 Wenxia Su
Wenxia Su Shuyi Deng
Shuyi Deng Zhifeng Gu
Zhifeng Gu Keli Yang
Keli Yang Hui Ding
Hui Ding Hui Chen
Hui Chen Zhaoyue Zhang
Zhaoyue Zhang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 28 February 2023
Sec. Computational Genomics
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1157021
Introduction: Apoptosis proteins play an important role in the process of cell apoptosis, which makes the rate of cell proliferation and death reach a relative balance. The function of apoptosis protein is closely related to its subcellular location, it is of great significance to study the subcellular locations of apoptosis proteins. Many efforts in bioinformatics research have been aimed at predicting their subcellular location. However, the subcellular localization of apoptotic proteins needs to be carefully studied.
Methods: In this paper, based on amphiphilic pseudo amino acid composition and support vector machine algorithm, a new method was proposed for the prediction of apoptosis proteins\x{2019} subcellular location.
Results and Discussion: The method achieved good performance on three data sets. The Jackknife test accuracy of the three data sets reached 90.5%, 93.9% and 84.0%, respectively. Compared with previous methods, the prediction accuracies of APACC_SVM were improved.
Apoptosis is a type of programmed cell death mechanism that eliminates unnecessary or damaged cells from the body for cellular homeostasis regulation. The apoptotic program is executed by multiple pathways and controlled by the interactions between several molecules. Apoptosis proteins, such as the inhibitor of apoptosis protein (IAP) family, are proteins involved in the process of cell apoptosis for various stress responses. The different functions of apoptosis proteins are related to their subcellular location (Reed and Paternostro, 1999). The subcellular location of apoptosis proteins will not only help us understand the life process and mechanism of programmed cell death but also provide a very important method for understanding the structure and function of proteins (Chou, 2001). It can provide a new perspective for subsequent protein-related tasks such as protein structure prediction and drug-protein relationship prediction (Li et al., 2022a; Li et al., 2022b). However, it is expensive and time-consuming to carry out various experiments to obtain location information (Koroleva et al., 2005). With the explosive growth of protein sequences in the post-genomic era, it is both challenging and necessary to develop an automatic method for quick and accurate prediction of the apoptosis proteins’ subcellular location.
In recent years, serval methods have been proposed for the prediction of apoptosis proteins’ subcellular location. Yu et al. (2012) proposed a prediction method called CELLO, which used multiple SVM classifiers based on N-peptide features. The overall accuracies for their two datasets achieve 87.1% and 90%, respectively. Zhou and Doctor, (2003) established a 98 apoptosis protein data set named ZD98 based on the SWISS-PROT database. They constructed the predictor based on the amino acid composition of the apoptosis protein sequences. The overall success rates of the self-consistent test and jackknife test were 90.8% and 72.5%, respectively. Bulashevska and Eils (2006) used the ZD98 dataset and the jackknife test overall prediction accuracy of the single Bayesian classifier (BC) and hierarchical Bayesian classifier (HensBC) was 85.7% and 89.8% respectively. Chen et al. (2021). proposed a new method to predict the subcellular location of apoptosis proteins by combining dipeptide composition and a discrete increment (ID) algorithm. They predicted the subcellular location of apoptosis proteins based on the main sequence of proteins and the measurement and increase of diversity. According to the latest SWISS-PROT database, they selected 317 apoptosis proteins to establish a data set CL317 and classified them into six subcellular locations (Chen and Li, 2007a). Subsequently, the self-consistent test and jackknife test were conducted, and the overall prediction success rates were 92.1% and 82.7%, respectively. At the same time, they applied this method to ZD98. The overall prediction success rates of the self-consistent test and jackknife test were 94.9% and 90.8%, respectively. Chen and Li, (2007) applied Discrete Incremental Fusion to the dataset. The overall prediction accuracy obtained by the Jackknife test reached 90.8%. For other classes with small samples, the sensitivity reached 91.7%. Later, they combined the ID with a support vector machine (SVM) to propose a new algorithm. For the database of 317 apoptosis proteins in six categories, the overall accuracy of the jackknife test was improved to 85.8%. Zhang et al. (2006) built a larger data set named ZW225. They adopted the feature extraction method based on grouping weight coding, and the overall prediction success rates of self-consistent and jackknife tests were 97.3% and 75.1% respectively. Then they combined the support vector machine with the encoding based on grouped weights feature extraction method, and the overall accuracy of the jackknife test rose to 83.1%.
In this article, we proposed a novel algorithm for apoptosis proteins’ subcellular location prediction. The amphiphilic pseudo amino acid components were used to extract the features from protein sequences. Then, the optimal features were inputted into a machine-learning method to train, test and build a model. The developed approach will be useful for studying apoptosis proteins’ localization and distribution.
