 Marco Corazzari
Marco Corazzari Licio Collavin
Licio Collavin
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Genet. , 20 March 2023
Sec. Cancer Genetics and Oncogenomics
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1148192
This article is part of the Research Topic Role of p53 in Cell Metabolism, Ferroptosis, and Stemness View all 12 articles
Cancer cells within tumor masses are chronically exposed to stress caused by nutrient deprivation, oxygen limitation, and high metabolic demand. They also accumulate hundreds of mutations, potentially generating aberrant proteins that can induce proteotoxic stress. Finally, cancer cells are exposed to various damages during chemotherapy. In a growing tumor, transformed cells eventually adapt to these conditions, eluding the death-inducing outcomes of signaling cascades triggered by chronic stress. One such extreme outcome is ferroptosis, a form of iron-dependent non-apoptotic cell death mediated by lipid peroxidation. Not surprisingly, the tumor suppressor p53 is involved in this process, with evidence suggesting that it acts as a pro-ferroptotic factor and that its ferroptosis-inducing activity may be relevant for tumor suppression. Missense alterations of the TP53 gene are extremely frequent in human cancers and give rise to mutant p53 proteins (mutp53) that lose tumor suppressive function and can acquire powerful oncogenic activities. This suggests that p53 mutation provides a selective advantage during tumor progression, raising interesting questions on the impact of p53 mutant proteins in modulating the ferroptotic process. Here, we explore the role of p53 and its cancer-related mutants in ferroptosis, using a perspective centered on the resistance/sensitivity of cancer cells to exogenous and endogenous stress conditions that can trigger ferroptotic cell death. We speculate that an accurate molecular understanding of this particular axis may improve cancer treatment options.
TP53 is possibly the most frequently altered gene in human cancers (Kandoth et al., 2013). The encoded p53 protein is a powerful tumor suppressor, and its loss-of-function is associated with cancer development and progression (Levine, 2020). Intriguingly, the majority of TP53 mutations are missense, encoding full-length proteins (mutp53) that are stably expressed in tumor cells. The pervasive retention of mutp53 in cancer suggests a selective advantage; indeed, missense p53 mutants have been reported to foster cancer cell proliferation, invasion, metastasis, and chemoresistance (Pilley et al., 2021; Dolma and Muller, 2022). Various oncogenic phenotypes and mechanisms of action, transcriptional and non-transcriptional, have been described for mutant p53 (Bellazzo et al., 2018; Kim and Lozano, 2018); nonetheless, our understanding of the real impact of mutp53 in cancer formation and progression remains incomplete.
An interesting hypothesis is that mutp53, similarly to its wild-type counterpart, may sense transformation-related cellular stresses and coordinate adaptive responses that help tumor progression (D’Orazi and Cirone, 2019; Mantovani et al., 2019). Such indirect action, dependent on multiple unpredictable circumstances, could explain why missense TP53 mutations are pervasively selected in tumors, but depletion of mutp53 in cancer cell lines and preclinical models gives variable and often contradictory results (Kennedy and Lowe, 2022; Wang et al., 2022).
Cancer cells within tumors experience multiple adverse conditions: nutrient and oxygen shortage, high metabolic demand, increased mutation rate, and chemotherapy-induced DNA damage. They eventually adapt to chronic stress, often hijacking stress-response pathways to favor homeostasis and survival. For instance, aberrant activation of the unfolded protein response can facilitate cancer progression by inducing epithelial mesenchymal transition, stimulating angiogenesis, and supporting tumor cell dormancy (Senft and Ronai, 2016; Limia et al., 2019). Some mechanisms by which mutp53 can help cancer cells adapt to cancer-related stress are beginning to emerge from tissue culture and animal models; characterizing such mechanisms may open new opportunities for targeted therapy.
Cancer-related stress conditions can directly or indirectly cause ferroptosis, a cell death process resulting from intracellular accumulation of lipid peroxides. Ferroptosis is under intense study due to its potential anti-cancer activity, especially in apoptosis-resistant tumors (Friedmann Angeli et al., 2019; Lei et al., 2022; Rodriguez et al., 2022). In fact, due to their altered metabolism, cancer cells are susceptible to ferroptosis and highly dependent on protective systems for survival; genes and pathways involved in such processes, therefore, could be targeted to improve chemotherapy. Not surprisingly, wild-type p53 has been reported to modulate ferroptosis in tumor models, possibly affecting response to treatment. The emerging relevance of the p53-ferroptosis axis inevitably raises important questions about the impact of cancer-associated mutant p53 in this phenomenon.
The term ‘ferroptosis’ describes a form of non-apoptotic cell death characterized by iron-dependent production of Lipid-ROS responsible for cell killing (Dixon et al., 2012). Since its first description, the number of papers studying ferroptosis has increased exponentially (Stockwell, 2022) confirming its involvement in both physiological and pathological events ranging from development, immune functions and tumor suppression, to neurodegeneration, autoimmunity and tumorigenesis (Jiang et al., 2021).
Lipid-ROS are the main executioners of ferroptosis, produced by intracellular iron accumulation, promoting peroxidation of PL-PUFA through Fenton reactions (Shah et al., 2018). The cellular labile iron pool required to stimulate ferroptosis can be the result of either increased iron import from the extracellular compartment, or released by autophagy-mediated degradation of ferritin (ferritinophagy) (Hou et al., 2016). Also iron-containing enzymes, such as ALOXs and POR, can promote lipid peroxidation, driving ferroptosis (Yang and Stockwell, 2016; Gagliardi et al., 2020; Zou et al., 2020).
On the other hand, biological processes protecting cells from Lipid-ROS must be concomitantly inhibited. GPX4 is the main intracellular factor responsible for Lipid-ROS reduction, using GSH as cofactor (Seiler et al., 2008). Thus, inhibition of GPX4 activity (e.g., through RSL3 administration), or impairment of GSH production through inhibition of the transmembrane glutamate/cystine antiporter “System Xc−”, will result in Lipid-ROS accumulation and ferroptosis (Dixon et al., 2012).
A key component of “System Xc−” is the solute transporter SLC7A11, frequently overexpressed in human malignancies, representing a potential target for ferroptosis-based therapies. In addition, Lipid-ROS can be detoxified by GPX4-independent factors such as FSP1 (Bersuker et al., 2019; Doll et al., 2019), DHODH (Mao et al., 2021), GCHI/BH4 (Kraft et al., 2020), and AKRs (Gagliardi et al., 2019; Gagliardi et al., 2020). The precise molecular mechanism(s) by which membrane-bound Lipid-ROS execute the death process remains unclear; one hypothesis is that they destabilize the plasma membrane structure, dysregulating its permeability (Figure 1).
