 Shiroh Miura1
Shiroh Miura1 Shigeyoshi Hiruki2
Shigeyoshi Hiruki2 Tomohisa Okada3Satoko Itani Takei1Kensuke Senzaki1Yoko Okada1Masayuki Ochi1
Tomohisa Okada3Satoko Itani Takei1Kensuke Senzaki1Yoko Okada1Masayuki Ochi1 Yuki Tanabe3
Yuki Tanabe3 Hirofumi Ochi4
Hirofumi Ochi4 Michiya Igase5
Michiya Igase5 Yasumasa Ohyagi1
Yasumasa Ohyagi1 Hiroki Shibata2*
Hiroki Shibata2*- 1Department of Neurology and Geriatric Medicine, Ehime University Graduate School of Medicine, Toon, Ehime, Japan
- 2Division of Genomics, Medical Institute of Bioregulation, Kyushu University, Higashi-ku, Fukuoka, Japan
- 3Department of Radiology, Ehime University Graduate School of Medicine, Toon, Ehime, Japan
- 4Department of Intractable Disease and Aging Science, Ehime University Graduate School of Medicine, Toon, Ehime, Japan
- 5Department of Anti-aging Medicine, Ehime University Graduate School of Medicine, Toon, Ehime, Japan
Frontotemporal dementia and/or amyotrophic lateral sclerosis 6, also known as amyotrophic lateral sclerosis 14, is an autosomal dominant, progressive neurodegenerative disorder caused by various mutations in the valosin-containing protein gene. In this report, we examined a 51-year-old female Japanese patient with frontotemporal dementia and amyotrophic lateral sclerosis. The patient began noticing gait disturbances at the age of 45 years. Neurological examination at the age of 46 years met the Awaji criteria for clinically probable amyotrophic lateral sclerosis. At the age of 49 years, she tended to have poor mood and an aversion to activity. Her symptoms gradually worsened. She required a wheelchair for transport and had difficulty communicating with others because of poor comprehension. She then began to frequently exhibit irritability. Eventually, she was admitted to the psychiatric hospital because uncontrollable violent behavior throughout the day. Longitudinal brain magnetic resonance imaging revealed progressive brain atrophy with temporal dominance, non-progressive cerebellar atrophy, and some non-specific white matter intensities. Brain single photon emission computed tomography showed hypoperfusion in the bilateral temporal lobes and cerebellar hemispheres. Clinical exome sequencing revealed the presence of a heterozygous nonsynonymous variant (NM_007126.5, c.265C>T; p.Arg89Trp) in the valosin-containing protein gene, which was absent in the 1000 Genomes Project, the Exome Aggregation Consortium Database, and the Genome Aggregation Database, and was predicted to be “damaging” by PolyPhen-2 and “deleterious” using SIFT with a Combined Annotation Dependent Depletion score of 35. We also confirmed the absence of this variant in 505 Japanese control subjects. Therefore, we concluded that the variant in the valosin-containing protein gene was responsible for the symptoms of this patient.
Introduction
The valosin-containing protein (VCP, MIM: 601023) gene is considered responsible for frontotemporal dementia (FTD) and/or amyotrophic lateral sclerosis (ALS) 6 (FTDALS6), also known as ALS14 (MIM: 613954), and inclusion body myopathy with early-onset Paget disease and frontotemporal dementia 1 (MIM: 167320) via a dominant-negative mechanism (Ayaki et al., 2014). It is known to exhibit large intrafamilial and interfamilial phenotypic variations among patients with VCP mutations (Al-Obeidi et al., 2018). Myopathy, Paget disease of bone (PDB), FTD, ALS, Parkinson’s disease, and Alzheimer’s disease occur in 89.8%–91.0%, 42.4%–51.7%, 29.6%–30.3%, 8.6%–8.9%, 3.4%–3.8%, and 1.6% of patients carrying VCP mutations, respectively (Al-Obeidi et al., 2018; Mehta et al., 2013). Regarding ALS, the frequency of VCP mutations in familial ALS patients, young-onset ALS patients (age of onset ≤45), and sporadic ALS patients are up to 2%, 3.7%, and 0.4%, respectively (González-Pérez et al., 2012; Johnson et al., 2010; Koppers et al., 2012; Deng et al., 2019; Abramzon et al., 2012). For FTD, 3.5% of patients carry VCP mutations (Saracino et al., 2018). The coexistence of ALS and FTD (ALS-FTD) is a rare manifestation among patients carrying VCP mutations. Here, we report a Japanese patient with ALS-FTD carrying a missense variant of the VCP gene. We also present brain magnetic resonance imaging (MRI) findings over time to demonstrate disease progression.
