
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet., 22 March 2023
Sec. Genetics of Common and Rare Diseases
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1140406
 Qinqin Xiang1,2,3†
Qinqin Xiang1,2,3† Jing Chen1,2,3†Xiao Xiao1,2,3Bocheng Xu1,2,3Hanbing Xie1,2,3He Wang1,2,3
Jing Chen1,2,3†Xiao Xiao1,2,3Bocheng Xu1,2,3Hanbing Xie1,2,3He Wang1,2,3 Mei Yang1,2,3*†
Mei Yang1,2,3*† Shanling Liu1,2,3*†
Shanling Liu1,2,3*†Background: Lymphedema is a local form of tissue swelling, which is caused by excessive retention of lymph fluid in interstitial compartment caused by impaired lymphatic drainage damage. Primary lymphedema is caused by developmental lymphatic vascular abnormalities. Most cases are inherited as autosomal dominant, with incomplete penetrance and variable expression. Here we report compound heterozygotes variants in FLT4 of a Chinese family associated with primary lymphedema display autosomal recessive inheritance.
Case presentation: Trio-whole-exome sequencing (Trio-WES) was performanced to analyse the underlying genetic cause of a proband with primary lymphedema in a Chinese family. Sanger sequencing was used to validate the variants in proband with primary lymphedema and members of the family with no clinical signs and symptoms. We reported compound heterozygotes for the Fms Related Receptor Tyrosine Kinase 4 (FLT4) gene detected in the proband, who carrying two different point variants. One was a missense variant (NM_182925.5; c.1504G>A, p.Glu502Lys), and the other was a recurrent variant (NM_182925.5; c.3323_3325del, p.Phe1108del). The missense variant c.1504G>A was detected in the proband, unaffected father, and unaffected paternal grandmother but not detected in unaffected paternal grandfather. The recurrent variant c.3323_3325del was detected in the proband, unaffected mother, and unaffected maternal grandfather but not detected in unaffected maternal grandmother. Our results suggests the possibility of an autosomal recessive inherited form of primary lymphedema resulting from variants of FLT4 encoding the vascular endothelial growth factor receptor-3.
Conclusion: The results of the present study identifed compound heterozygotes FLT4 variants in a family with primary lymphedema which provides more information for autosomal recessive primary lymphedema caused by FLT4.
Lymphedema is a local form of tissue swelling, which is caused by excessive retention of lymph fluid in interstitial compartment caused by impaired lymphatic drainage damage (Brouillard et al., 2014; Grada and Phillips, 2017). Lymphedema is classified as primary or secondary. Primary lymphedema is caused by developmental lymphatic vascular abnormalities. Secondary lymphedema is acquired and caused by disease processes, recurrent infection, trauma or surgery (Kerchner et al., 2008). Primary lymphedema is rare. Due to the difficulty of diagnosis, its epidemiology is not accurate, but the incidence of primary lymphedema in children is predicted to be 1 in 100,000 (Smeltzer et al., 1985; Fitzpatrick, 2008).
Primary lymphedema is caused by anatomic or functional defects in the lymphatic system, resulting in chronic swelling of body parts, and can be an isolated disease or part of a complex syndrome. Most cases are inherited as autosomal dominant, with incomplete penetrance and variable expression. Gene variants can be identified in nearly 30% of patients with primary lymphedema. Primary lymphedema shows a high degree of genetic heterogeneity, more than 20 genes are related to primary lymphedema (Brouillard et al., 2014; Grada and Phillips, 2017). Isolated lymphedema is mainly associated with Fms Related Receptor Tyrosine Kinase 4 (FLT4) (also known as vascular endothelial growth factor receptor 3, VEGFR3) and vascular endothelial growth factors C (VEGFC) (Brouillard et al., 2014).
In this study, genetic analysis of a Chinese family with autosomal recessive primary lymphedema was performed. The Trio-WES analysis revealed that the proband had compound heterozygotes variants for the FLT4 gene (OMIM:136352; c.1504G>A, p.Glu502Lys and c.3323_3325del, p.Phe1108del).