Reliable data is the basis of model construction (Su et al., 2021). Three datasets extracted from the Uniprot (https://www.uniprot.org/) were used to construct the benchmark dataset. The dataset CL317 provided by Chen and Li (2007) consists of 317 apoptosis proteins divided into six subcellular locations with 112 cytoplasmic proteins (Cyto), 55 plasma membrane-bound proteins (Memb), 52 nuclear proteins (Nucl), 47 endoplasmic reticulum proteins (Endo), 34 mitochondrial proteins (Mito) and 17 secreted proteins (Secr). All the accession numbers can be found in the literature (Zhou and Doctor, 2003; Chen and Li, 2007; Zhang et al., 2006). ZW225 is a larger dataset provided by Zhang et al. (2006). It contains 225 apoptosis proteins divided into four subcellular locations of which 41 are Nucl, 70 Cyto, 25 Mito and 89 Memb. The dataset ZD98 was generated by Zhou and Doctor, 2003. The 98 apoptosis proteins were classified into four location categories, of which 43 are Cyto, 30 Memb, 13 Mito and 12 other proteins (Other). In this study, the jackknife test was applied to build the prediction model and examine the effectiveness of these three datasets.
We need to convert sequences into vectors in mathematical representation (Amanatidou, and Dedoussis, 2021; Dao et al., 2022a; Jeon et al., 2022; Li H et al., 2022; Nidhi et al., 2022; Sun et al., 2022; Tran and Nguyen, 2022; Wang et al., 2022; Yang et al., 2022; Zhang H et al., 2022). The amino acid composition (ACC) of the protein has a great impact on its subcellular location (Chou and Elrod, 1999a; Awais et al., 2021; Chou and Elrod, 1999b; Rout et al., 2022; Naseer et al., 2021; Manavalan and Patra, 2022; Shoombuatong et al., 2022). By using the ACC to extract features of the protein sequences. a protein sequence can be represented as a 20-D (dimension) vector as follows:
In Eq. 1,
where the first 20 numbers in Eq. 2 are the classic AAC features, and the next 2λ discrete numbers are sequence-correlation factors, which can be calculated according to the literature (Chou, 2005). For different problems, the optimal value of λ is variable. In this study, the optimal value of λ was selected as the one that yielded the highest overall accuracy through the jackknife test. The APAAC features were generated by the iLearnPlus (Chen, 2021) web server (https://ilearnplus.erc.monash.edu/).
Support vector machine (SVM) is a powerful supervised machine learning method based on statistical learning theory (Manavalan et al., 2019a). It was originally designed for solving binary classification problems. The basic idea of the generalized linear classifier is as follows: 1) mapping input vector to feature space (possibly high-dimensional space); 2) In the mapped feature space, a separating hyperplane is constructed to separate the two categories (Vapnik, 2019). To sidestep the expensive calculations, the mapping function only involves the relatively low dimensional vector in the input space and the dot product in the feature space. SVM always seeks solutions for global optimization and avoids overfitting. SVM has been successfully applied to many bioinformatic problems (Wei et al., 2017; Wei et al., 2018; Manayalan et al., 2019a; Manayalan et al., 2019b; Ao et al., 2021; Basith et al., 2021; Zeng et al., 2021; Basith et al., 2022; Zhang Q et al., 2022), such as the disease development prediction (Zhang et al., 2020; Zhang et al., 2021a; Ren et al., 2022; Yu et al., 2022), protein prediction (Tang et al., 2018; Tao et al., 2020; Zou et al., 2021; Ao et al., 2022), etc. In this paper, a widely used software LIBSVM (http://www.csie.ntu.edu.tw/∼cjlin/libsvm) (Chang and Lin, 2011) was used to implement the support vector machine. The radial basis function which is defined as
At present, there are three main test methods to evaluate the prediction results: the re-substitution test, the Jackknife test and the k-fold cross-validation test (Zhang et al., 2020; Zhang et al., 2021b; Deng et al., 2021; Liu et al., 2021; Tabaie et al., 2021; Ao et al., 2022a; Dai et al., 2022; Dao et al., 2022; Jin et al., 2022; Wei et al., 2022; Xiao et al., 2022; Zhou et al., 2022). Chou and Zhang have discussed in depth the classification performance estimation in bioinformatics and found the Jackknife test and k-fold cross-validation test have extrapolation ability in statistics (Malik et al., 2021; Hasan et al., 2022). In this article, we used the Jackknife test to evaluate the prediction results. The sensitivity (Sn), specificity (Sp), overall prediction accuracy (OA) and Matthew’s correlation coefficient (MCC) were used to evaluate the prediction performance of the algorithm (Jiang et al., 2013; Guo et al., 2020; Lv et al., 2020; Xu et al., 2021; Yang et al., 2021; Yu et al., 2021; Han et al., 2022; Zhang Z Y et al., 2022), which are defined as follows:
where TP represents the number of the positive sample correctly identified, FN represents the positive sample wrongly identified as a negative sample, FP represents the negative sample wrongly identified as a positive sample, and TN represents the negative sample correctly identified (Jia et al., 2020; Li et al., 2021).