Clinical report
A 51-year-old woman, who had no family history of neuropsychiatric diseases, began noticing gait disturbance at the age of 45 years and upper limb weakness at the age of 46 years. These symptoms gradually worsened. Her personality showed a mild change. She developed depression associated with obsessive behavior and difficulty understanding everyday speech at the age of 49 years. She began using a wheelchair for transport at the age of 50 years. Finally, she was admitted to a psychiatric hospital because she was easily distracted and behaved violently throughout the day. Her past medical history revealed a transient ischemic attack at the age of 41 years. Neurological examination at the age of 46 years showed that the patient had mild muscular weakness of the four limbs with left distal dominance. Amyotrophy was unremarkable. Her hand grip was 18 kg in the right hand and 11 kg in the left hand. She showed hyperreflexia and spasticity in all four extremities with left dominance, and her jaw reflex was increased. Furthermore, her Babinski reflexes were positive bilaterally, and her gait was spastic. There were no abnormalities in the cranial nerves. Sensory, autonomic, and cerebellar functions were normal. Nerve conduction studies revealed that F-wave occurrences were reduced in the median nerve bilaterally (right: 56%, left: 25%; normal values: 70% <). Amplitudes of compound muscle action potentials, motor conduction velocities, amplitudes of sensory nerve action potentials, and sensory conduction velocities were normal in all nerves tested (i.e., the bilateral median, ulnar, tibial, and sural nerves). Neurogenic changes were detected in the biceps brachii muscle, the first interossei dorsalis muscle, the thoracic paraspinal muscle, the quadriceps femoris muscle, and the tibialis anterior muscle using needle electromyography. Upon neurological examination at the age of 50 years, the patient showed attention disturbances, transcortical sensory aphasia, and a positive snout reflex. Hyperreflexia, spasticity, and weakness in all four extremities had worsened, and bilateral ankle contracture was observed. She was unable to maintain a standing position. Her hand grip was 16 kg in the right hand and 6 kg in the left hand. There were no abnormalities in the cranial nerves or sensory, autonomic, and cerebellar functions. Her Mini-Mental State Examination score was 16/30, Frontal Assessment Battery score was 5/18, Alzheimer’s Disease Assessment Scale was 29.3/70, and Raven’s Colored Progressive Matrices was 32/36.
The cerebrospinal fluid examination was negative, as were the test results for oligoclonal bands and immunoglobulin G indices at the ages of 41, 46, 48, and 50 years. Furthermore, the levels of serum creatine kinase and alkaline phosphatase were normal throughout her clinical course. Longitudinal brain MRI revealed progressive brain atrophy with temporal dominance (Figure 1A), non-progressive cerebellar atrophy (Figure 1A), and some non-specific white matter intensities (data not shown). Brain N-isopropyl-p-[123I] iodoamphetamine single photon emission computed tomography (SPECT) at the age of 50 years showed hypoperfusion in the bilateral temporal lobe with left dominance and the cerebellar hemispheres (Figure 1B). Neither the cervical MRI acquired at the age of 41 years nor the whole spinal MRI acquired at the age of 46 years showed abnormalities.