A Chinese family with primary lymphedema was investigated in this study. The proband was a male infant who present abnormal ultrasound with bilateral lower legs and feet subcutaneous thickening in fetal period. After birth, both lower legs and feet were obviously swollen and show pitting edema (lower leg circumference:left lateral 14.5 cm, right lateral 15 cm; foot circumference:left lateral 12.8 cm, right lateral 14 cm), and ultrasound targeted swelling location indicated that the subcutaneous layer were thickened. The clinical diagnosis was suspicious primary lymphedema (Figure 1; Supplementary Figure S1). The proband’s mother has an adverse pregnancy history, ultrasound in the second trimester showed bilateral lower legs and feet subcutaneous thickening, so the pregnancy was terminated, however, no genetic test was performed with any samples of fetus and no samples were retained. In order to further clarify the diagnosis, Trio-whole-exome sequencing (Trio-WES) was performed to analyse the underlying genetic cause of the family. Suspected variants detected by next-generation sequencing (NGS) were validated by Sanger sequencing.
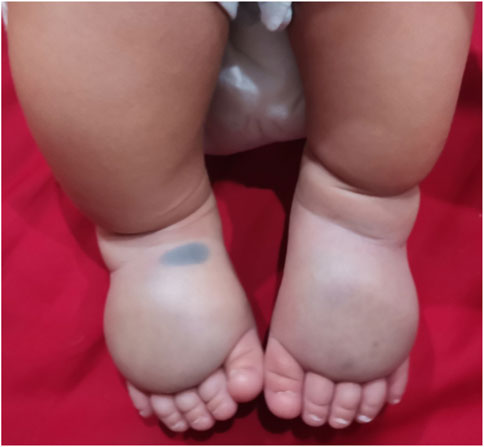
FIGURE 1. Congenital lymphedema of the lower extremities of the proband.
This study was approved by the Medical Ethics Committee of West China Second University Hospital, Sichuan University, and written informed consent was obtained from all participants.
Total genomic DNA was extracted from the whole blood of the proband, parents, and grandparents using a DNeasy Blood and Tissue DNA kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. For detecting the variation carried by the proband, the Nano WES Human Exome V2 (Berry Genomics) was used to capture the sequences. Then the enriched library was sequenced on the Novaseq 6,000 with 150 paired-end reads. The reads were mapped to the human reference genome (hg38) with BWA (v0.7.17). Variant calling was performed by Verita Trekker (v2.1.1). The 1,000 Genomes Project database, and the Genome Aggregation Database (gnomAD, http://gnomad.broadinstitute.org/) were used for minor allele frequency (MAF). For the pathogenicity prediction, CADD (https://cadd.gs.washington.edu), DANN (https://cbcl.ics.uci.edu/public_data/DANN/), dbNSFP (https://sites.google.com/site/jpopgen/dbNSFP), SIFT (http://sift.jcvi.org), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2), M-CAP (http://bejerano.stanford.edu/mcap/), Mutation Taster (http://www.mutationtaster.org) were used. To select disease-causing variants, we referred to the information from the OMIM (http://www.omim.org), ClinVar (http://www.ncbi.nlm.nih.gov/clinvar) and Human Gene Mutation Database (http://www.hgmd.org). The detailed process for identifying candidate variants is shown in Supplementary Figure S2 and the detailed information of quality control of the proband and parentsis shown in supplemental excel sheet. The 3D structure of the wild-type and mutant FLT4 homology domains was constructed with SWISS-MODEL (https://swissmodel.expasy.org/).
Sanger sequencing was performed using specific PCR primers designed with Primer Premier 6. The sequences of FLT4 primers used were FLT4-1-F: 5′-AACCACCTGCTTCAGAAC-3′ and FLT4-1-R: 5′-AGACAGACCCAGGAGAAC-3’; FLT4-2-F: 5′-CATGTCAGCTTCCTTGTCT-3′ and FLT4-2-R: 5′-CTTGCCTCTTCTGGTCCT-3’. The PCR products were separated and verified by electrophoresis in an 2% agarose gel and characterized by direct Sanger sequencing.