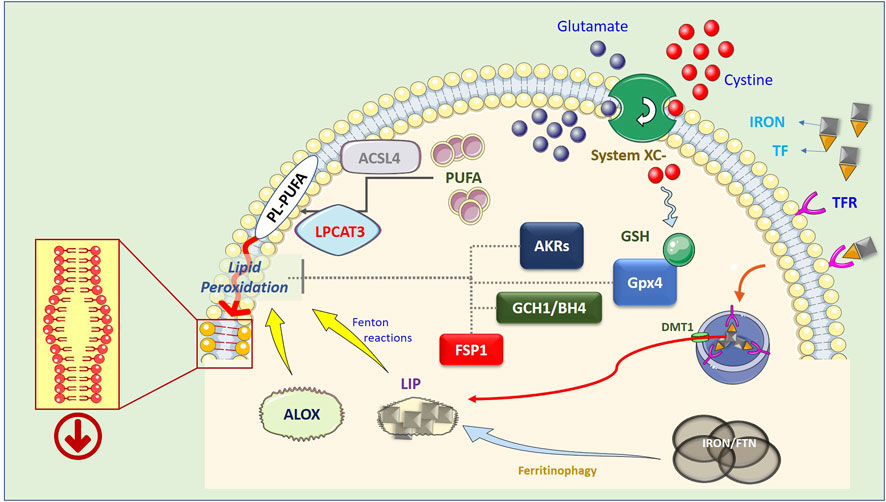
FIGURE 1. Schematic representation of the ferroptotic process. Lipid peroxidation resulting in the generation of Lipid-ROS is considered the point of no return in the execution of ferroptosis. However, the precise mechanism(s) by which these highly reactive molecules execute the cell death process is still not completely clear. The current hypothesis is that peroxidized PL-PUFAs destabilize the membrane thus compromising its barrier functions. PUFA are introduced into cell membranes, as PL-PUFA, through the combined activity of ACSL4 and LPCAT3, while lipid peroxidation is catalyzed by increased available iron (LIP) through Fenton reactions, or by lipoxygenases (ALOX), which use iron as a cofactor. In turn, LIP can be generated by both ferritinophagy, which degrades intracellular and ferritin-based iron stores, or increased uptake of extracellular iron, through the iron/TF interaction with membrane TFRs, endosomal release of iron, and DMT1-mediated relocation in the cytosol. On the other hand, Lipid-ROS can be actively degraded by the GSH-dependent activity of GPX4, by increased expression/activity of AKRs, or can be reduced by the FSP1- or GCH1/BH4- dependent cycles.
P53 behaves primarily as a pro-ferroptotic factor, since it negatively regulates SLC7A11, increasing sensitivity to ferroptosis (Jiang et al., 2015b). P53 also controls the expression of enzymes involved in polyamine, glutamine, and iron metabolism, facilitating cell death by ferroptosis inducers. Importantly, using mouse models, the pro-ferroptotic activity of p53 was elegantly demonstrated to be sufficient for tumor suppression in vivo (Wang et al., 2016). Under certain conditions, however, p53 can also inhibit ferroptosis facilitating ROS detoxification and lipid homeostasis, limiting their pro-oncogenic action (Liu et al., 2020; Liu and Gu, 2022b).
Less is known on the impact of mutant p53 in the ferroptotic process. The consensus is that it increases sensitivity to ferroptosis, since mutp53 efficiently represses SLC7A11 (Gnanapradeepan et al., 2018; Magri et al., 2021). However, there is contradictory evidence. For instance, the drug APR-246 can induce ferroptosis more efficiently in blood cancer cells with mutp53 (Birsen et al., 2021; Fujihara et al., 2022; Hong et al., 2022). Although the pro-ferroptotic action of APR-246 is independent of p53 (Liu et al., 2017; Magri et al., 2021; Fujihara et al., 2022), the drug is a powerful inhibitor of mutant p53 (Hassin and Oren, 2022; Levine, 2022), and this may contribute to its efficacy. Similarly, the quinolinol MMRi62 was shown to induce ferroptosis in pancreatic cancer cells by inducing ferritinophagy (see below), but also by mutp53 destabilization (Li et al., 2022). Thus, we speculate that mutant p53 can modulate the sensitivity of cancer cells to ferroptosis not only directly, e.g., controlling ferroptotic genes, but also indirectly, by facilitating cellular adaptation to cancer-related stress.
Hypoxia is chronic in most tumors, and this condition is often exploited by cancer cells to sustain proliferation, metabolism, tumor invasion, and metastasis (Yang et al., 2020). In this context, a key role is played by HIF1, a transcription factor activated by low oxygen and frequently overexpressed in cancer (Su et al., 2022). Interestingly, HIF1 inhibits ferroptosis by: i) upregulating SCD1 to increase MUFA synthesis; ii) inhibiting the expression of ACSL4 to reduce Lipid-ROS generation, and iii) inhibiting the degradation of SLC7A11 (Su et al., 2022). Therefore, the reduced efficacy of radiation or drug-based therapies in solid tumors has been, at least in part, associated with HIF1-mediated inhibition of ferroptosis (Wang et al., 2019; Su et al., 2022).
p53 is activated by hypoxia, driving a cellular response that also involves modulation of cell metabolism (Liu and Gu, 2022a). In particular, p53 has a complex relationship with HIF1ɑ. The two proteins interact, and both wild-type and mutp53 potentiate the transcriptional activity of HIF1ɑ (Sermeus and Michiels, 2011; Eriksson et al., 2019). Reciprocally, activated HIF1ɑ stimulates p53 expression by binding to its promoter (Madan et al., 2019). Such positive feedback may be relevant for aberrant accumulation of highly stable mutp53 proteins in hypoxic cancer cells. In turn, mutp53 interacts with HIF1ɑ, stabilizing it, and promoting its DNA binding, increasing expression of genes that contribute to hypoxia-induced cell growth and survival (Madan et al., 2019). Mutant p53 can enhance angiogenesis by HIF1/VEGF signaling, and many HIF1-target genes are also targets of NRF2, linking hypoxic response to redox homeostasis (Eriksson et al., 2019). It would be interesting to establish to what extent the interaction of mutp53 with HIF1ɑ contributes to determine the sensitivity to ferroptosis of hypoxic cancer cells. Of note, mutp53/HIF1ɑ complexes drive expression of miR-30d, that reshapes the structure of Golgi apparatus, promoting cancer cells secretory activity. This impacts on the tumor microenvironment, with implications for hormonal and mechanical signaling pathways (Capaci et al., 2020), but also affects ER homeostasis and UPR signaling that may affect ferroptosis (see below).
