.1016/s0888-7543(03)00010-7
Kelsell, R. E., Gregory-Evans, K., Payne, A. M., Perrault, I., Kaplan, J., Yang, R. B., et al. (1998). Mutations in the retinal guanylate cyclase (RETGC-1) gene in dominant cone-rod dystrophy. Hum. Mol. Genet. 7 (7), 1179–1184. doi:10.1093/hmg/7.7.1179
Kerr, T. P., Sewry, C. A., Robb, S. A., and Roberts, R. G. (2001). Long mutant dystrophins and variable phenotypes: evasion of nonsense mediated decay? Hum. Genet. 109, 402–407. doi:10.1007/s004390100598
Khajavi, M., Inoue, K., Wiszniewski, W., Ohyama, T., Snipes, G. J., and Lupski, J. R. (2005). Curcumin treatment abrogates endoplasmic reticulum retention and aggregation-induced apoptosis associated with neuropathy-causing myelin protein zero-truncating mutants. Am. J. Hum. Genet. 77, 841–850. doi:10.1086/497541
Khajavi, M., Inoue, K., and Lupski, J. R. (2006). Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur. J. Hum. Genet. 14 (10), 1074–1081. doi:10.1038/sj.ejhg.5201649
Kobayashi, A., Higashide, T., Hamasaki, D., Kubota, S., Sakuma, H., An, W., et al. (2000). HRG4 (UNC119) mutation found in cone-rod dystrophy causes retinal degeneration in a transgenic model. Invest. Ophthalmol. Vis. Sci. 41 (11), 3268–3277.
Kohl, S., Zobor, D., Chiang, W. C., Weisschuh, N., Staller, J., Gonzalez Menendez, I., et al. (2015). Mutations in the unfolded protein response regulator ATF6 cause the cone dysfunction disorder achromatopsia. Nat. Genet. 47 (7), 757–765. doi:10.1038/ng.3319
Le Hir, H., Gatfifield, D., Izaurralde, E., and Moore, M. J. (2001). The exon-exon junction complex provides a binding platform for factors involved in mRNA export and nonsense-mediated mRNA decay. Embo J. 20, 4987–4997. doi:10.1093/emboj/20.17.4987
Littink, K. W., Koenekoop, R. K., van den Born, L. I., Collin, R. W. J., Moruz, L., Veltman, J. A., et al. (2010). Homozygosity mapping in patients with cone-rod dystrophy: novel mutations and clinical characterizations. Invest. Ophthalmol. Vis. Sci. 51, 5943–5951. doi:10.1167/iovs.10-5797
Logan, N. S., Gilmartin, B., Marr, J. E., Stevenson, M. R., and Ainsworth, J. R. (2004). Community-based study of the association of high myopia in children with ocular and systemic disease. Optom. Vis. Sci. 81, 11–13. doi:10.1097/00006324-200401000-00004
Mahimkar, R. M., Visaya, O., Pollock, A. S., and Lovett, D. H. (2005). The disintegrin domain of ADAM9: a ligand for multiple beta1 renal integrins. Biochem. J. 385, 461–468. doi:10.1042/BJ20041133
Marr, J. E., Halliwell-Ewen, J., Fisher, B., Soler, L., and Ainsworth, J. R. (2001). Associations of high myopia in childhood. Eye 15, 70–74. doi:10.1038/eye.2001.17
Maw, M. A., Corbeil, D., Koch, J., Hellwig, A., Wilson-Wheeler, J. C., Bridges, R. J., et al. (2000). A frameshift mutation in prominin(mouse)-like 1 causes human retinal degeneration. Hum. Mol. Genet. 9 (1), 27–34. doi:10.1093/hmg/9.1.27
McRory, J. E., Hamid, J., Doering, C. J., Garcia, E., Parker, R., Hamming, K., et al. (2004). The CACNA1F gene encodes an L-type calcium channel with unique biophysical properties and tissue distribution. J. Neurosci. 24, 1707–1718. doi:10.1523/JNEUROSCI.4846-03.2004