Compound heterozygotes for the FLT4 gene was detected, the proband carrying two different variants. One was a missense variant (NM_182925.5; c.1504G>A, p.Glu502Lys) derived from his father (II1), and the other was a recurrent variant (NM_182925.5, c.3323_3325del, p.Phe1108del) derived from his mother (II2).
The presence of the variants were further validated by Sanger sequencing in proband, parents, and grandparents. The variant c.1504G>A was detected in the proband (III2), unaffected father (II1), and unaffected paternal grandmother (I2) but not detected in unaffected paternal grandfather (I1) (Figures 2, 3; Supplementary Figure S3). The recurrent variant c.3323_3325del was detected in the proband (III2), unaffected mother (II2), and unaffected maternal grandfather (I3) but not detected in unaffected maternal grandmother (I4) (Figures 2, 3; Supplementary Figure S3). These results further support that, in addition to dominantly inherited primary lymphedema, certain FLT4 variants may lead to recessive primary lymphedema.
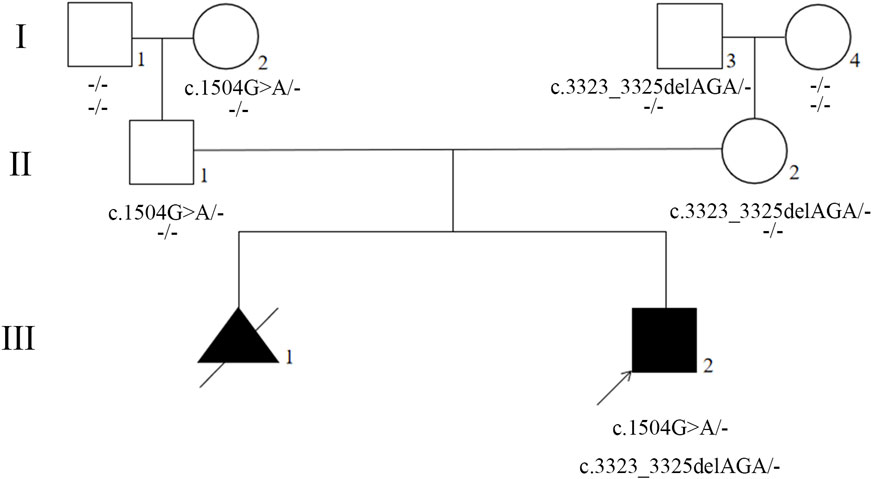
FIGURE 2. Pedigree of the proband’s family. Generations are shown as I–III. Squares indicate male, and circles indicate female. Empty symbols indicate unaffected individuals and filled symbols indicate affected individuals. Deceased individuals are indicated by a slash (/), the arrow shows the proband.
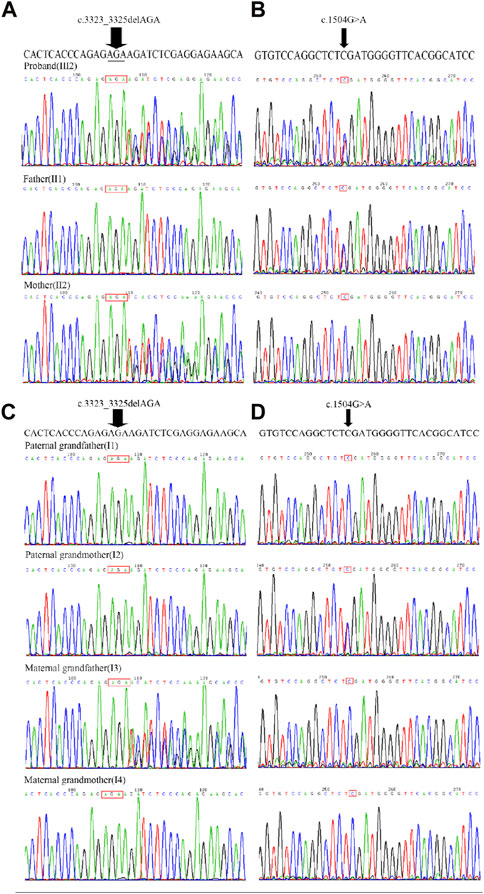
FIGURE 3. Sanger sequencing chromatograms of FLT4, c.3323_3325del and c.1504G>A. (A) The recurrent variant c.3323_3325del (indicated by an arrow) was detected in the proband (III2), unaffected mother (II2), but not detected in unaffected father (II1). (B) The missense variant c.1504G>A (indicated by an arrow) was detected in the proband (III2), unaffected father (II1) but not detected in unaffected mother (II2). (C) The recurrent variant c.3323_3325del (indicated by an arrow) was detected in the unaffected maternal grandfather (I3) but not detected in unaffected maternal grandmother (I4), unaffected paternal grandmother (I2) and unaffected paternal grandfather (I1). (D) The missense variant c.1504G>A was detected in the unaffected paternal grandmother (I2), but not detected in unaffected paternal grandfather (I1), unaffected maternal grandmother (I4)and unaffected maternal grandfather (I3).