The proposed algorithm based on APACC and SVM was named APACC_SVM. APAAC was generated by the iLearnPlus, with two parameters to be determined, λ and ω namely. In order to obtain ideal results, the selected values of ω were 0.05, 0.1, 0.2, 0.3, 0.4 and 0.5. The selected values of λ were the integers from 2 to 9. The jackknife test was applied to examine APAAC_SVM model. The predictive results for the three apoptosis protein datasets were enumerated in Table 1. When ω = 0.1 and λ = 7, the overall prediction effect was the best for the CL317 dataset. For CL317, the predictive results showed that the overall accuracy was 90.5% in the jackknife test. We noticed the prediction result on the Secr was far lower than the other which may be due to the small subset (17 proteins). To improve the accuracy of prediction, it is necessary to collect enough proteins in the dataset.
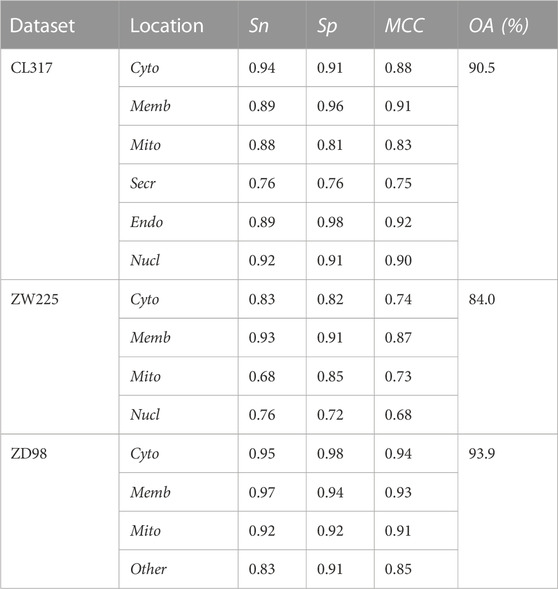
TABLE 1. The predictive results of the three datasets.
When ω = 0.3 and λ = 7, the overall prediction effect was the best for the ZW225 dataset. For ZW225, the jackknife test showed the overall accuracy was 84.0%. According to the prediction results obtained from the training of the ZW225 dataset, although the prediction effect was not as good as CL317, the overall appearance was similar. In the subsets Mito and Nucl (25 and 41 proteins, respectively) with fewer sequences, the prediction accuracies were significantly lower than the others. It showed that expanding the data scale was important for prediction improvement.
When ω = 0.2 and λ = 7, the overall prediction effect was the best for the ZD98 dataset. The predictive results for ZD98 apoptosis protein sets showed that the overall accuracy was 93.9% in the jackknife test.
To prove the prediction ability of our APAAC_SVM algorithm, we compared our model with previous algorithms. For the CL317 dataset, Chen and Li proposed the ID method and ID-SVM method, Zhang Li et al. used the DF-SVM method for the apoptosis proteins’ subcellular location prediction, respectively. The comparison results were shown in Table 2. It can be seen from the table that our APAAC-SVM method significantly improved the prediction results in both the overall prediction accuracy and in each subcellular location, especially in Cyto, Mito and Endo.
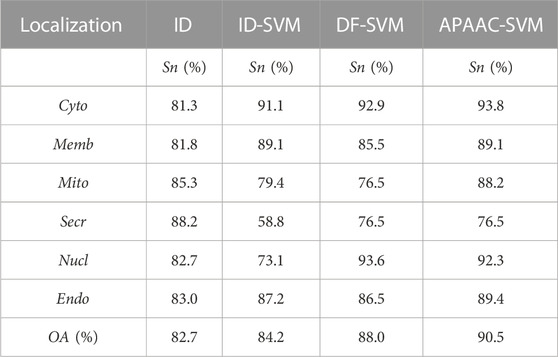
TABLE 2. Comparison of prediction performance for different methods on the CL317 dataset.
For the ZW225 dataset, Zhang and Wang used the EBGW-SVM and DF-SVM methods, and Chen and Li used the ID-SVM method for prediction. The prediction model performances were shown in Table 3. It can be seen from Table 3 that the overall prediction accuracy of each method was relatively close. However, the APAAC-SVM algorithm achieved good prediction accuracy in both the Memb and Nucl. It indicated that our algorithm was relatively ideal.
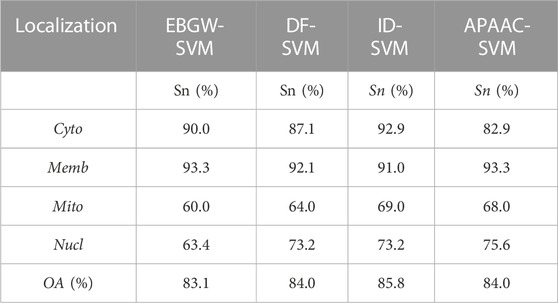
TABLE 3. Comparison of prediction performance for different methods on the ZW225 dataset.