Michaelides, M., Johnson, S., Poulson, A., Bradshaw, K., Bellmann, C., Hunt, D. M., et al. (2003). An autosomal dominant bull’s-eye macular dystrophy (MCDR2) that maps to the short arm of chromosome 4. Invest. Ophthalmol. Vis. Sci. 44 (4), 1657–1662. doi:10.1167/iovs.02-0941
Michaelides, M., Aligianis, I. A., Ainsworth, J. R., Good, P., Mollon, J. D., Maher, E. R., et al. (2004). Progressive cone dystrophy associated with mutation in CNGB3. Invest. Ophthalmol. Vis. Sci. 45 (6), 1975–1982. doi:10.1167/iovs.03-0898
Michaelides, M., Gaillard, M. C., Escher, P., Tiab, L., Bedell, M., Borruat, F. X., et al. (2010). The PROM1 mutation p.R373C causes an autosomal dominant bull’s eye maculopathy associated with rod, rod-cone, and macular dystrophy. Invest. Ophthalmol. Vis. Sci. 51, 4771–4780. doi:10.1167/iovs.09-4561
Miraglia, S., Godfrey, W., Yin, A. H., Atkins, K., Warnke, R., Holden, J. T., et al. (1997). A novel five-transmembrane hematopoietic stem cell antigen: isolation, characterization, and molecular cloning. Blood 90 (12), 5013–5021. PMID: 9389721. doi:10.1182/blood.v90.12.5013.5013_5013_5021
Morgans, C. W. (2001). Localization of the alpha(1F) calcium channel subunit in the rat retina. Invest. Ophthalmol. Vis. Sci. 42, 2414–2418. PMID: 11527958.
Nagy, E., and Maquat, L. E. (1998). A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends biochem. Sci. 23, 198–199. doi:10.1016/s0968-0004(98)01208-0
Nakamura, M., Hotta, Y., Tanikawa, A., Terasaki, H., and Miyake, Y. (2000). A high association with cone dystrophy in Fundus albipunctatus caused by mutations of the RDH5 gene. Invest. Ophthalmol. Vis. Sci. 41 (12), 3925–3932.
Nikopoulos, K., Farinelli, P., Giangreco, B., Tsika, C., Royer-Bertrand, B., Mbefo, M. K., et al. (2016). Mutations in CEP78 cause cone-rod dystrophy and hearing loss associated with primary-cilia defects. Am. J. Hum. Genet. 99 (3), 770–776. doi:10.1016/j.ajhg.2016.07.009
Ostergaard, E., Batbayli, M., Duno, M., Vilhelmsen, K., and Rosenberg, T. (2010). Mutations in PCDH21 cause autosomal recessive cone-rod dystrophy. J. Med. Genet. 47 (10), 665–669. doi:10.1136/jmg.2009.069120
Parry, D. A., Toomes, C., Bida, L., Danciger, M., Towns, K. V., McKibbin, M., et al. (2009). Loss of the metalloprotease ADAM9 leads to cone-rod dystrophy in humans and retinal degeneration in mice. Am. J. Hum. Genet. 84 (5), 683–691. doi:10.1016/j.ajhg.2009.04.005
Peng, G. H., Ahmad, O., Ahmad, F., Liu, J., and Chen, S. (2005). The photoreceptor-specific nuclear receptor Nr2e3 interacts with Crx and exerts opposing effects on the transcription of rod versus cone genes. Hum. Mol. Genet. 14, 747–764. doi:10.1093/hmg/ddi070
Piri, N., Gao, Y. Q., Danciger, M., Mendoza, E., Fishman, G. A., and Farber, D. B. (2005). A substitution of G to C in the cone cGMP-phosphodiesterase gamma subunit gene found in a distinctive form of cone dystrophy. Ophthalmology 112 (1), 159–166. doi:10.1016/j.ophtha.2004.07.011
Roosing, S., Rohrschneider, K., Beryozkin, A., Sharon, D., Weisschuh, N., Staller, J., et al. (2013). Mutations in RAB28, encoding a farnesylated small GTPase, are associated with autosomal-recessive cone-rod dystrophy. Am. J. Hum. Genet. 93 (1), 110–117. doi:10.1016/j.ajhg.2013.05.005