According to the ACMG Guidelines, neither the proband nor his parents had other pathogenic or likely pathogenic variants about second findings.
The variant c.1504G>A in FLT4 results in replacement of a glutamic acid (acidic amino acid) by lysine (basic amino acid) at position 502. In silico analysis of the c.1504G>A reveals this substitution may be disease causing, and predicted deleterious by CADD, SIFT, DANN, MetaSVM, MetaLR, and M-CAP. In addition, multiple sequence alignment of the FLT4 from different species showed the evolutionary conservation of the glutamic acid at position 502 (Figure 4A). The 3D model of wild-type and mutant homologous domains constructed by SWISS-MODEL shows that replacing glutamic acid with lysine at position 502 will not cause a change in the structure, but will cause a change in polarity (Figure 4A).
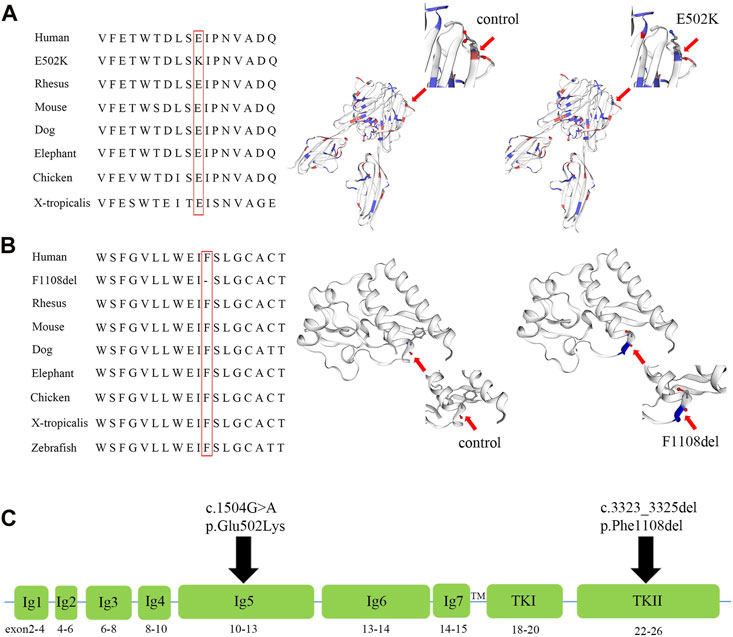
FIGURE 4. Amino acid alignment and prediction of the protein conformational changes of p.F1108del and p.E502K of the FLT4 gene, and the diagram of the FLT4 protein. (A) Multiple sequence alignment of the FLT4 homeodomain from different species of vertebrates and X-tropicalis showing the evolutionary conservation of the glutamic residue at position 502 (highlighted in red box), and models of the wild-type and mutant E502K homeodomains of FLT4 were constructed with SWISS-MODEL. (B) Multiple sequence alignment of the FLT4 homeodomain from different species of vertebrates,X-tropicalis and Zebrafish showing the evolutionary conservation of the phenylalanine acid at position 1,108 (highlighted in red box), and models of the wild-type and mutant F1108del homeodomains of FLT4 were constructed with SWISS-MODEL. (C) Diagram of the FLT4 protein showing the functional domains (Ig1 to Ig7 domains and TKI, TKII), and the location of p. E502K and p.F1108del.