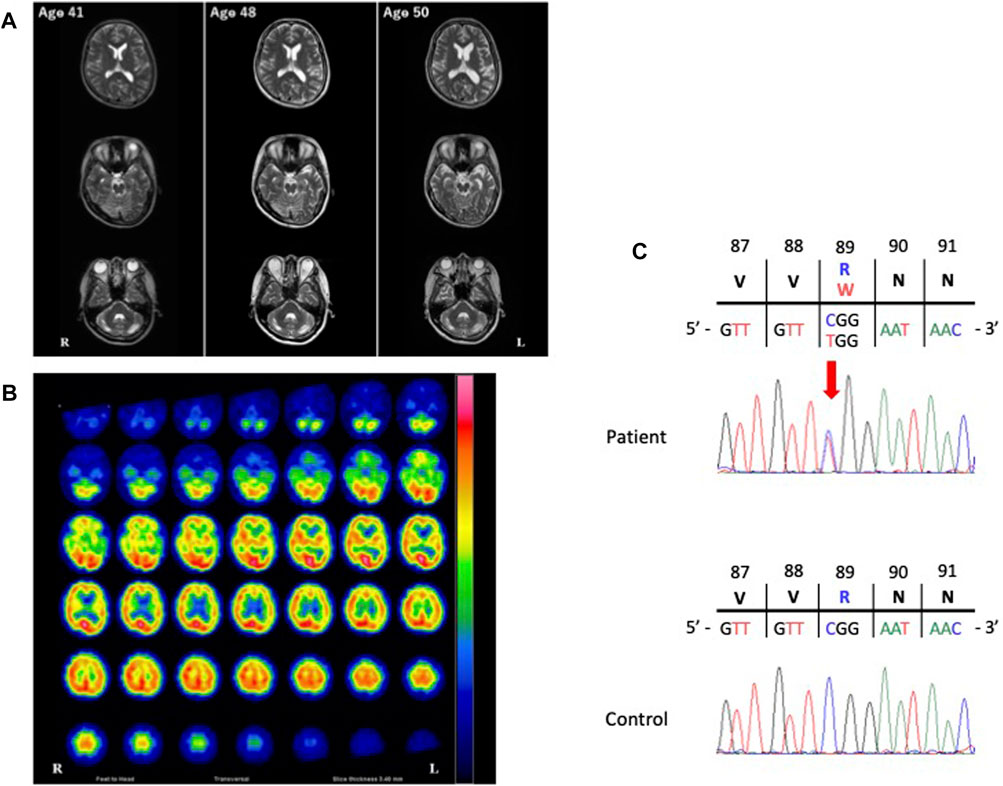
FIGURE 1. Images of the present patient and the nucleotide variant. (A), The course of T2-weighted brain magnetic resonance imaging (MRI). Longitudinal brain MRI showed progressive brain atrophy with left temporal dominance and non-progressive cerebellar atrophy. (B), Brain N-isopropyl-p-[123I] iodoamphetamine single photon emission computed tomography (123I-IMP SPECT). The patient’s123I-IMP SPECT image acquired at the age of 50 years showed hypoperfusion in the bilateral temporal lobes with left dominance and the bilateral cerebellar hemispheres. (C) Electropherogram of the region of the variant of the valosin-containing protein gene (NM_007126.5: exon 9: c.265C>T [Arg89Trp]) in our patient and an unaffected control. The location of the variant is indicated by a red arrow.
Preliminary genetic analyses confirmed the absence of repeat expansions in 11 genes known to be associated with cerebellar atrophy: ATXN1, ATXN2, ATXN3, CACNA1A, ATXN7, ATXN8, ATXN10, PPP2R2B, TBP, NOP56, and ATN1.
Her clinical or electrophysiological findings showed upper and lower motor neuron signs which met clinically probable ALS of the Awaji criteria (Table 1). Also, she did not exhibit any clinical or laboratory findings suggestive of myopathy or PDB throughout her clinical course.

TABLE 1. Distribution of lower and upper motor neuron signs in the present case.
Materials and methods
Patient information
The patient was Japanese and originated in Ehime Prefecture of Shikoku Island in Japan. A peripheral blood sample was collected. Unfortunately, peripheral blood samples were unavailable from any other family members due to the private reasons.
Whole-exome sequencing
We performed whole-exome sequencing. Genomic DNA was extracted from the peripheral blood using a QIAamp DNA Blood Kit (Qiagen, Hilden, Germany). Exonic regions were enriched using SureSelect Human All Exon v6 (Agilent Technologies, Santa Clara, CA, United States). Paired-end sequencing at 150 bp was performed using the Illumina NovaSeq6000 platform (Illumina, Inc., CA, United States). Raw sequencing reads were analyzed using Trimmomatic v0.36, BWA v0.7.16, Samtools v1.5, and Genome Analysis Toolkit v3.8.0. Variants were annotated using wAnnovar (https://wannovar.wglab.org/).