For the ZD98 dataset, Zhou and Doctor, Huang Jing, Bulashevska, Eils, Chen and Li have all conducted research. They have respectively applied covariant discrimination algorithm, SVM algorithm, Bayesian discrimination algorithm and discrete incremental fusion algorithm. The predicted results were shown in Table 4. The overall prediction accuracy of the APAAC-SVM method was 93.9% for the ZD98 dataset, which was higher than other methods. When the Jackknife test was used, the overall prediction accuracy was improved by 21.3% compared with the covariant discriminant algorithm of Zhou and Doctor. Compared with the Bayesian discriminant method of Bulashevska Eils, the overall prediction accuracy was increased by about 8.1%. For a small sample of other apoptosis proteins in the data set, the sensitivity of these two methods was only 25% and 50%, while the sensitivity of this method can reach 83.33%. Compared with Huang Jing’s SVM algorithm, this method had a higher overall prediction success rate, which was increased by about 3%; Moreover, the sensitivity of Cyto was higher, which reached 95.3%. Compared with the discrete incremental fusion method of Chen Yingli and Li Qianzhong, the overall prediction success rate of this method was also higher.
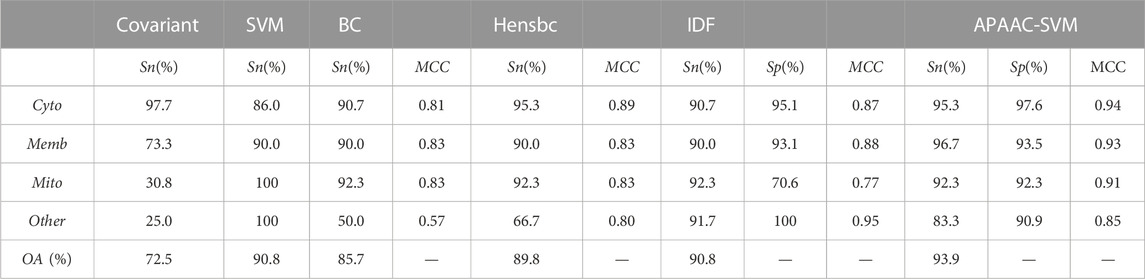
TABLE 4. Comparison of prediction performance for different methods on the ZD98 dataset.
By compared with previous studies, it can be found that the APAAC-SVM method was better for category prediction with more sequence data. It showed that this method was more suitable for the prediction of apoptosis protein subcellular locations in the case of increasing sequence data, and it also had an optimistic application prospect in future research.
Previous apoptosis proteins’ subcellular location analysis demonstrated that information in protein sequence has a great influence on its subcellular localization. However, the performance of the proposed algorithms for apoptosis proteins’ subcellular location prediction is inadequate. This study selected three apoptosis protein sequence datasets CL317, ZD98 and ZW225 to develop a new prediction algorithm. The APAAC feature extraction method and SVM were combined to predict the subcellular location of apoptosis proteins. Through the reasonable selection of parameters, our algorithm APAAC_SVM achieved jackknife test prediction accuracy of 90.5%, 93.9% and 84.0% on CL317, ZD98 and ZW225, respectively. Compared with other methods, APAAC-SVM improved the prediction performance.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Project design and oversight: ZZ, HC, and HD; Sample collection and curation: WS and KY; Experiment conduction and data analysis: WS, SD, and ZG; Table preparation: WS and SD; Result interpretation and discussion: WS, HC, and ZG; Manuscript writing and revision: WS and ZZ; Funding acquisition: WS and ZZ. All authors have read and approved the final version of this manuscript.
This work was supported by a grant from the National Natural Science Foundation of China (Nos. 62201299, 62102067), Natural Science Foundation of the Inner Mongolia of China (No. 2021BS06003), Science and Technology Research Project of Colleges and Universities in Inner Mongolia of China (No. NJZY21473), and Basic Scientific Research Foundation of Colleges and Universities directly under Inner Mongolia of China (No. BR220505).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Amanatidou, A. I., and Dedoussis, G. V. (2021). Construction and analysis of protein-protein interaction network of non-alcoholic fatty liver disease. Comput. Biol. Med. 131, 104243. doi:10.1016/j.compbiomed.2021.104243
Ao, C., Jiao, S., Wang, Y., Yu, L., and Zou, Q. (2022). Biological sequence classification: A review on data and general methods. Research 2022, 0011. doi:10.34133/research.0011
Ao, C., Yu, L., and Zou, Q. (2021). Prediction of bio-sequence modifications and the associations with diseases. Briefings Funct. genomics 20, 1–18. doi:10.1093/bfgp/elaa023
Ao, C., Zou, Q., and Yu, L. (2022a). NmRF: Identification of multispecies RNA 2'-O-methylation modification sites from RNA sequences. Briefings Bioinforma. 23, bbab480. doi:10.1093/bib/bbab480