The variant c.3323_3325del in FLT4 results in deletion of a phenylalanine at position 1,108. MutationTaster predicted that c.3323_3325del in FLT4 gene was a disease-causing variant. A change in the same codon that results in p. Phe1108del had been previously reported in patients with primary congenital lymphedema. In addition, multiple sequence alignment of the FLT4 from different species showed the evolutionary conservation of the glutamic acid at position 1,108 (Figure 4B). The 3D model of wild-type and mutant homologous domains constructed by SWISS-MODEL shows that deletion of a phenylalanine at position 1,108 will cause structure change with destruction of α-helix (Figure 4B).
Here we describe the compound heterozygous variants in the FLT4 gene (NM_182925.5; c.1504G>A, p.Glu502Lys and c.3323_3325del, p.Phe1108del). Our family is the fourth reported to display autosomal recessive inheritance associated with primary lymphedema type I or lymphatic malformation 1 (also known as Milroy disease, OMIM:153100) (Ghalamkarpour et al., 2009; Melikhan-Revzin et al., 2015; Kim and Lim, 2022). Lymphatic malformation 1 is caused by anatomical or functional defects of the lymphatic system, leading to chronic swelling of parts of the body (Gordon et al., 2013a; Balboa-Beltran et al., 2014). The onset is usually at birth or early childhood, but it can also occur later (Gordon et al., 2013a; Balboa-Beltran et al., 2014). Lymphatic malformation 1 is generally due to autosomal dominant variants in the FLT4 gene, but usually with variable expression and severity (Ferrell et al., 1998; Brouillard et al., 2014; Grada and Phillips, 2017). In previous studies, the autosomal dominant mode of inheritance did not account for all observed familial correlations, suggesting that shared environmental or additional genetic factors may also be important in explaining the observed familial aggregation (Holberg et al., 2001). Three cases of recessive inheritance, caused by variants in the FLT4 gene, was reported associated with primary lymphedema (Ghalamkarpour et al., 2009; Melikhan-Revzin et al., 2015; Kim and Lim, 2022). In this case, the parents and grandparents with only one variant were phenotypically normal, while the proband with two variants presented with lymphedema. Although there were no samples of fetuses induced by subcutaneous thickening, this adverse pregnancy history also suggests the presence of a family history and suggests the possibility of autosomal recessive inheritance in this family. In general, our research provides more information for autosomal recessive primary lymphedema caused by FLT4.
The FLT4 gene encodes VEGFR3, which regulates the development and maintenance of lymphatic system (Monaghan et al., 2021). The VEGFR3 acts as a cell-surface tyrosine kinase receptor for vascular endothelial growth factors C and D (VEGFC and VEGFD) (Iljin et al., 2001; Leppanen et al., 2011; Gordon et al., 2013b). When VEGFC and VEGFD bind to VEGFR3, downstream signaling docking sites are produced, regulating the proliferation, migration and survival of lymphatic endothelial cells (LEC) (Lohela et al., 2003). The Chy mouse possesses a heterozygous FLT4 variant in the tyrosine kinase domain, preventing phosphorylation and resulting in early developmental deficiencies in some lymphatic vessels (Rutkowski et al., 2010). The FLT4 gene contains 30 exons and encodes seven Ig-homology domains (I-VII), a transmembrane region (TM), and tyrosine kinase domains (TK I and TK II) (Iljin et al., 2001) (Figure 4C). One hundred and forty-three FLT4 variants have been reported so far worldwide. Most of the variants associated with primary lymphedema were missense variants or single amino acid deletions located in the TK domain of the protein (Evans et al., 2003; Connell et al., 2009; Gordon et al., 2013b; Mendola et al., 2013), while non-sense or frameshift variants in FLT4 were mainly related to congenital heart defects characterized mainly by tetralogy of Fallot (Jin et al., 2017; Page et al., 2019; Reuter et al., 2019).