Because of cerebellar atrophy and the ALS symptoms exhibited by the patient, we prioritized the analysis of variants located in genes 77 and 72, which are known to be associated with hereditary spinocerebellar ataxias (SCAs) and hereditary ALSs, respectively (Supplementary Table S1). We examined the frequencies of candidate variants in control populations using the 1000 Genomes Project (1000G; http://www.1000genomes.org), the Exome Aggregation Consortium (ExAC) database (http://exac.broadinstitute.org), the Genome Aggregation Database (gnomAD; https://gnomad.broadinstitute.org/) and the Japanese Multi-Omics Reference Panel (jMorp; https://jmorp.megabank.tohoku.ac.jp). We further removed non-pathogenic local variants found in the in-house exome data of 56 unrelated Japanese individuals who were matched geographically. The functional consequences of the variants were evaluated in silico using PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (https://sift.bii.a-star.edu.sg/), and Combined Annotation Dependent Depletion (CADD; http://cadd.gs.washington.edu/home).
Sanger sequencing
The region that included the variation site in exon 9 of the VCP gene was amplified using the following primers: 5′-TTTCTTTCCTCAGCCCAAGA-3′ (forward) and 5′-ATCGACAGGTGCCAAGAACT-3′ (reverse). Polymerase chain reaction (PCR) conditions were 35 cycles of 94°C for 30 s, 58°C for 30 s, and 72°C for 30 s. PCR products were sequenced using the ABI PRISM Big Dye Terminator (v 3.1) Cycle Sequencing Kit (Thermo Fisher Scientific, Waltham, MA, United States) and the ABI PRISM 3130-Avant Genetic Analyzer (Thermo Fisher Scientific, Waltham, MA, United States).
Results
Exome sequencing
A mean read depth of 106.5× was achieved for the target regions. From 23,766 variants initially called, we identified 75 single nucleotide variants (SNVs) located in genes known to be responsible for hereditary SCAs (Supplementary Table S1). None of the SNVs met our filtering criteria of a minor allele frequency of <0.001 in the public databases. We also identified 74 SNVs located in genes known to be responsible for hereditary ALSs (Supplementary Table S1), which comprised 32 homozygous and 42 heterozygous variants. Of the 74 variants, two SNVs met the criteria of a minor allele frequency of <0.001 in the public databases. We further excluded one of these variants, which was predicted to be “benign” and “tolerated” by PolyPhen-2 and SIFT, respectively. Thus, only one heterozygous non-synonymous variant located on exon 9 of the VCP gene was retained (NM_007126.5, c.265C>T [Arg89Trp]). This variant was confirmed to be absent in the exome data of 56 unrelated in-house controls.
Validation of the VCP variant using Sanger sequencing
The VCP variant (NM_007126.5, c.265C>T [Arg89Trp]) was absent in 1000G, ExAC, gnomAD and jMorp, and was predicted to be “damaging” by PolyPhen-2 and “deleterious” by SIFT, with a CADD score of 35 (Figure 1C). We further confirmed the absence of the variant in 505 healthy, unrelated Japanese individuals using Sanger sequencing, which indicated that the variant is extremely rare in the Japanese population, with a frequency of <0.001.
Discussion
We described a Japanese FTDALS6 case carrying a rare variant (c.265C>T) in the VCP gene that resulted in an amino acid substitution (Arg89Trp). The variant has been registered in ClinVar with no clinical information (Allele ID: 993443). The patient’s neurological findings at the age of 46 years met the Awaji ALS criteria for clinically probable ALS (de Carvalho et al., 2008). Subsequently, a diagnosis of ALS complicated with FTD was made according to the revised diagnostic criteria (Strong et al., 2017). To date, at least 10 cases of ALS complicated with FTD have been reported to be associated with VCP variants (Miller et al., 2009; Kumar et al., 2010; Johnson et al., 2010; Koppers et al., 2012; Spina et al., 2013; Hirano et al., 2015; Hirano et al., 2017; Matsubara et al., 2021). Clinical and/or pathological information of previously reported patients with ALS complicated with FTD carrying VCP variants are summarized in Table 2, which includes our patient. In most cases, the initial symptom of ALS preceded that of FTD. Six out of 11 cases showed no neurological signs except for ALS and FTD. Other neurological signs observed were myopathy (two cases), hearing impairment (two cases), PDB (two cases), parkinsonism (one case), and urinary urge incontinence (one case). Neither serum alkaline phosphatase nor creatine kinase was elevated in most cases. Moreover, no specific variants were correlated with the incidence of clinical features of ALS complicated with FTD. In the current case, as well as previously reported cases, no abnormalities were observed on brain MRI before the onset of dementia and/or psychotic symptoms (Johnson et al., 2010; Matsubara et al., 2021). However, once dementia and/or psychotic symptoms appeared, frontotemporal atrophy seemed to progress. This phenomenon is compatible with the fact that ALS usually precedes FTD, although one report showed that modest temporal atrophy was observed before the onset of dementia-associated symptoms (Hirano et al., 2017). In general, frontotemporal atrophy has been shown to progress as dementia and/or psychotic symptoms worsen. However, the degree of brain atrophy varies among individuals with the same symptom severity. In addition, some non-specific cerebral white matter intensities were associated with the disease. Indeed, SPECT examinations have revealed that hypoperfusion in the left temporal lobe may be a distinct feature of the disease.