Awais, M., Hussain, W., Rasool, N., and Khan, Y. D. (2021). iTSP-PseAAC: Identifying tumor suppressor proteins by using fully connected neural network and PseAAC. Curr. Bioinforma. 16, 700–709. doi:10.2174/1574893615666210108094431
Basith, S., Hasan, M. M., Lee, G., Wei, L., and Manavalan, B. (2021). Integrative machine learning framework for the identification of cell-specific enhancers from the human genome. Briefings Bioinforma. 22, bbab252. bbab252. doi:10.1093/bib/bbab252
Basith, S., Lee, G., and Manavalan, B. (2022). Stallion: A stacking-based ensemble learning framework for prokaryotic lysine acetylation site prediction. Briefings Bioinforma. 23, bbab376. doi:10.1093/bib/bbab376
Bulashevska, A., and Eils, R. (2006). Predicting protein subcellular locations using hierarchical ensemble of Bayesian classifiers based on Markov chains. BMC Bioinforma. 7, 298. doi:10.1186/1471-2105-7-298
Chang, C. C., and Lin, C. J. (2011). Libsvm: A library for support vector machines. Acm Trans. Intelligent Syst. Technol. 2, 1–27. doi:10.1145/1961189.1961199
Chen, Y. L., and Li, Q. Z. (2007a). Prediction of apoptosis protein subcellular location using improved hybrid approach and pseudo-amino acid composition. J. Theor. Biol. 248, 377–381. doi:10.1016/j.jtbi.2007.05.019
Chen, Y. L., and Li, Q. Z. (2007). Prediction of the subcellular location of apoptosis proteins. J. Theor. Biol. 245, 775–783. doi:10.1016/j.jtbi.2006.11.010
Chen, Z., Zhao, P., Li, C., Li, F., Xiang, D., Chen, Y. Z., et al. (2021). iLearnPlus: a comprehensive and automated machine-learning platform for nucleic acid and protein sequence analysis, prediction and visualization. Nucleic acids Res. 49, e60. doi:10.1093/nar/gkab122
Chou, K. C., and Elrod, D. W. (1999b). Prediction of membrane protein types and subcellular locations. Proteins 34, 137–153. doi:10.1002/(sici)1097-0134(19990101)34:1<137::aid-prot11>3.0.co;2-o
Chou, K. C., and Elrod, D. W. (1999a). Protein subcellular location prediction. Protein Eng. 12, 107–118. doi:10.1093/protein/12.2.107
Chou, K. C. (2001). Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins 43, 246–255. doi:10.1002/prot.1035
Chou, K. C. (2005). Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinformatics 21, 10–19. doi:10.1093/bioinformatics/bth466
Dai, C., Jiang, Y., Yin, C., Su, R., Zeng, X., Zou, Q., et al. (2022). scIMC: a platform for benchmarking comparison and visualization analysis of scRNA-seq data imputation methods. Nucleic acids Res. 50, 4877–4899. doi:10.1093/nar/gkac317
Dao, F. Y., Lv, H., Fullwood, M. J., and Lin, H. (2022). Accurate identification of DNA replication origin by fusing epigenomics and chromatin interaction information. Research 2022, 9780293. doi:10.34133/2022/9780293
Dao, F. Y., Lv, H., Zhang, Y., and Lin, H. (2022a). BDselect: A package for k-mer selection based on the binomial distribution. Curr. Bioinform 17, 238–244. doi:10.2174/1574893616666211007102747
Deng, L., Huang, Y., Liu, X., and Liu, H. (2021). Graph2MDA: A multi-modal variational graph embedding model for predicting microbe-drug associations. Bioinformatics 38, 1118–1125. btab792. doi:10.1093/bioinformatics/btab792
Guo, Z., Wang, P., Liu, Z., and Zhao, Y. (2020). Discrimination of thermophilic proteins and non-thermophilic proteins using feature dimension reduction. Front. Bioeng. Biotechnol. 8, 584807. doi:10.3389/fbioe.2020.584807
Han, Y. M., Yang, H., Huang, Q. L., Sun, Z. J., Li, M. L., Zhang, J. B., et al. (2022). Risk prediction of diabetes and pre-diabetes based on physical examination data. Math. Biosci. Eng. 19, 3597–3608. doi:10.3934/mbe.2022166
Hasan, M. M., Tsukiyama, S., Cho, J. Y., Kurata, H., Alam, M. A., Liu, X., et al. (2022). Deepm5C: A deep-learning-based hybrid framework for identifying human rna N5-methylcytosine sites using a stacking strategy. Mol. Ther. 30, 2856–2867. doi:10.1016/j.ymthe.2022.05.001
Heijnen, H. F., Waaijenborg, S., Crapo, J. D., Bowler, R. P., Akkerman, J. W., and Slot, J. W. (2004). Colocalization of eNOS and the catalytic subunit of PKA in endothelial cell junctions: A clue for regulated NO production. J. Histochem. Cytochem. 52, 1277–1285. doi:10.1177/002215540405201004