ROS production is associated with both physiological and pathological conditions. Proper ROS production contributes to differentiation, immunity, and cell signaling, but uncontrolled accumulation leads to damage of proteins, lipids, and nucleic acids, causing “oxidative stress”, involved in cardiovascular and neurodegenerative diseases, obesity, aging, and cancer (Pizzino et al., 2017; Szewczyk-Golec et al., 2018).
Oxidative DNA damage is one of the stimuli driving tumorigenesis (Pizzino et al., 2017), and was detected in cells dying through ferroptosis (Erlanson et al., 2019; Liu J. et al., 2021). Therefore, in addition to being an integral part of the molecular mechanism of ferroptotic death, oxidative stress might regulate the process itself (Liu J. et al., 2021).
p53 is activated by oxidative stress, and can reduce ROS to promote cell survival, or increase ROS to facilitate cell death, depending on its gene targets or binding partners (Eriksson et al., 2019). The cellular response to oxidative stress is mainly regulated by NRF2, a transcription factor that controls expression of several antioxidant proteins (Rojo de la Vega et al., 2018). Notably, depending on cellular context, p53 can increase NRF2 levels by preventing its degradation, or reduce NRF2 levels by repressing its transcription (Eriksson et al., 2019; Liu and Gu, 2022a). Oncogenic mutp53 apparently has opposite effects. For instance, in lung and breast epithelial cells wt p53 suppressed NOX4 reducing ROS levels and cell migration, while mutp53 was shown to stimulate ROS production and metastasis (Boudreau et al., 2014). Mutp53 binds NRF2 on the SLC7A11 promoter, repressing transcription; this renders mutant p53 cells more sensitive to oxidative assaults and prone to ferroptosis (Liu et al., 2017). However, in breast cancer models, mutant p53 cooperates with NRF2 to transcribe proteasome components, alleviating proteotoxic stress and enhancing cell survival and cancer aggressiveness (Walerych et al., 2016; Lisek et al., 2018). Intriguingly, expression of transactivation-defective p53(3K), or ROS generation alone, could not induce ferroptosis, but their combination induced massive ferroptotic cell death (Jiang et al., 2015a; Jiang et al., 2015b); this indicates that p53-dependent ferroptosis may be a crucial tumor-suppressive response to oxidative stress. Similarly, the deacetylase SIRT3 represses p53-mediated ferroptosis in various cancer cells (Jin et al., 2021). SIRT3 expression is altered in several tumors (Chen et al., 2014; Ansari et al., 2017), and may cooperate with p53 mutation to increase cancer cell resistance to ferroptosis upon oxidative stress.
Oxidative stress can also trigger ferroptosis by enhancing peroxidation of membrane lipids. Interestingly, p53 can upregulate iPLA2β, a Ca-independent phospholipase that cleaves oxidized fatty acids, promoting their cytosolic detoxification, and thus limiting ferroptosis. Notably, p53 upregulates iPLA2β only under conditions of moderate lipid damage, facilitating adaptation to oxidative stress (Chen et al., 2021; Liu and Gu, 2022b). Loss of p53 function would cut this modulatory feedback, sensitizing p53-null cancer cells to ROS-induced lipid damage. Cells with oncogenic p53 mutations also lack this adaptive circuit, but may compensate with enhanced activity of NRF2 (see above).
Nutrient deprivation, proteasome dysfunction, sustained secretory activity, and somatic mutations in ER client proteins cause dysregulated proteostasis in proliferating tumor cells, thus triggering activation of the unfolded protein response (UPR) (Corazzari et al., 2017; Chen and Cubillos-Ruiz, 2021). Accumulation of unfolded/misfolded proteins in the ER is sensed by the receptors PERK, IRE1, and ATF6, that trigger activation/upregulation of transcription factors: ATF4, induced by PERK activation, XBP1s, produced by IRE1-dependent cytoplasmic splicing of XBP1 mRNA, and ATF6f, generated by proteolytic cleavage of activated ATF6. These factors orchestrate a transcriptional response aimed to: i) increase ER folding capacity; ii) inhibit cap-dependent translation; iii) degrade misfolded/unfolded ER client proteins (ERAD). Overall these activities sustain cell survival (“adaptation phase” of UPR), but acute or unresolved ER stress stimulates apoptosis (“cell death phase”) (Pagliarini et al., 2015; Corazzari et al., 2017). A potential link between ER stress and ferroptosis has been proposed due to the identification of CHAC1 as a ferroptotic marker (Dixon et al., 2014); indeed CHAC1 is upregulated upon ER stress and contributes to GSH degradation (Galluzzi et al., 2012), thus connecting the two pathways (Dixon et al., 2014). However, we observed that UPR is not required for ferroptosis in metastatic melanoma cells, despite a clear and early upregulation of CHAC1, that could be be abrogated by inhibiting NRF2, suggesting that CHAC1 is under control of both UPR and NRF2 (Gagliardi et al., 2019; Gagliardi et al., 2020). Clearly, further studies are required to unveil the real involvement of ER stress in ferroptosis.
Evidence linking wt p53 to ER stress is scarce, but various observations implicate mutant p53 in protein homeostasis. First, mutp53 cooperates with NRF2 to upregulate proteasome components, thus increasing protein turnover in cancer cells (Walerych et al., 2016; Lisek et al., 2018). This accelerates degradation of tumor-suppressors, promoting cell proliferation; at the same time it can help reduce or resolve ER stress, promoting cell survival. Second, mutp53 enhances expression of ENTPD5, an ER enzyme involved in folding of N-glycosylated proteins (Vogiatzi et al., 2016). This facilitates the maturation and secretion of growth-factor receptors, promoting cell proliferation; it may also alleviate ER stress by enhancing protein folding. Third, mutp53 induces Golgi remodeling and increases protein secretion; this could alter ER protein homeostasis and favor adaptation to ER stress (Capaci et al., 2020). Finally, we found that mutp53 protects cancer cells from drug-induced ER stress by modulating the UPR, in particular by enhancing activation of ATF6 (Sicari et al., 2019). Although the impact of ER stress in ferroptosis remains to be defined, it is conceivable that alterations in p53 function may affect sensitivity to ferroptosis at least in part by modulating protein homeostasis and the UPR.