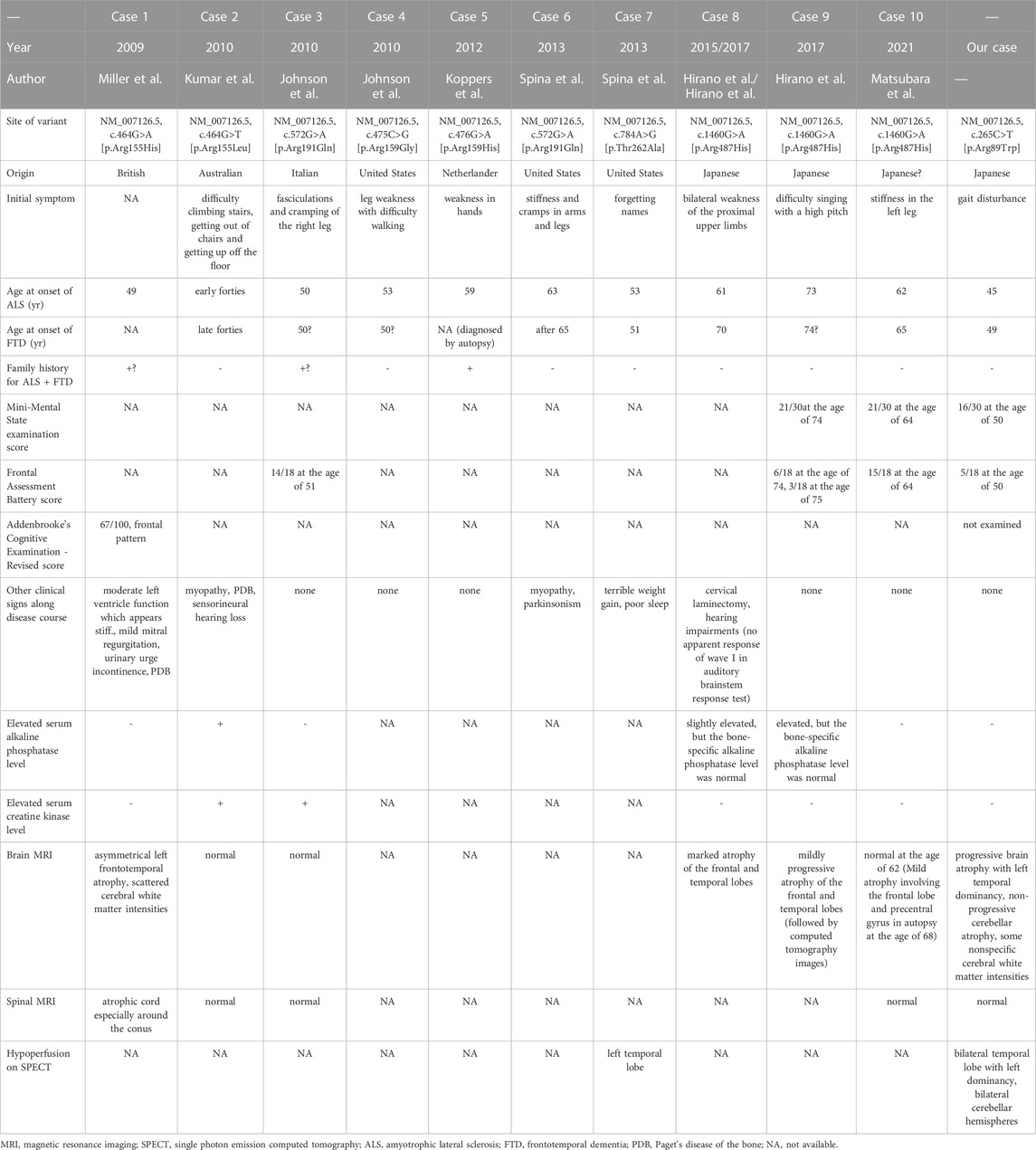
TABLE 2. Clinical characteristics of the present case and previously reported cases with amyotrophic lateral sclerosis plus frontotemporal dementia with valosin-containing protein variants.