Schwettmann, L., and Tschesche, H. (2001). Cloning and expression in Pichia pastoris of metalloprotease domain of ADAM 9 catalytically active against fibronectin. Protein Expr. Purif. 21 (1), 65–70. doi:10.1006/prep.2000.1374
Seals, D. F., and Courtneidge, S. A. (2003). The ADAMs family of metalloproteases: multidomain proteins with multiple functions. Genes Dev. 17, 7–30. doi:10.1101/gad.1039703
Sergouniotis, P. I., Chakarova, C., Murphy, C., Becker, M., Lenassi, E., Arno, G., et al. (2014). Biallelic variants in TTLL5, encoding a tubulin glutamylase, cause retinal dystrophy. Am. J. Hum. Genet. 94 (5), 760–769. doi:10.1016/j.ajhg.2014.04.003
Sohocki, M. M., Sullivan, L. S., Mintz-Hittner, H. A., Birch, D., Heckenlively, J. R., Freund, C. L., et al. (1998). A range of clinical phenotypes associated with mutations in CRX, a photoreceptor transcription-factor gene. Am. J. Hum. Genet. 63 (5), 1307–1315. doi:10.1086/302101
Sohocki, M., Daiger, S., Bowne, S., Rodriquez, J. A., Northrup, H., Heckenlively, J. R., et al. (2001). Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum. Mutat. 17, 42–51. doi:10.1002/1098-1004(2001)17:1<42::AID-HUMU5>3.0.CO;2-K
Solaguren-Beascoa, M., Bujakowska, K. M., Méjécase, C., Emmenegger, L., Orhan, E., Neuillé, M., et al. (2021). WDR34, a candidate gene for non-syndromic rod-cone dystrophy. Clin. Genet. 99 (2), 298–302. doi:10.1111/cge.13872
Sun, W., Huang, L., Xu, Y., Xiao, X., Li, S., Jia, X., et al. (2015). Exome sequencing on 298 probands with early-onset high myopia: approximately one-fourth show potential pathogenic mutations in RetNet genes. Invest. Ophthalmol. Vis. Sci. 56, 8365–8372. doi:10.1167/iovs.15-17555
Thiadens, A. A., den Hollander, A. I., Roosing, S., Nabuurs, S. B., Zekveld-Vroon, R. C., Collin, R. W., et al. (2009). Homozygosity mapping reveals PDE6C mutations in patients with early-onset cone photoreceptor disorders. Am. J. Hum. Genet. 85 (2), 240–247. doi:10.1016/j.ajhg.2009.06.016
Thiadens, A. A., Phan, T. M., Zekveld-Vroon, R. C., Leroy, B. P., van den Born, L. I., Hoyng, C. B., et al. (2012). Clinical course, genetic etiology, and visual outcome in cone and cone-rod dystrophy. Ophthalmology 119, 819–826. doi:10.1016/j.ophtha.2011.10.011
Tran, N., and Chen, S. (2014). Mechanisms of blindness: animal models provide insight into distinct CRX-associated retinopathies. Dev. Dyn. 243, 1153–1166. doi:10.1002/dvdy.24151
van Huet, R. A., Estrada-Cuzcano, A., Banin, E., Rotenstreich, Y., Hipp, S., Kohl, S., et al. (2013). Clinical characteristics of rod and cone photoreceptor dystrophies in patients with mutations in the C8orf37 gene. Invest. Ophthalmol. Vis. Sci. 54 (7), 4683–4690. doi:10.1167/iovs.12-11439
Wang, Q. L., Chen, S., Esumi, N., Swain, P. K., Haines, H. S., Peng, G., et al. (2004). QRX, a novel homeobox gene, modulates photoreceptor gene expression. Hum. Mol. Genet. 13 (10), 1025–1040. doi:10.1093/hmg/ddh117
Wawrocka, A., Skorczyk-Werner, A., Wicher, K., Niedziela, Z., Ploski, R., Rydzanicz, M., et al. (2018). Novel variants identified with next-generation sequencing in Polish patients with cone-rod dystrophy. Mol. Vis. 24, 326–339.