The recurrent variant (c.3323_3325del, p. Phe1108del) was identified to be located in the tyrosine kinase II domain (TK II) of the FLT4 gene (Figure 4C). Evans et al. (2003) identified the same heterozygous variant of FLT4 in a family with an autosomal dominant form of primary congenital lymphedema, the affected members of this family were variable degrees of painless pitting lower limb edema of congenital onset. In this study, the unaffected members, mother and maternal grandfather, also carried the variant p.Phe1108del, possibly reflecting some residual receptor activity, and suggesting that environmental or other genetic factors may play an important role in the pathogenicity of this variant. In this family, the proband carried another missense variant (c.1504G>A, p.Glu502Lys), which was identified to be located in the Ig-homology V domain (Figure 4C). To date, the variants located in the Ig-homology domains were mainly related to tetralogy of Fallot (Jin et al., 2017; Page et al., 2019), and no disease causing variants in this domain have been reported to be related to primary lymphedema. Combining the in silico analysis and the history of this family, we speculate that p.Glu502Lys may be the first reported variant in the Ig-homology domain of VEGFR3 protein that causes primary lymphedema with an autosomal recessive inheritance mode. However, the pathogenicity of these two missense variants needs further functional studies to verify.
Previous molecular analysis of primary lymphedema or Milroy disease was mainly through PCR and direct sequencing of FLT4 gene encoding the “tyrosine-kinase domain,” or all exons of FLT4 gene, or screening of a few lymphedema-related genes, which cannot exclude other genetic variants that may explain the clinical presentation of the patients (Kitsiou-Tzeli et al., 2010; Michelini et al., 2012; Michelini et al., 2016). At present, Trio-WES based on next-generation sequencing technology is one of the most effective tools for diagnosis of genetic diseases, which can detect exons of all known gene. Therefore, our results provide accurate genetic information for targeted treatment, genetic counseling and subsequent prenatal diagnosis of the patients in this family.
In conclusion, we successfully identified compound heterozygous variants for the FLT4 gene (OMIM:136352; c.1504G>A, p.Glu502Lys and c.3323_3325del, p.Phe1108del) in this family. The missense variant p.Glu502Lys is the first reported disease-causing variant in the Ig-homology domain of VEGFR3 protein that causes primary lymphedema. Furthermore, our study provides more information for autosomal recessive primary lymphedema caused by FLT4.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by West China Second University Hospital, Sichuan University, Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
MY, SL, and HW designed the study. XX, BX, and HX performed the experiments. QX, JC, and MY conducted data analysis. QX and JC wrote the article. All authors read and approved the final manuscript.
This work was supported by the National Key Research and Development Program of China (2021YFC1005303) and the Science and Technology Department of Sichuan Province, China (2021YFS0078). The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1140406/full#supplementary-material
FLT4, Fms Related Receptor Tyrosine Kinase 4; VEGFR3, vascular endothelial growth factor receptor 3; VEGFC, vascular endothelial growth factors C; Trio-WES, Trio-whole-exome sequencing; NGS, next-generation sequencing; VEGFD, vascular endothelial growth factors D; LEC, lymphatic endothelial cells; TM, transmembrane region; TK, tyrosine kinase.