Although we examined variants in the genes known to be associated with hereditary SCAs based on the cerebellar atrophy observed in the current case, no functional variant survived our filtering criteria. Moreover, there was neither progression of cerebellar atrophy nor cerebellar symptoms. Thus, we concluded that the patient’s cerebellar atrophy was either congenital or in a premature state. The association between cerebellar atrophy and the variant in the VCP gene remains unclear.
Table 3 summarizes the patients with missense variants at residue 89 in the VCP gene. The p.Arg89Trp variant has been previously reported in one Portuguese patient with distal myopathy and FTD (de Campos and de Carvalho, 2019). Another missense variant at the same residue (NM_007126.5, c.266G>A; p.Arg89Gln) in the VCP gene has been reported in a Chinese patient with young-onset ALS, who died of respiratory failure 5 months after the onset of initial symptoms (Deng et al., 2019). The clinical features varied among the three cases. Thus, there is no correlation between clinical phenotype and variants at residue 89 in the VCP gene. According to the American College of Medical Genetics and Genomics (ACMG)/Association for Molecular Pathology (AMP)/College of American Pathologists (CAP) guidelines, the non-synonymous variant in the VCP gene is classified as “pathogenic” because it meets the criteria of PS1, PM1, and PM2 (Richards et al., 2015). Therefore, we concluded that the missense variant in the VCP gene, NM_007126.5, c.265C>T; p.Arg89Trp, is the variant that causes the disease. Accordingly, we highlight the importance of examining VCP variations in patients with ALS and FTD, even in those with no family history of these diseases.
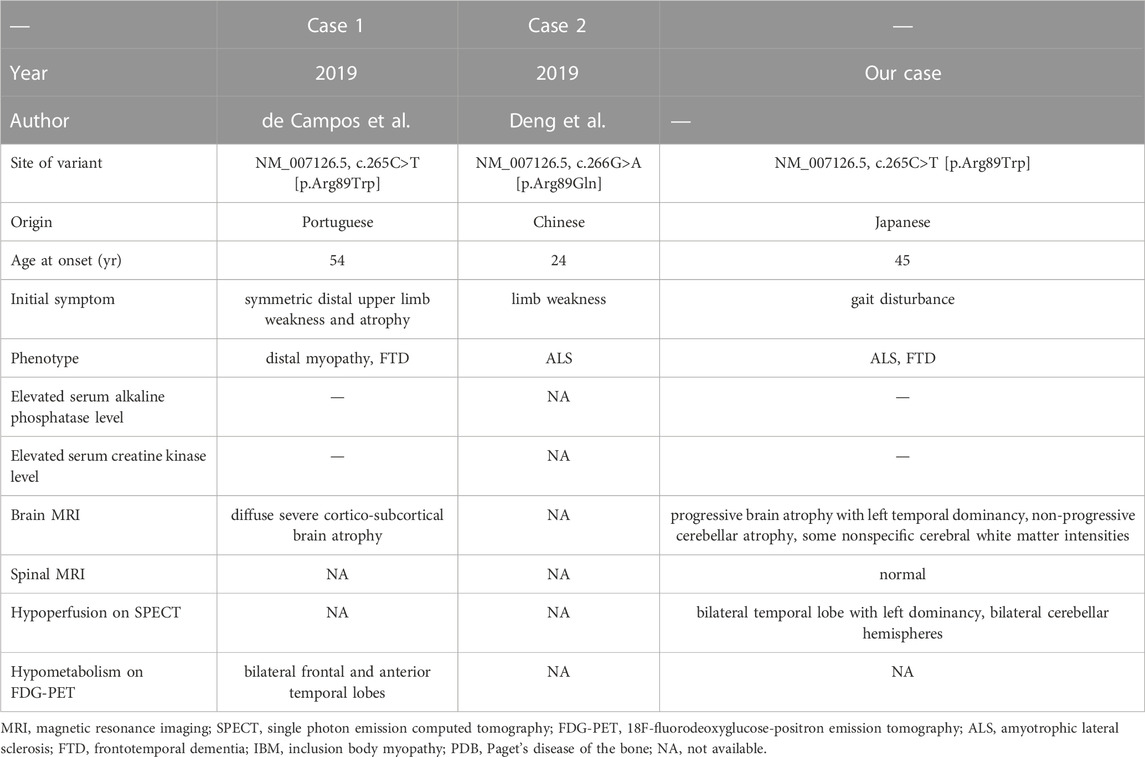
TABLE 3. Summary of missense variants reported at residue 89 in the valosin-containing protein gene.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
This study was approved by the Ethics Committees of Ehime University Graduate School of Medicine (#31-1-R3-K1, #31-15-R3-K2) and Kyushu University, Faculty of Medicine (#818-00, #819-00). Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
SM, SIT, KS, YoO, and MO collected patient samples and patient data. SM, SH, and TO prepared the initial draft of the manuscript. YT assisted in developing figures and images. SM, SH, and HS carried out laboratory analyses and analyzed the data. HO, MI, YaO, and HS contributed to the writing of the manuscript. YaO and HS supervised the study. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Cooperative Research Project Program of the Medical Institute of Bioregulation, Kyushu University.
Acknowledgments
We thank Sarina Iwabuchi, PhD, from Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1155998/full#supplementary-material
References