Hosen, M. F., Mahmud, S. M. H., Ahmed, K., Chen, W., Moni, M. A., Deng, H. W., et al. (2022). DeepDNAbP: A deep learning-based hybrid approach to improve the identification of deoxyribonucleic acid-binding proteins. Comput. Biol. Med. 145, 105433. doi:10.1016/j.compbiomed.2022.105433
Jeon, Y. J., Hasan, M. M., Park, H. W., Lee, K. W., and Manavalan, B. (2022). Tacos: A novel approach for accurate prediction of cell-specific long noncoding RNAs subcellular localization. Briefings Bioinforma. 23, bbac243. doi:10.1093/bib/bbac243
Jia, C., Bi, Y., Chen, J., Leier, A., Li, F., and Song, J. (2020). Passion: An ensemble neural network approach for identifying the binding sites of RBPs on circRNAs. Bioinformatics 36, 4276–4282. doi:10.1093/bioinformatics/btaa522
Jiang, Q., Wang, G., Jin, S., Li, Y., and Wang, Y. (2013). Predicting human microRNA-disease associations based on support vector machine. Int. J. data Min. Bioinforma. 8, 282–293. doi:10.1504/ijdmb.2013.056078
Jin, J., Yu, Y., Wang, R., Zeng, X., Pang, C., Jiang, Y., et al. (2022). iDNA-ABF: multi-scale deep biological language learning model for the interpretable prediction of DNA methylations. Genome Biol. 23, 219. doi:10.1186/s13059-022-02780-1
Koroleva, O. A., Tomlinson, M. L., Leader, D., Shaw, P., and Doonan, J. H. (2005). High-throughput protein localization in Arabidopsis using Agrobacterium-mediated transient expression of GFP-ORF fusions. Plant J. 41, 162–174. doi:10.1111/j.1365-313X.2004.02281.x
Li, F., Chen, J., Ge, Z., Wen, Y., Yue, Y., Hayashida, M., et al. (2021). Computational prediction and interpretation of both general and specific types of promoters in Escherichia coli by exploiting a stacked ensemble-learning framework. Briefings Bioinforma. 22, 2126–2140. doi:10.1093/bib/bbaa049
Li, H., Pang, Y., Liu, B., and Yu, L. (2022). MoRF-FUNCpred: Molecular recognition feature function prediction based on multi-label learning and ensemble learning. Front. Pharmacol. 13, 856417. doi:10.3389/fphar.2022.856417
Li, Y., Qiao, G., Gao, X., and Wang, G. (2022b). Supervised graph co-contrastive learning for drug-target interaction prediction. Bioinformatics 38, 2847–2854. doi:10.1093/bioinformatics/btac164
Li, Y., Qiao, G., Wang, K., and Wang, G. (2022a). Drug-target interaction predication via multi-channel graph neural networks. Briefings Bioinforma. 23, bbab346. doi:10.1093/bib/bbab346
Liu, D., Huang, Y., Nie, W., Zhang, J., and Deng, L. (2021). Smalf: miRNA-disease associations prediction based on stacked autoencoder and XGBoost. BMC Bioinforma. 22, 219. doi:10.1186/s12859-021-04135-2
Lv, Z., Wang, P., Zou, Q., and Jiang, Q. (2020). Identification of Sub-Golgi protein localization by use of deep representation learning features. Bioinformatics 36, 5600–5609. doi:10.1093/bioinformatics/btaa1074
Malik, A., Subramaniyam, S., Kim, C. B., and Manavalan, B. (2021). SortPred: The first machine learning based predictor to identify bacterial sortases and their classes using sequence-derived information. Comput. Struct. Biotechnol. J. 20, 165–174. doi:10.1016/j.csbj.2021.12.014
Manavalan, B., Basith, S., Shin, T. H., Wei, L., and Lee, G. (2019a). mAHTPred: a sequence-based meta-predictor for improving the prediction of anti-hypertensive peptides using effective feature representation. Bioinformatics 35, 2757–2765. doi:10.1093/bioinformatics/bty1047
Manavalan, B., Basith, S., Shin, T. H., Wei, L., and Lee, G. (2019b). Meta-4mCpred: A sequence-based meta-predictor for accurate dna 4mC site prediction using effective feature representation. Mol. Ther. nucleic acids 16, 733–744. doi:10.1016/j.omtn.2019.04.019
Manavalan, B., and Patra, M. C. (2022). Mlcpp 2.0: An updated cell-penetrating peptides and their uptake efficiency predictor. J. Mol. Biol. 434, 167604. doi:10.1016/j.jmb.2022.167604
Naseer, S., Hussain, W., Khan, Y. D., and Rasool, N. (2021). NPalmitoylDeep-pseaac: A predictor of N-palmitoylation sites in proteins using deep representations of proteins and PseAAC via modified 5-steps rule. Curr. Bioinforma. 16, 294–305. doi:10.2174/1574893615999200605142828
Nidhi, M. B. K., Ganapathy, R., Subbiah, P., Suvaiyarasan, S., and Karuppasamy, M. P. (2022). GenNBPSeq: Online web server to generate never born protein sequences using toeplitz matrix approach with structure analysis. Curr. Bioinforma. 17, 565–577. doi:10.2174/1574893617666220519110154