Autophagy is an evolutionarily-conserved process responsible for lysosomal degradation of intracellular cargoes, sustaining cell survival under nutrient shortage conditions (Corazzari et al., 2013). Autophagy plays a paradoxical role in tumorigenesis, depending on the stage of tumor development; it is suppressive in early stages, mainly through degradation of potentially oncogenic molecules, but becomes oncogenic in advanced stages, promoting cell survival and ameliorating stress in the microenvironment (Galluzzi et al., 2015). Evidence of autophagy has been detected in cancer cells dying by ferroptosis, suggesting a potential connection between the two pathways (Liu L. et al., 2021). Indeed, NCOA4 mediates autophagy-dependent degradation of FTH, thus releasing iron (ferritinophagy) and triggering lipid peroxidation and ferroptosis (Mancias et al., 2014). Recently, other factors linking ferroptosis to specific autophagic processes have been identified, in particular affecting Lipid-ROS generation: for instance RAB7A (lipophagy) (Bai et al., 2019), ARNTL (clockophagy) (Yang et al., 2019), and HSP90/HSC70 (CMA) (Wu et al., 2019). In fact, it has been suggested that ferroptosis may be considered an autophagy-based type of cell death (Zhou et al., 2020), although this concept is still debated.
Wild-type p53 modulates autophagy both directly and indirectly (Maiuri et al., 2010; D’Orazi and Cirone, 2019; Liu and Gu, 2022a). When activated by DNA-damage, nuclear p53 upregulates autophagy-associated genes, contributing to cancer cell death upon chemotherapy (Broz and Attardi, 2013). In contrast, cytoplasmic/mitochondrial p53 can suppress autophagy (Green and Kroemer, 2009). Additionally, p53 controls autophagy via interaction with key metabolic pathways, for instance positively modulating AMPK activity and negatively regulating AKT and mTOR (Mrakovcic and Fröhlich, 2018; Liu and Gu, 2022a).
Tumor-associated p53 mutants cannot transactivate autophagy genes and may acquire a suppressive role in autophagy (Cordani et al., 2017; Shi et al., 2021); especially mutp53 proteins with a pervasive cytoplasmic localization (Morselli et al., 2008). Mutp53 can also bind and inhibit AMPK (Zhou et al., 2014), and promote mTOR activation, indirectly suppressing autophagy (Liu and Gu, 2022a). So, although autophagy can help cancer cells overcome nutrient stress, evidence indicates that mutp53 inhibits autophagy to foster cancer aggressiveness. It is plausible that the p53 status may determine the sensitivity of cancer cells to ferroptosis also by modulating stress-induced autophagy.
Although cancer-related stress originates from a relatively small number of conditions—nutrient imbalance, hypoxia, reactive oxygen or nitrogen compounds, DNA damage, somatic mutations—the multiple pathways involved and the variable conditions that a tumor experiences during its clinical evolution generate an extremely complex scenario. Within this framework, the p53 pathway plays a central role in the response to stress, in particular determining whether cancer cells adapt or succumb to it via regulated cell death—including ferroptosis (Figure 2).
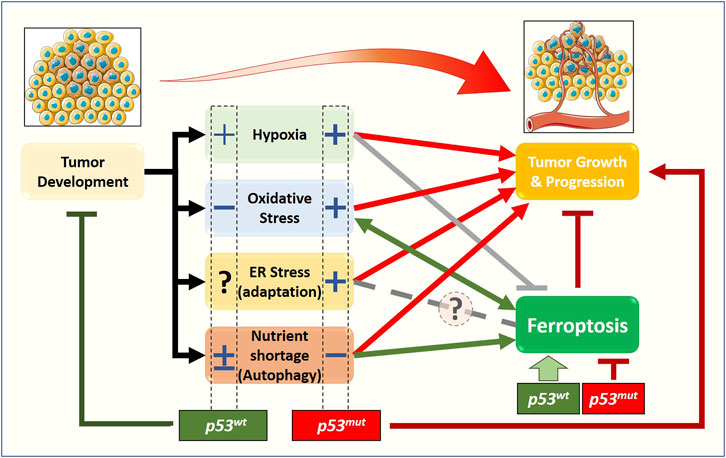
FIGURE 2. Relationship between stress-related signaling pathways, ferroptosis, and tumor growth, from a p53 status-centered (wt or mut) perspective. In the early stages of solid tumor development, cancer cells are subjected to oxygen and nutrient shortage, oxidative stress, and dysfunctional proteostasis. The molecular pathways activated in response to those stimuli will define the fate of the early tumor: survival (red arrows) or death. Beyond apoptosis, very recently, the new form of cell death named ferroptosis has been described to have a role in preventing/limiting the early tumor formation and growth, although the precise molecular mechanisms are still elusive. Emerging data show that stress-related signaling pathways have a non-negligible impact on ferroptosis induction/execution, with some of them stimulating (green arrows) while other preventing or inhibiting (gray lines) the process, thus enhancing or weakening the impact of ferroptotic cell death on tumor growth and progression. The complexity of this scenario is further amplified by the cellular response to stress-induced activation of the p53 tumor suppressor, and by the fact that a large fraction of tumors express gain-of-function p53 mutants. Accumulating data suggest that the p53 status (wt vs. mut) might have a significant impact on ferroptosis and tumor growth through a positive (+) or negative (−) effect on cancer-associated stress-related signaling pathways.
We suggest that mutant p53 can provide a selective advantage to tumors by facilitating adaptation to stress. This effect may not be evident under all conditions, but may become relevant under specific circumstances; for instance, at a given stage during cancer evolution, in response to a certain therapy, or in selected subpopulations of the tumor mass. Currently, there is a lack of experimental studies aimed to test this hypothesis, and we encourage research in this direction. Similarly, it may be important to define the specific stress conditions associated with a given tumor and/or chemotherapeutic drug; a better comprehension of this complexity may help predict the efficacy of treatments, in particular those inducing ferroptosis, in cancers with or without p53 mutation.