Abramzon, Y., Johnson, J. O., Scholz, S. W., Taylor, J. P., Brunetti, M., Calvo, A., et al. (2012). Valosin-containing protein (VCP) mutations in sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 33, 2231.e1–2231. doi:10.1016/j.neurobiolaging.2012.04.005
Al-Obeidi, E., Al-Tahan, S., Surampalli, A., Goyal, N., Wang, A., Hermann, A., et al. (2018). Genotype-phenotype study in patients with valosin-containing protein mutations associated with multisystem proteinopathy. Clin. Genet. 93, 119–125. doi:10.1111/cge.13095
Ayaki, T., Ito, H., Fukushima, H., Inoue, T., Kondo, T., Ikemoto, A., et al. (2014). Immunoreactivity of valosin-containing protein in sporadic amyotrophic lateral sclerosis and in a case of its novel mutant. Acta Neuropathol. Commun. 2, 172. doi:10.1186/s40478-014-0172-0
de Campos, C. F., and de Carvalho, M. (2019). Distal myopathy and rapidly progressive dementia associated with a novel mutation in the VCP gene: Expanding inclusion body myopathy with early-onset Paget disease and frontotemporal dementia spectrum. J. Clin. Neurosci. 64, 8–10. doi:10.1016/j.jocn.2019.03.063
de Carvalho, M., Dengler, R., Eisen, A., England, J. D., Kaji, R., Kimura, J., et al. (2008). Electrodiagnostic criteria for diagnosis of ALS. Clin. Neurophysiol. 119, 497–503. doi:10.1016/j.clinph.2007.09.143
Deng, J., Wu, W., Xie, Z., Gang, Q., Yu, M., Liu, J., et al. (2019). Novel and recurrent mutations in a cohort of Chinese patients with young-onset amyotrophic lateral sclerosis. Front. Neurosci. 13, 1289. doi:10.3389/fnins.2019.01289
González-Pérez, P., Cirulli, E. T., Drory, V. E., Dabby, R., Nisipeanu, P., Carasso, R. L., et al. (2012). Novel mutation in VCP gene causes atypical amyotrophic lateral sclerosis. Neurology 79, 2201–2208. doi:10.1212/WNL.0b013e318275963b
Hirano, M., Nakamura, Y., Saigoh, K., Sakamoto, H., Ueno, S., Isono, C., et al. (2015). VCP gene analyses in Japanese patients with sporadic amyotrophic lateral sclerosis identify a new mutation. Neurobiol. Aging 36, 1604.e1–e6. doi:10.1016/j.neurobiolaging.2014.10.012
Hirano, M., Yamagishi, Y., Yanagimoto, S., Saigoh, K., Nakamura, Y., and Kusunoki, S. (2017). Time course of radiological imaging and variable interindividual symptoms in amyotrophic lateral sclerosis and frontotemporal dementia associated with p.Arg487His mutation in the VCP gene. Eur. Neurol. 78, 78–83. doi:10.1159/000478906
Johnson, J. O., Mandrioli, J., Benatar, M., Abramzon, Y., van Deerlin, V. M., Trojanowski, J. Q., et al. (2010). Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68, 857–864. doi:10.1016/j.neuron.2010.11.036
Koppers, M., van Blitterswijk, M. M., Vlam, L., Rowicka, P. A., van Vught, P. W. J., Groen, E. J. N., et al. (2012). VCP mutations in familial and sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 33, 837.e7–13. doi:10.1016/j.neurobiolaging.2011.10.006