Qian, Y. Q., Meng, H., Lu, W. Z., Liao, Z. J., Ding, Y. J., and Wu, H. J. (2022). Identification of DNA-binding proteins via hypergraph based laplacian support vector machine. Curr. Bioinforma. 17, 108–117. doi:10.2174/1574893616666210806091922
Reed, J. C., and Paternostro, G. (1999). Postmitochondrial regulation of apoptosis during heart failure. Proc. Natl. Acad. Sci. U. S. A. 96, 7614–7616. doi:10.1073/pnas.96.14.7614
Ren, L., Xu, Y., Ning, L., Pan, X., Li, Y., Zhao, Q., et al. (2022). TCM2COVID: A resource of anti-COVID-19 traditional Chinese medicine with effects and mechanisms. iMeta e42, e42. doi:10.1002/imt2.42
Rout, R. K., Hassan, S. S., Sheikh, S., Umer, S., Sahoo, K. S., and Gandomi, A. H. (2022). Feature-extraction and analysis based on spatial distribution of amino acids for SARS-CoV-2 Protein sequences. Comput. Biol. Med. 141, 105024. doi:10.1016/j.compbiomed.2021.105024
Shoombuatong, W., Basith, S., Pitti, T., Lee, G., and Manavalan, B. (2022). Throne: A new approach for accurate prediction of human rna N7-methylguanosine sites. J. Mol. Biol. 434, 167549. doi:10.1016/j.jmb.2022.167549
Su, W., Liu, M. L., Yang, Y. H., Wang, J. S., Li, S. H., Lv, H., et al. (2021). Ppd: A manually curated database for experimentally verified prokaryotic promoters. J. Mol. Biol. 433, 166860. doi:10.1016/j.jmb.2021.166860
Sun, Z., Huang, Q., Yang, Y., Li, S., Lv, H., Zhang, Y., et al. (2022). PSnoD: Identifying potential snoRNA-disease associations based on bounded nuclear norm regularization. Briefings Bioinforma. 23, bbac240. doi:10.1093/bib/bbac240
Tabaie, A., Orenstein, E. W., Nemati, S., Basu, R. K., Kandaswamy, S., Clifford, G. D., et al. (2021). Predicting presumed serious infection among hospitalized children on central venous lines with machine learning. Comput. Biol. Med. 132, 104289. doi:10.1016/j.compbiomed.2021.104289
Tang, H., Zhao, Y. W., Zou, P., Zhang, C. M., Chen, R., Huang, P., et al. (2018). HBPred: A tool to identify growth hormone-binding proteins. Int. J. Biol. Sci. 14, 957–964. doi:10.7150/ijbs.24174
Tao, Z., Li, Y., Teng, Z., and Zhao, Y. (2020). A method for identifying vesicle transport proteins based on LibSVM and MRMD. Comput. Math. methods Med. 2020, 8926750. doi:10.1155/2020/8926750
Tran, H. V., and Nguyen, Q. H. (2022). iAnt: Combination of convolutional neural network and random forest models using PSSM and BERT features to identify antioxidant proteins. Curr. Bioinforma. 17, 184–195. doi:10.2174/1574893616666210820095144
Vapnik, V. N. (2019). Complete statistical theory of learning. Autom. Remote Control 80, 1949–1975. doi:10.1134/S000511791911002X
Wang, J., Liu, X., Shen, S., Deng, L., and Liu, H. (2022). DeepDDS: Deep graph neural network with attention mechanism to predict synergistic drug combinations. Briefings Bioinforma. 23, bbab390. doi:10.1093/bib/bbab390
Wei, L., Tang, J., and Zou, Q. (2017). Local-DPP: An improved DNA-binding protein prediction method by exploring local evolutionary information. Inf. Sci. 384, 135–144. doi:10.1016/j.ins.2016.06.026
Wei, L., Ye, X., Sakurai, T., Mu, Z., and Wei, L. (2022). ToxIBTL: Prediction of peptide toxicity based on information bottleneck and transfer learning. Bioinforma. btac006 38, 1514–1524. doi:10.1093/bioinformatics/btac006
Wei, L., Zhou, C., Chen, H., Song, J., and Su, R. (2018). ACPred-FL: A sequence-based predictor using effective feature representation to improve the prediction of anti-cancer peptides. Bioinformatics 34, 4007–4016. doi:10.1093/bioinformatics/bty451
Xiao, J., Liu, M., Huang, Q., Sun, Z., Ning, L., Duan, J., et al. (2022). Analysis and modeling of myopia-related factors based on questionnaire survey. Comput. Biol. Med. 150, 106162. doi:10.1016/j.compbiomed.2022.106162
Xu, Z., Luo, M., Lin, W., Xue, G., Wang, P., Jin, X., et al. (2021). DLpTCR: An ensemble deep learning framework for predicting immunogenic peptide recognized by T cell receptor. Briefings Bioinforma. 22, bbab335. bbab335. doi:10.1093/bib/bbab335
Yang, H., Luo, Y., Ren, X., Wu, M., He, X., Peng, B., et al. (2021). Risk Prediction of Diabetes: Big data mining with fusion of multifarious physical examination indicators. Inf. Fusion 75, 140–149. doi:10.1016/j.inffus.2021.02.015