Research in the past decades led to development of several drugs that specifically target mutant p53, either by destabilizing the protein to reduce its levels, or by modulating its conformation to restore p53 tumor-suppressive functions (Dolma and Muller, 2022; Hassin and Oren, 2022; Levine, 2022). Such molecules are being tested for clinical use in combination with chemotherapy in p53 mutated cancers, with variable results. Many chemotherapeutic drugs can induce ferroptosis in addition to their primary action (e.g., cisplatin, gemcitabine, sorafenib); in preclinical cancer models their action is increased by co-treatment with ferroptosis inducers, such as drugs that inhibit System Xc−, reduce GSH, inhibit GPX4, or alter intracellular iron levels (Su et al., 2020; Wu et al., 2020; Lei et al., 2022). In this scenario, we hypothesize that targeting mutant p53 may increase the efficacy of pro-ferroptotic drugs under specific stress conditions, thus improving the clinical response of p53 mutated tumors.
Both authors conceptualized the paper and wrote the manuscript. MC conceived and drew the figures.
Research in the LC lab is supported by an AIRC (Italian Association for Cancer Research) Investigator Grant (IG 21803) and by the Italian Ministry of Research (PRIN2017 protocol 20174PLLYN_004). Research in the MC lab is supported by the Italian Ministry of Research (MUR) program “Departments of Excellence 2018-2022”, FOHN Project—Department of Health Sciences, Università del Piemonte Orientale; FAR 2019 (Progetti di Ateneo), and the EU grants “PREMUROSA” (ID#860462) and “ExcellMater” (ID #952033) H2020.
The authors apologize to the many colleagues whose relevant work has not been cited, or has been mentioned only marginally, due to space limitations.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Ansari, A., Rahman, M. S., Saha, S. K., Saikot, F. K., Deep, A., and Kim, K. H. (2017). Function of the SIRT3 mitochondrial deacetylase in cellular physiology, cancer, and neurodegenerative disease. Aging Cell 16 (1), 4–16. doi:10.1111/acel.12538
Bai, Y., Meng, L., Han, L., Jia, Y., Zhao, Y., Gao, H., et al. (2019). Lipid storage and lipophagy regulates ferroptosis. Biochem. biophysical Res. Commun. 508 (4), 997–1003. doi:10.1016/J.BBRC.2018.12.039
Bellazzo, A., Sicari, D., Valentino, E., Del Sal, G., and Collavin, L. (2018). Complexes formed by mutant p53 and their roles in breast cancer. Breast Cancer (Dove Med. Press) 10, 101–112. doi:10.2147/BCTT.S145826
Bersuker, K., Hendricks, J. M., Li, Z., Magtanong, L., Ford, B., Tang, P. H., et al. (2019). The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575 (7784), 688–692. doi:10.1038/s41586-019-1705-2
Birsen, R., Larrue, C., Decroocq, J., Johnson, N., Guiraud, N., Gotanegre, M., et al. (2021). APR-246 induces early cell death by ferroptosis in acute myeloid leukemia. Haematologica 107 (2), 403–416. doi:10.3324/haematol.2020.259531
Boudreau, H. E., Casterline, B. W., Burke, D. J., and Leto, T. L. (2014). Wild-type and mutant p53 differentially regulate NADPH oxidase 4 in TGF-β-mediated migration of human lung and breast epithelial cells. Br. J. Cancer 110 (10), 2569–2582. doi:10.1038/bjc.2014.165
Broz, D. K., and Attardi, L. D. (2013). TRP53 activates a global autophagy program to promote tumor suppression. Autophagy 9 (9), 1440–1442. doi:10.4161/auto.25833
Capaci, V., Bascetta, L., Fantuz, M., Beznoussenko, G. V., Sommaggio, R., Cancila, V., et al. (2020). Mutant p53 induces Golgi tubulo-vesiculation driving a prometastatic secretome. Nat. Commun. 11 (1), 3945. doi:10.1038/s41467-020-17596-5
Chen, D., Chu, B., Yang, X., Liu, Z., Jin, Y., Kon, N., et al. (2021). iPLA2β-mediated lipid detoxification controls p53-driven ferroptosis independent of GPX4. Nat. Commun. 12 (1), 3644. doi:10.1038/s41467-021-23902-6
Chen, X., and Cubillos-Ruiz, J. R. (2021). Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat. Rev. Cancer 21 (2), 71–88. doi:10.1038/s41568-020-00312-2
Chen, Y., Fu, L. L., Wen, X., Wang, X. Y., Liu, J., Cheng, Y., et al. (2014). Sirtuin-3 (SIRT3), a therapeutic target with oncogenic and tumor-suppressive function in cancer. Cell Death Dis. 5 (2), e1047. doi:10.1038/cddis.2014.14
Corazzari, M., Fimia, G. M., Lovat, P., and Piacentini, M. (2013). Why is autophagy important for melanoma? Molecular mechanisms and therapeutic implications. Semin. Cancer Biol. 23 (5), 337–343. doi:10.1016/j.semcancer.2013.07.001
Corazzari, M., Gagliardi, M., Fimia, G. M., and Piacentini, M. (2017). Endoplasmic reticulum stress, unfolded protein response, and cancer cell fate. Front. Oncol. 7 (APR), 78. doi:10.3389/fonc.2017.00078
Cordani, M., Butera, G., Pacchiana, R., and Donadelli, M. (2017). Molecular interplay between mutant p53 proteins and autophagy in cancer cells. Biochimica biophysica acta 1867 (1), 19–28. doi:10.1016/j.bbcan.2016.11.003
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 149 (5), 1060–1072. doi:10.1016/j.cell.2012.03.042
Dixon, S. J., Patel, D. N., Welsch, M., Skouta, R., Lee, E. D., Hayano, M., et al. (2014). Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 3, e02523. doi:10.7554/eLife.02523
Doll, S., Freitas, F. P., Shah, R., Aldrovandi, M., da Silva, M. C., Ingold, I., et al. (2019). FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575 (7784), 693–698. doi:10.1038/s41586-019-1707-0