Kumar, K. R., Needham, M., Mina, K., Davis, M., Brewer, J., Staples, C., et al. (2010). Two Australian families with inclusion-body myopathy, Paget’s disease of bone and frontotemporal dementia: Novel clinical and genetic findings. Neuromuscul. Disord. 20, 330–334. doi:10.1016/j.nmd.2010.03.002
Matsubara, T., Izumi, Y., Oda, M., Takahashi, M., Maruyama, H., Miyamoto, R., et al. (2021). An autopsy report of a familial amyotrophic lateral sclerosis case carrying VCP Arg487His mutation with a unique TDP-43 proteinopathy. Neuropathology 41, 118–126. doi:10.1111/neup.12710
Mehta, S. G., Khare, M., Ramani, R., Watts, G. D. J., Simon, M., Osann, K. E., et al. (2013). Genotype-phenotype studies of VCP-associated inclusion body myopathy with Paget disease of bone and/or frontotemporal dementia. Clin. Genet. 83, 422–431. doi:10.1111/cge.12000
Miller, T. D., Jackson, A. P., Barresi, R., Smart, C. M., Eugenicos, M., Summers, D., et al. (2009). Inclusion body myopathy with Paget disease and frontotemporal dementia (IBMPFD): Clinical features including sphincter disturbance in a large pedigree. J. Neurol. Neurosurg. Psychiatry 80, 583–584. doi:10.1136/jnnp.2008.148676
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of medical genetics and Genomics and the association for Molecular Pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Saracino, D., Clot, F., Camuzat, A., Anquetil, V., Hannequin, D., Guyant-Maréchal, L., et al. (2018). Novel VCP mutations expand the mutational spectrum of frontotemporal dementia. Neurobiol. Aging 72, 187.e11–187.187.e14. doi:10.1016/j.neurobiolaging.2018.06.037
Spina, S., van Laar, A. D., Murrell, J. R., Hamilton, R. L., Kofler, J. K., Epperson, F., et al. (2013). Phenotypic variability in three families with valosin-containing protein mutation. Eur. J. Neurol. 20, 251–258. doi:10.1111/j.1468-1331.2012.03831.x
Keywords: frontotemporal dementia and/or amyotrophic lateral sclerosis-6 (FTDALS6), amyotrophic lateral sclerosis 14 (ALS14), VCP, cerebellar atrophy, missense variant
Citation: Miura S, Hiruki S, Okada T, Takei SI, Senzaki K, Okada Y, Ochi M, Tanabe Y, Ochi H, Igase M, Ohyagi Y and Shibata H (2023) Case report: Frontotemporal dementia and amyotrophic lateral sclerosis caused by a missense variant (p.Arg89Trp) in the valosin-containing protein gene. Front. Genet. 14:1155998. doi: 10.3389/fgene.2023.1155998
Received: 01 February 2023; Accepted: 12 May 2023;
Published: 26 May 2023.
Edited by:
Jun Mitsui, The University of Tokyo, JapanReviewed by:
Magdalena Mroczek, Psychiatric University Hospital Zurich, SwitzerlandHiroya Naruse, The University of Tokyo, Japan
Copyright © 2023 Miura, Hiruki, Okada, Takei, Senzaki, Okada, Ochi, Tanabe, Ochi, Igase, Ohyagi and Shibata. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hiroki Shibata, aHNoaWJhdGFAZ2VuLmt5dXNodS11LmFjLmpw