Wissinger, B., Jägle, H., Kohl, S., Broghammer, M., Baumann, B., Hanna, D. B., et al. (1998). Human rod monochromacy: linkage analysis and mapping of a cone photoreceptor expressed candidate gene on chromosome 2q11. Genomics 51 (3), 325–331. doi:10.1006/geno.1998.5390
Wu, H., Cowing, J. A., Michaelides, M., Wilkie, S. E., Jeffery, G., Jenkins, S. A., et al. (2006). Mutations in the gene KCNV2 encoding a voltage-gated potassium channel subunit cause “cone dystrophy with supernormal rod electroretinogram” in humans. Am. J. Hum. Genet. 79 (3), 574–579. doi:10.1086/507568
Wycisk, K. A., Zeitz, C., Feil, S., Wittmer, M., Forster, U., Neidhardt, J., et al. (2006). Mutation in the auxiliary calcium-channel subunit CACNA2D4 causes autosomal recessive cone dystrophy. Am. J. Hum. Genet. 79 (5), 973–977. doi:10.1086/508944
Yang, Z., Peachey, N. S., Moshfeghi, D. M., Thirumalaichary, S., Chorich, L., Shugart, Y. Y., et al. (2002). Mutations in the RPGR gene cause X-linked cone dystrophy. Hum. Mol. Genet. 11 (5), 605–611. doi:10.1093/hmg/11.5.605
Yang, Z., Chen, Y., Lillo, C., Chien, J., Yu, Z., Michaelides, M., et al. (2008). Mutant prominin 1 found in patients with macular degeneration disrupts photoreceptor disk morphogenesis in mice. J. Clin. Invest. 118, 2908–2916. doi:10.1172/JCI35891
Zacchigna, S., Oh, H., Wilsch-Bräuninger, M., Missol-Kolka, E., Jászai, J., Jansen, S., et al. (2009). Loss of the cholesterol-binding protein prominin-1/CD133 causes disk dysmorphogenesis and photoreceptor degeneration. JNeurosci 29 (7), 2297–2308. doi:10.1523/JNEUROSCI.2034-08.2009
Zeitz, C., Robson, A. G., and Audo, I. (2015). Congenital stationary night blindness: an analysis and update of genotype-phenotype correlations and pathogenic mechanisms. Prog. Retin Eye Res. 45, 58–110. doi:10.1016/j.preteyeres.2014.09.001
Zeitz, C., Michiels, C., Neuillé, M., Friedburg, C., Condroyer, C., Boyard, F., et al. (2019). Where are the missing gene defects in inherited retinal disorders? Intronic and synonymous variants contribute at least to 4% of CACNA1F-mediated inherited retinal disorders. Hum. Mutat. 40, 765–787. doi:10.1002/humu.23735
Keywords: cone–rod dystrophy, whole-exome sequencing, genotype, clinical phenotype, variant
Citation: Li Z, Cheng W, Zi F, Wang J, Huang X, Sheng X and Rong W (2023) Four different gene-related cone–rod dystrophy: clinical and genetic findings in six Chinese families with diverse modes of inheritance. Front. Genet. 14:1157156. doi: 10.3389/fgene.2023.1157156
Received: 02 February 2023; Accepted: 11 October 2023;
Published: 02 November 2023.
Edited by:
Musharraf Jelani, Islamia College University, PakistanReviewed by:
Leen Abu-Safieh, King Fahad Medical City, Saudi ArabiaSaid El Shamieh, Beirut Arab University, Lebanon
Copyright © 2023 Li, Cheng, Zi, Wang, Huang, Sheng and Rong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weining Rong, cm9uZ3dlaW5pbmc0MjZAMTI2LmNvbQ==; Xunlun Sheng, c2hlbmd4dW5sdW5AMTYzLmNvbQ==
†These authors have contributed equally to this work and share first authorship