Dolma, L., and Muller, P. A. J. (2022). GOF mutant p53 in cancers: A therapeutic challenge. Cancers 14 (20), 5091. doi:10.3390/cancers14205091
D’Orazi, G., and Cirone, M. (2019). Mutant p53 and cellular stress pathways: A criminal alliance that promotes cancer progression. Cancers 11 (5), 614. doi:10.3390/cancers11050614
Eriksson, S. E., Ceder, S., Bykov, V. J. N., and Wiman, K. G. (2019). p53 as a hub in cellular redox regulation and therapeutic target in cancer. J. Mol. Cell Biol. 11 (4), 330–341. doi:10.1093/jmcb/mjz005
Erlanson, D. A., Davis, B. J., and Jahnke, W. (2019). Fragment-based drug discovery: Advancing fragments in the absence of crystal structures. Cell Chem. Biol. 26 (1), 9–15. doi:10.1016/J.CHEMBIOL.2018.10.001
Friedmann Angeli, J. P., Krysko, D. V., and Conrad, M. (2019). Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat. Rev. Cancer 19 (7), 405–414. doi:10.1038/S41568-019-0149-1
Fujihara, K. M., Zhang, B. Z., Jackson, T. D., Ogunkola, M. O., Nijagal, B., Milne, J. V., et al. (2022). Eprenetapopt triggers ferroptosis, inhibits NFS1 cysteine desulfurase, and synergizes with serine and glycine dietary restriction. Sci. Adv. 8 (37), eabm9427. doi:10.1126/sciadv.abm9427
Gagliardi, M., Cotella, D., Santoro, C., Cora, D., Barlev, N. A., Piacentini, M., et al. (2019). Aldo-keto reductases protect metastatic melanoma from ER stress-independent ferroptosis. Cell Death Dis. 10 (12), 902. doi:10.1038/s41419-019-2143-7
Gagliardi, M., Saverio, V., Monzani, R., Ferrari, E., Piacentini, M., and Corazzari, M. (2020). Ferroptosis: A new unexpected chance to treat metastatic melanoma? Cell cycleGeorget. Tex.) 19 (19), 2411–2425. doi:10.1080/15384101.2020.1806426
Galluzzi, L., de Santi, M., Crinelli, R., de Marco, C., Zaffaroni, N., Duranti, A., et al. (2012). Induction of endoplasmic reticulum stress response by the indole-3-carbinol cyclic tetrameric derivative CTet in human breast cancer cell lines. PLoS ONE 7 (8), e43249. doi:10.1371/JOURNAL.PONE.0043249
Galluzzi, L., Pietrocola, F., Bravo-San Pedro, J. M., Amaravadi, R. K., Baehrecke, E. H., Cecconi, F., et al. (2015). Autophagy in malignant transformation and cancer progression. EMBO J. 34 (7), 856–880. doi:10.15252/embj.201490784
Gnanapradeepan, K., Basu, S., Barnoud, T., Budina-Kolomets, A., Kung, C.-P., and Murphy, M. E. (2018). The p53 tumor suppressor in the control of metabolism and ferroptosis. Front. Endocrinol. 9, 124. doi:10.3389/fendo.2018.00124
Green, D. R., and Kroemer, G. (2009). Cytoplasmic functions of the tumour suppressor p53. Nature 458 (7242), 1127–1130. doi:10.1038/nature07986
Hassin, O., and Oren, M. (2022). Drugging p53 in cancer: One protein, many targets. Nat. Rev. Drug Discov. 22, 127–144. doi:10.1038/s41573-022-00571-8
Hong, Y., Ren, T., Wang, X., Liu, X., Fei, Y., Meng, S., et al. (2022). APR-246 triggers ferritinophagy and ferroptosis of diffuse large B-cell lymphoma cells with distinct TP53 mutations. Leukemia 36 (9), 2269–2280. doi:10.1038/s41375-022-01634-w
Hou, W., Xie, Y., Song, X., Sun, X., Lotze, M. T., Zeh, H. J., et al. (2016). Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12 (8), 1425–1428. doi:10.1080/15548627.2016.1187366
Jiang, L., Hickman, J. H., Wang, S.-J., and Gu, W. (2015a). Dynamic roles of p53-mediated metabolic activities in ROS-induced stress responses. Cell Cycle 14 (18), 2881–2885. doi:10.1080/15384101.2015.1068479
Jiang, L., Kon, N., Li, T., Wang, S.-J., Su, T., Hibshoosh, H., et al. (2015b). Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520 (7545), 57–62. doi:10.1038/nature14344
Jiang, X., Stockwell, B. R., and Conrad, M. (2021). Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 22 (4), 266–282. doi:10.1038/S41580-020-00324-8
Jin, Y., Gu, W., and Chen, W. (2021). Sirt3 is critical for p53-mediated ferroptosis upon ROS-induced stress. J. Mol. Cell Biol. 13 (2), 151–154. doi:10.1093/jmcb/mjaa074
Kandoth, C., McLellan, M. D., Vandin, F., Ye, K., Niu, B., Lu, C., et al. (2013). Mutational landscape and significance across 12 major cancer types. Nature 502 (7471), 333–339. doi:10.1038/nature12634
Kennedy, M. C., and Lowe, S. W. (2022). Mutant p53: it’s not all one and the same. Cell Death Differ. 29 (5), 983–987. doi:10.1038/s41418-022-00989-y
Kim, M. P., and Lozano, G. (2018). Mutant p53 partners in crime. Cell death Differ. 25, 161–168. doi:10.1038/cdd.2017.185
Kraft, V. A. N., Bezjian, C. T., Pfeiffer, S., Ringelstetter, L., Müller, C., Zandkarimi, F., et al. (2020). GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Central Sci. 6 (1), 41–53. doi:10.1021/acscentsci.9b01063
Lei, G., Zhuang, L., and Gan, B. (2022). Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 22 (7), 381–396. doi:10.1038/S41568-022-00459-0
Levine, A. J. (2020). p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer 20 (8), 471–480. doi:10.1038/s41568-020-0262-1
Levine, A. J. (2022). Targeting the P53 protein for cancer therapies: The translational impact of P53 research. Cancer Res. 82 (3), 362–364. doi:10.1158/0008-5472.can-21-2709
Li, J., Lama, R., Galster, S. L., Inigo, J. R., Wu, J., Chandra, D., et al. (2022). Small molecule MMRi62 induces ferroptosis and inhibits metastasis in pancreatic cancer via degradation of ferritin heavy chain and mutant p53. Mol. Cancer Ther. 21 (4), 535–545. doi:10.1158/1535-7163.mct-21-0728