Balboa-Beltran, E., Fernandez-Seara, M. J., Perez-Munuzuri, A., Lago, R., Garcia-Magan, C., Couce, M. L., et al. (2014). A novel stop mutation in the vascular endothelial growth factor-C gene (VEGFC) results in Milroy-like disease. J. Med. Genet. 51 (7), 475–478. doi:10.1136/jmedgenet-2013-102020
Brouillard, P., Boon, L., and Vikkula, M. (2014). Genetics of lymphatic anomalies. J. Clin. Invest. 124 (3), 898–904. doi:10.1172/JCI71614
Connell, F. C., Ostergaard, P., Carver, C., Brice, G., Williams, N., Mansour, S., et al. (2009). Analysis of the coding regions of VEGFR3 and VEGFC in Milroy disease and other primary lymphoedemas. Hum. Genet. 124 (6), 625–631. doi:10.1007/s00439-008-0586-5
Evans, A. L., Bell, R., Brice, G., Comeglio, P., Lipede, C., Jeffery, S., et al. (2003). Identification of eight novel VEGFR-3 mutations in families with primary congenital lymphoedema. J. Med. Genet. 40 (9), 697–703. doi:10.1136/jmg.40.9.697
Ferrell, R. E., Levinson, K. L., Esman, J. H., Kimak, M. A., Lawrence, E. C., Barmada, M. M., et al. (1998). Hereditary lymphedema: Evidence for linkage and genetic heterogeneity. Hum. Mol. Genet. 7 (13), 2073–2078. doi:10.1093/hmg/7.13.2073
Fitzpatrick, T. B. (2008). Fitzpatrick’s dermatology in general medicine. 7th ed. New York: McGraw-Hill Medical.
Ghalamkarpour, A., Holnthoner, W., Saharinen, P., Boon, L. M., Mulliken, J. B., Alitalo, K., et al. (2009). Recessive primary congenital lymphoedema caused by a VEGFR3 mutation. J. Med. Genet. 46 (6), 399–404. doi:10.1136/jmg.2008.064469
Gordon, K., Schulte, D., Brice, G., Simpson, M. A., Roukens, M. G., van Impel, A., et al. (2013a). Mutation in vascular endothelial growth factor-C, a ligand for vascular endothelial growth factor receptor-3, is associated with autosomal dominant milroy-like primary lymphedema. Circ. Res. 112 (6), 956–960. doi:10.1161/CIRCRESAHA.113.300350
Gordon, K., Spiden, S. L., Connell, F. C., Brice, G., Cottrell, S., Short, J., et al. (2013b). FLT4/VEGFR3 and milroy disease: Novel mutations, a review of published variants and database update. Hum. Mutat. 34 (1), 23–31. doi:10.1002/humu.22223
Grada, A. A., and Phillips, T. J. (2017). Lymphedema: Pathophysiology and clinical manifestations. J. Am. Acad. Dermatol 77 (6), 1009–1020. doi:10.1016/j.jaad.2017.03.022
Holberg, C. J., Erickson, R. P., Bernas, M. J., Witte, M. H., Fultz, K. E., Andrade, M., et al. (2001). Segregation analyses and a genome-wide linkage search confirm genetic heterogeneity and suggest oligogenic inheritance in some Milroy congenital primary lymphedema families. Am. J. Med. Genet. 98 (4), 303–312. doi:10.1002/1096-8628(20010201)98:4<303::aid-ajmg1113>3.0.co;2-9
Iljin, K., Karkkainen, M. J., Lawrence, E. C., Kimak, M. A., Uutela, M., Taipale, J., et al. (2001). VEGFR3 gene structure, regulatory region, and sequence polymorphisms. FASEB J. 15 (6), 1028–1036. doi:10.1096/fj.00-0383com
Jin, S. C., Homsy, J., Zaidi, S., Lu, Q., Morton, S., DePalma, S. R., et al. (2017). Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat. Genet. 49 (11), 1593–1601. doi:10.1038/ng.3970
Kerchner, K., Fleischer, A., and Yosipovitch, G. (2008). Lower extremity lymphedema update: Pathophysiology, diagnosis, and treatment guidelines. J. Am. Acad. Dermatol 59 (2), 324–331. doi:10.1016/j.jaad.2008.04.013
Kim, J., and Lim, S. Y. (2022). Compound heterozygosity for a variably penetrant variant and a variant of unknown significance in FLT4 causes fully penetrant Milroy's lymphedema. Lymphology 55 (2), 41–46. doi:10.2458/lymph.5264
Kitsiou-Tzeli, S., Vrettou, C., Leze, E., Makrythanasis, P., Kanavakis, E., and Willems, P. (2010). Milroy's primary congenital lymphedema in a male infant and review of the literature. Vivo 24 (3), 309–314.