Yang, Y., Gao, D., Xie, X., Qin, J., Li, J., Lin, H., et al. (2022). DeepIDC: A prediction framework of injectable drug combination based on heterogeneous information and deep learning. Clin. Pharmacokinet. 61, 1749–1759. doi:10.1007/s40262-022-01180-9
Yu, L., Wang, M., Yang, Y., Xu, F., Zhang, X., Xie, F., et al. (2021). Predicting therapeutic drugs for hepatocellular carcinoma based on tissue-specific pathways. PLoS Comput. Biol. 17, e1008696. doi:10.1371/journal.pcbi.1008696
Yu, L., Zheng, Y., and Gao, L. (2022). MiRNA-disease association prediction based on meta-paths. Briefings Bioinforma. 23, bbab571. bbab571. doi:10.1093/bib/bbab571
Yu, X., Zheng, X., Liu, T., Dou, Y., and Wang, J. (2012). Predicting subcellular location of apoptosis proteins with pseudo amino acid composition: Approach from amino acid substitution matrix and auto covariance transformation. Amino acids 42, 1619–1625. doi:10.1007/s00726-011-0848-8
Zeng, R., Lu, Y., Long, S., Wang, C., and Bai, J. (2021). Cardiotocography signal abnormality classification using time-frequency features and Ensemble Cost-sensitive SVM classifier. Comput. Biol. Med. 130, 104218. doi:10.1016/j.compbiomed.2021.104218
Zhang, D., Xu, Z. C., Su, W., Yang, Y. H., Lv, H., Yang, H., et al. (2021). iCarPS: a computational tool for identifying protein carbonylation sites by novel encoded features. Bioinformatics 37, 171–177. doi:10.1093/bioinformatics/btaa702
Zhang H, H., Zou, Q., Ju, Y., Song, C., and Chen, D. (2022). Distance-based support vector machine to predict DNA N6-methyladenine modification. Curr. Bioinforma. 17, 473–482. doi:10.2174/1574893617666220404145517
Zhang, Q., Li, H., Liu, Y., Li, J., Wu, C., and Tang, H. (2022). Exosomal non-coding RNAs: New insights into the biology of hepatocellular carcinoma. Curr. Oncol. 29, 5383–5406. doi:10.3390/curroncol29080427
Zhang, Y., Liu, T., Hu, X., Wang, M., Wang, J., Zou, B., et al. (2021a). CellCall: Integrating paired ligand-receptor and transcription factor activities for cell-cell communication. Nucleic acids Res. 49, 8520–8534. doi:10.1093/nar/gkab638
Zhang, Y., Liu, T., Wang, J., Zou, B., Li, L., Yao, L., et al. (2021b). Cellinker: A platform of ligand-receptor interactions for intercellular communication analysis. Bioinforma. Btab036. 37, 2025–2032. doi:10.1093/bioinformatics/btab036
Zhang Z Y, Z. Y., Ning, L., Ye, X., Yang, Y. H., Futamura, Y., Sakurai, T., et al. (2022). iLoc-miRNA: extracellular/intracellular miRNA prediction using deep BiLSTM with attention mechanism. Briefings Bioinforma. 23, bbac395. doi:10.1093/bib/bbac395
Zhang, Z. H., Wang, Z. H., Zhang, Z. R., and Wang, Y. X. (2006). A novel method for apoptosis protein subcellular localization prediction combining encoding based on grouped weight and support vector machine. FEBS Lett. 580, 6169–6174. doi:10.1016/j.febslet.2006.10.017
Zhang, Z. M., Wang, J. S., Zulfiqar, H., Lv, H., Dao, F. Y., and Lin, H. (2020). Early diagnosis of pancreatic ductal adenocarcinoma by combining relative expression orderings with machine-learning method. Front. Cell Dev. Biol. 8, 582864. doi:10.3389/fcell.2020.582864
Zhou, G. P., and Doctor, K. (2003). Subcellular location prediction of apoptosis proteins. Proteins 50, 44–48. doi:10.1002/prot.10251
Zhou, H., Wang, H., Ding, Y., and Tang, J. (2022). Multivariate information fusion for identifying antifungal peptides with hilbert-schmidt independence criterion. Curr. Bioinforma. 17, 89–100. doi:10.2174/1574893616666210727161003
Keywords: apoptosis protein, subcellular location, amphiphilic pseudo amino acid composition, support vector machine, jackknife test
Citation: Su W, Deng S, Gu Z, Yang K, Ding H, Chen H and Zhang Z (2023) Prediction of apoptosis protein subcellular location based on amphiphilic pseudo amino acid composition. Front. Genet. 14:1157021. doi: 10.3389/fgene.2023.1157021
Received: 02 February 2023; Accepted: 20 February 2023;
Published: 28 February 2023.
Edited by:
Lin Zhang, China University of Mining and Technology, ChinaReviewed by:
Lei Yang, Harbin Medical University, ChinaCopyright © 2023 Su, Deng, Gu, Yang, Ding, Chen and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Chen, Y2hlbmh1aUBuc3UuZWR1LmNu; Zhaoyue Zhang, enl6aGFuZ0B1ZXN0Yy5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.