Limia, C. M., Sauzay, C., Urra, H., Hetz, C., Chevet, E., and Avril, T. (2019). Emerging roles of the endoplasmic reticulum associated unfolded protein response in cancer cell migration and invasion. Cancers (Basel) 11 (5), 631. doi:10.3390/cancers11050631
Lisek, K., Campaner, E., Ciani, Y., Walerych, D., and Del Sal, G. (2018). Mutant p53 tunes the NRF2-dependent antioxidant response to support survival of cancer cells. Oncotarget 9 (29), 20508–20523. doi:10.18632/oncotarget.24974
Liu, D. S., Duong, C. P., Haupt, S., Montgomery, K. G., House, C. M., Azar, W. J., et al. (2017). Inhibiting the system xC(-)/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 8, 14844. doi:10.1038/ncomms14844
Liu, J., Kang, R., and Tang, D. (2021a). Signaling pathways and defense mechanisms of ferroptosis. FEBS J. 289, 7038–7050. doi:10.1111/FEBS.16059
Liu, J., Zhang, C., Wang, J., Hu, W., and Feng, Z. (2020). The regulation of ferroptosis by tumor suppressor p53 and its pathway. Int. J. Mol. Sci. 21 (21), 8387. doi:10.3390/ijms21218387
Liu, L., Li, L., Li, M., and Luo, Z. (2021b). Autophagy-dependent ferroptosis as a therapeutic target in cancer. ChemMedChem 16 (19), 2942–2950. doi:10.1002/CMDC.202100334
Liu, Y., and Gu, W. (2022b). p53 in ferroptosis regulation: the new weapon for the old guardian. Cell Death Differ. 29 (5), 895–910. doi:10.1038/s41418-022-00943-y
Liu, Y., and Gu, W. (2022a). The complexity of p53-mediated metabolic regulation in tumor suppression. Seminars Cancer Biol. 85, 4–32. doi:10.1016/j.semcancer.2021.03.010
Madan, E., Parker, T. M., Pelham, C. J., Palma, A. M., Peixoto, M. L., Nagane, M., et al. (2019). HIF-transcribed p53 chaperones HIF-1α. Nucleic Acids Res. 47 (19), 10212–10234. doi:10.1093/nar/gkz766
Magri, J., Gasparetto, A., Conti, L., Calautti, E., Cossu, C., Ruiu, R., et al. (2021). Tumor-associated antigen xCT and mutant-p53 as molecular targets for new combinatorial antitumor strategies. Cells 10 (1), 108. doi:10.3390/cells10010108
Maiuri, M. C., Galluzzi, L., Morselli, E., Kepp, O., Malik, S. A., and Kroemer, G. (2010). Autophagy regulation by p53. Curr. Opin. Cell Biol. 22 (2), 181–185. doi:10.1016/j.ceb.2009.12.001
Mancias, J. D., Wang, X., Gygi, S. P., Harper, J. W., and Kimmelman, A. C. (2014). Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509 (7498), 105–109. doi:10.1038/NATURE13148
Mantovani, F., Collavin, L., and Del Sal, G. (2019). Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 26 (2), 199–212. doi:10.1038/s41418-018-0246-9
Mao, C., Liu, X., Zhang, Y., Lei, G., Yan, Y., Lee, H., et al. (2021). DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 593:(7860), 586–590. doi:10.1038/s41586-021-03539-7
Morselli, E., Tasdemir, E., Maiuri, M. C., Galluzzi, L., Kepp, O., Criollo, A., et al. (2008). Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell cycleGeorget. Tex) 7 (19), 3056–3061. PMID - 18818522 7. doi:10.4161/cc.7.19.6751
Mrakovcic, M., and Fröhlich, L. F. (2018). p53-Mediated molecular control of autophagy in tumor cells. Biomolecules 8 (2), 14. doi:10.3390/biom8020014
Pagliarini, V., Giglio, P., Bernardoni, P., De Zio, D., Fimia, G. M., Piacentini, M., et al. (2015). Downregulation of E2F1 during ER stress is required to induce apoptosis. J. Cell Sci. 128 (6), 1166–1179. doi:10.1242/jcs.164103
Pilley, S., Rodriguez, T. A., and Vousden, K. H. (2021). Mutant p53 in cell-cell interactions. Genes and Dev. 35 (7-8), 433–448. doi:10.1101/gad.347542.120
Pizzino, G., Irrera, N., Cucinotta, M., Pallio, G., Mannino, F., Arcoraci, V., et al. (2017). Oxidative stress: Harms and benefits for human Health. Oxidative Med. Cell. Longev. 2017, 8416763. doi:10.1155/2017/8416763
Rodriguez, R., Schreiber, S. L., and Conrad, M. (2022). Persister cancer cells: Iron addiction and vulnerability to ferroptosis. Mol. Cell 82 (4), 728–740. doi:10.1016/J.MOLCEL.2021.12.001
Rojo de la Vega, M., Chapman, E., and Zhang, D. D. (2018). NRF2 and the hallmarks of cancer. Cancer Cell 34 (1), 21–43. doi:10.1016/j.ccell.2018.03.022
Seiler, A., Schneider, M., Förster, H., Roth, S., Wirth, E. K., Culmsee, C., et al. (2008). Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 8 (3), 237–248. doi:10.1016/J.CMET.2008.07.005
Senft, D., and Ronai, Z. E. (2016). Adaptive stress responses during tumor metastasis and dormancy. Trends Cancer 2 (8), 429–442. doi:10.1016/j.trecan.2016.06.004
Sermeus, A., and Michiels, C. (2011). Reciprocal influence of the p53 and the hypoxic pathways. Cell Death Dis. 2 (5), e164. doi:10.1038/cddis.2011.48
Shah, R., Shchepinov, M. S., and Pratt, D. A. (2018). Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Central Sci. 4 (3), 387–396. doi:10.1021/acscentsci.7b00589
Shi, Y., Norberg, E., and Vakifahmetoglu-Norberg, H. (2021). Mutant p53 as a regulator and target of autophagy. Front. Oncol. 10, 607149. doi:10.3389/fonc.2020.607149
Sicari, D., Fantuz, M., Bellazzo, A., Valentino, E., Apollonio, M., Pontisso, I., et al. (2019). Mutant p53 improves cancer cells’ resistance to endoplasmic reticulum stress by sustaining activation of the UPR regulator ATF6. Oncogene 38 (34), 6184–6195. doi:10.1038/s41388-019-0878-3
Stockwell, B. R. (2022). Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 185 (14), 2401–2421. doi:10.1016/J.CELL.2022.06.003