Leppanen, V. M., Jeltsch, M., Anisimov, A., Tvorogov, D., Aho, K., Kalkkinen, N., et al. (2011). Structural determinants of vascular endothelial growth factor-D receptor binding and specificity. Blood 117 (5), 1507–1515. doi:10.1182/blood-2010-08-301549
Lohela, M., Saaristo, A., Veikkola, T., and Alitalo, K. (2003). Lymphangiogenic growth factors, receptors and therapies. Thromb. Haemost. 90 (2), 167–184. doi:10.1160/TH03-04-0200
Melikhan-Revzin, S., Kurolap, A., Dagan, E., Mory, A., and Gershoni-Baruch, R. (2015). A novel missense mutation in FLT4 causes autosomal recessive hereditary lymphedema. Lymphat. Res. Biol. 13 (2), 107–111. doi:10.1089/lrb.2014.0044
Mendola, A., Schlogel, M. J., Ghalamkarpour, A., Irrthum, A., Nguyen, H. L., Fastre, E., et al. (2013). Mutations in the VEGFR3 signaling pathway explain 36% of familial lymphedema. Mol. Syndromol. 4 (6), 257–266. doi:10.1159/000354097
Michelini, S., Degiorgio, D., Cestari, M., Corda, D., Ricci, M., Cardone, M., et al. (2012). Clinical and genetic study of 46 Italian patients with primary lymphedema. Lymphology 45 (1), 3–12.
Michelini, S., Vettori, A., Maltese, P. E., Cardone, M., Bruson, A., Fiorentino, A., et al. (2016). Genetic screening in a large cohort of Italian patients affected by primary lymphedema using a next generation sequencing (NGS) approach. Lymphology 49 (2), 57–72.
Monaghan, R. M., Page, D. J., Ostergaard, P., and Keavney, B. D. (2021). The physiological and pathological functions of VEGFR3 in cardiac and lymphatic development and related diseases. Cardiovasc Res. 117 (8), 1877–1890. doi:10.1093/cvr/cvaa291
Page, D. J., Miossec, M. J., Williams, S. G., Monaghan, R. M., Fotiou, E., Cordell, H. J., et al. (2019). Whole exome sequencing reveals the major genetic contributors to nonsyndromic tetralogy of Fallot. Circ. Res. 124 (4), 553–563. doi:10.1161/CIRCRESAHA.118.313250
Reuter, M. S., Jobling, R., Chaturvedi, R. R., Manshaei, R., Costain, G., Heung, T., et al. (2019). Haploinsufficiency of vascular endothelial growth factor related signaling genes is associated with tetralogy of Fallot. Genet. Med. 21 (4), 1001–1007. doi:10.1038/s41436-018-0260-9
Rutkowski, J. M., Markhus, C. E., Gyenge, C. C., Alitalo, K., Wiig, H., and Swartz, M. A. (2010). Dermal collagen and lipid deposition correlate with tissue swelling and hydraulic conductivity in murine primary lymphedema. Am. J. Pathol. 176 (3), 1122–1129. doi:10.2353/ajpath.2010.090733
Keywords: FLT4, primary lymphedema, autosomal recessive, trio-whole-exome sequencing, case report
Citation: Xiang Q, Chen J, Xiao X, Xu B, Xie H, Wang H, Yang M and Liu S (2023) Case Report: The compound heterozygotes variants in FLT4 causes autosomal recessive hereditary lymphedema in a Chinese family. Front. Genet. 14:1140406. doi: 10.3389/fgene.2023.1140406
Received: 09 January 2023; Accepted: 13 March 2023;
Published: 22 March 2023.
Edited by:
Mohammad Athar, Umm al-Qura University, Saudi ArabiaReviewed by:
Samira Kalayinia, Shaheed Rajaei Cardiovascular Medical and Research Center, IranCopyright © 2023 Xiang, Chen, Xiao, Xu, Xie, Wang, Yang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mei Yang, eWFuZ21laTAzMTgyMDE2QDE2My5jb20=; Shanling Liu, c3Vubnk2MzBAMTI2LmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.