Su, X., Xie, Y., Zhang, J., Li, M., Zhang, Q., Jin, G., et al. (2022). HIF-α activation by the prolyl hydroxylase inhibitor roxadustat suppresses chemoresistant glioblastoma growth by inducing ferroptosis. Cell death Dis. 13 (10), 861. doi:10.1038/S41419-022-05304-8
Su, Y., Zhao, B., Zhou, L., Zhang, Z., Shen, Y., Lv, H., et al. (2020). Ferroptosis, a novel pharmacological mechanism of anti-cancer drugs. Cancer Lett. 483, 127–136. doi:10.1016/j.canlet.2020.02.015
Szewczyk-Golec, K., Czuczejko, J., Tylzanowski, P., and Lecka, J. (2018). Strategies for modulating oxidative stress under diverse physiological and pathological conditions. Oxidative Med. Cell. Longev. 2018, 3987941. doi:10.1155/2018/3987941
Vogiatzi, F., Brandt, D. T., Schneikert, J., Fuchs, J., Grikscheit, K., Wanzel, M., et al. (2016). Mutant p53 promotes tumor progression and metastasis by the endoplasmic reticulum UDPase ENTPD5. Proc. Natl. Acad. Sci. 113 (52), E8433–E8442. doi:10.1073/pnas.1612711114
Walerych, D., Lisek, K., Sommaggio, R., Piazza, S., Ciani, Y., Dalla, E., et al. (2016). Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nat. Cell Biol. 18 (8), 897–909. doi:10.1038/ncb3380
Wang, H., Jiang, H., Van De Gucht, M., and De Ridder, M. (2019). Hypoxic radioresistance: Can ROS Be the key to overcome it? Cancers 11 (1), 112. doi:10.3390/CANCERS11010112
Wang, S. J., Li, D., Ou, Y., Jiang, L., Chen, Y., Zhao, Y., et al. (2016). Acetylation is crucial for p53-mediated ferroptosis and tumor suppression. Cell Rep. 17 (2), 366–373. doi:10.1016/j.celrep.2016.09.022
Wang, Z., Strasser, A., and Kelly, G. L. (2022). Should mutant TP53 be targeted for cancer therapy? Cell Death Differ. 29 (5), 911–920. doi:10.1038/s41418-022-00962-9
Wu, Y., Yu, C., Luo, M., Cen, C., Qiu, J., Zhang, S., et al. (2020). Ferroptosis in cancer treatment: Another way to rome. Front. Oncol. 10, 571127. doi:10.3389/fonc.2020.571127
Wu, Z., Geng, Y., Lu, X., Shi, Y., Wu, G., Zhang, M., et al. (2019). Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc. Natl. Acad. Sci. U. S. A. 116 (8), 2996–3005. doi:10.1073/PNAS.1819728116
Yang, G., Shi, R., and Zhang, Q. (2020). Hypoxia and oxygen-sensing signaling in gene regulation and cancer progression. Int. J. Mol. Sci. 21 (21), 8162–8224. doi:10.3390/IJMS21218162
Yang, M., Chen, P., Liu, J., Zhu, S., Kroemer, G., Klionsky, D. J., et al. (2019). Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci. Adv. 5 (7), eaaw2238. doi:10.1126/SCIADV.AAW2238
Yang, W. S., and Stockwell, B. R. (2016). Ferroptosis: Death by lipid peroxidation. Trends Cell Biol. 26 (3), 165–176. doi:10.1016/j.tcb.2015.10.014
Zhou, B., Liu, J., Kang, R., Klionsky, D. J., Kroemer, G., and Tang, D. (2020). Ferroptosis is a type of autophagy-dependent cell death. Seminars cancer Biol. 66, 89–100. doi:10.1016/J.SEMCANCER.2019.03.002
Zhou, G., Wang, J., Zhao, M., Xie, T.-X., Tanaka, N., Sano, D., et al. (2014). Gain-of-Function mutant p53 promotes cell growth and cancer cell metabolism via inhibition of AMPK activation. Mol. Cell 54, 960–974. doi:10.1016/j.molcel.2014.04.024
Zou, Y., Li, H., Graham, E. T., Deik, A. A., Eaton, J. K., Wang, W., et al. (2020). Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat. Chem. Biol. 16, (3), 302–309. doi:10.1038/s41589-020-0472-6
ACSL4 acyl-CoA synthetase long-chain family member 4
AKRs aldo-keto reductases
ALOXs arachidonate lipoxygenases
ARNTL aryl hydrocarbon receptor nuclear translocator-like
ATF6 Activating Transcription Factor 6
ATF4 Activating Transcription Factor 4
CHAC1 ChaC Glutathione Specific Gamma-Glutamylcyclotransferase 1
CMA chaperone-mediated autophagy
DHODH dihydroorotate dehydrogenase
ER endoplasmic reticulum
ERAD endoplasmic-reticulum-associated protein degradation
FTH ferritin heavy chain
FSP1 Ferroptosis suppressor protein 1, or AIFM2 or AMID
GCHI/BH4 GTP cyclohydrolase 1/tetrahydrobiopterin
GPX4 Glutathione peroxidase 4
GSH Glutathione
HIF1 Hypoxia Inducible Factor 1
Hsp90 heat shock protein 90
IRE1 Inositol-Requiring Enzyme 1
Lipid-ROS lipid peroxides
MUFA monounsaturated fatty acids
NCOA4 nuclear receptor coactivator 4
NOX4 NADPH oxidase 4
NRF2 nuclear factor erythroid 2-related factor 2
PERK PKR-Like ER Kinase
PUFA Polyunsaturated fatty acids
PL-PUFA PUFA-containing membrane-bound phospholipids
POR cytochrome P450 oxidoreductase
RAB7A member RAS oncogene family
RSL3 RAS-selective-lethal-3
ROS reactive oxygen species
System Xc- cystine/glutamate antiporter system
SLC7A11 solute carrier family 7 member 11
SCD1 stearoyl-CoA desaturase 1
SIRT3 Sirtuin 3
TFR Transferrin Receptor
UPR unfolded protein response
XBP1 X-Box Binding Protein 1
Keywords: hypoxia, autophagy, UPR, unfolded protein response, p53 tumor suppressor, ER stress, ferroptosis, stress response pathways
Citation: Corazzari M and Collavin L (2023) Wild-type and mutant p53 in cancer-related ferroptosis. A matter of stress management?. Front. Genet. 14:1148192. doi: 10.3389/fgene.2023.1148192
Received: 19 January 2023; Accepted: 09 March 2023;
Published: 20 March 2023.
Edited by:
Yanqing Liu, Columbia University, United StatesReviewed by:
Jialin Shang, Cornell University, United StatesCopyright © 2023 Corazzari and Collavin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marco Corazzari, bWFyY28uY29yYXp6YXJpQHVuaXVwby5pdA==; Licio Collavin, bGNvbGxhdmluQHVuaXRzLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.