 Hanrui Yu
Hanrui Yu Jie Wu
Jie Wu Jinju Cong4†
Jinju Cong4† Liqiang Wang
Liqiang Wang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 20 March 2023
Sec. Genetics of Common and Rare Diseases
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1139161
This article is part of the Research Topic Exploiting Genetics and Genomics to Improve the Understanding of Eye Diseases View all 14 articles
Background: PRDM12 is a newly discovered gene responsible for congenital insensitivity to pain (CIP). Its clinical manifestations are various and not widely known.
Methods: The clinical data of two infants diagnosed with CIP associated with PRDM12 mutation were collected. A literature review was performed, and the clinical characteristics of 20 cases diagnosed with a mutation of PRDM12 were summarized and analyzed.
Results: Two patients had pain insensitivity, tongue and lip defects, and corneal ulcers. The genomic analysis results showed that variants of PRDM12 were detected in the two families. The case 1 patient carried heterozygous variations of c.682+1G > A and c.502C > T (p.R168C), which were inherited from her father and mother, respectively. We enrolled 22 patients diagnosed with CIP through a literature review together with our cases. There were 16 male (72.7%) and 6 female (27.3%) patients. The age of onset ranged from 6 months to 57 years. The prevalence of clinic manifestation was 14 cases with insensitivity to pain (63.6%), 19 cases with self-mutilation behaviors (86.4%), 11 cases with tongue and lip defects (50%), 5 cases with mid-facial lesions (22.7%), 6 cases with distal phalanx injury (27.3%), 11 cases of recurrent infection (50%), 3 cases (13.6%) with anhidrosis, and 5 cases (22.7%) with global developmental delay. The prevalence of ocular symptoms was 11 cases (50%) with reduced tear secretion, 6 cases (27.3%) with decreased corneal sensitivity, 7 cases (31.8%) with disappeared corneal reflexes, 5.5 cases (25%, 0.5 indicated a single eye) with corneal opacity, 5 cases (22.7%) with corneal ulceration, and 1 case (4.5%) with a corneal scar.
Conclusion: The syndrome caused by PRDM12 mutation is a clinically distinct and diagnosable disease that requires joint multidisciplinary management to control the development of the disease and minimize the occurrence of complications.
Pain is a protective perception response to most harmful stimuli. Insensitivity to pain leads to an unguarded body, vulnerable to damage. Congenital insensitivity to pain (CIP) is a group of rare genetic pain loss disorders defined by its congenital onset. Hereditary sensory and autonomic neuropathy (HSAN) is also a genetic pain loss disorder that tends to develop gradually over time. Occasionally CIP and HSAN can overlap as the difference is not clear (Lischka et al., 2022). HSAN has been classified into types I–VIII according to the main mutation genes: SPTLC1, SPTLC2, ALT1, WNK1, SCN9A, NTRK1, NGFβ, SCN11A, and PRDM12 (Rotthier et al., 2012; Schwartzlow and Kazamel, 2019). Types Ⅵ, VII, and VIII of HSAN correspond to types I, II, and III of CIP (Schwartzlow and Kazamel, 2019).
PRDM12 is a newly identified causative gene for CIP. Members of the PRDM protein family have a PR domain and differing numbers of Zn-finger repeats. PRDM proteins regulate gene expression by either directly altering the chromatin structure through intrinsic methyltransferase activity or indirectly by attracting chromatin remodeling complexes (Di Zazzo et al., 2013). The human PRDM12 gene is localized on chromosome 9 at 9q33-q34, according to the Entrez Gene [Gene ID: 59335]. It has been proved that PRDM12 plays an important role in human pain perception (Nahorski et al., 2015; Drissi et al., 2020). The nerve growth factor (NGF)/tyrosine receptor kinase A (TrkA) signaling pathway is required for the survival and specification of nociceptors and plays a major role in pain processing. PRDM12 regulates the expression of NGF receptor TrkA to guide the development of nociceptive sensory neurons (Desiderio et al., 2019).
Mutations in PRDM12 are currently believed to cause HSAN-VIII and midface toddler excoriation syndrome (MiTES). HSAN is a rare hereditary neuropathy classified into types I–VIII (Rotthier et al., 2012; Schwartzlow and Kazamel, 2019). The mutation of the PRDM12 gene leads to the autosomal recessive HSAN-VIII type, also known as CIP3. The clinical symptoms vary, including growth delays, anhidrosis, self-mutilation behaviors, and self-injury-induced oral and corneal ulcers. Some injuries could be cured with treatment, while others could lead to lifelong tissue defects if left untreated. For instance, the patient reported by Gaur et al. (2018) suffered from frequent self-mutilation behaviors due to insensitivity to pain. He was only 1 year old when he had severe corneal scarring, a lip defect, and distal phalangeal injury caused by self-mutilation behaviors. Likewise, Moss et al. (2018) also reported a disease associated with PRDM12 mutations called MiTES. Unlike CIP3, MiTES is a relatively singular clinical presence with mostly scarring in the midface rather than a widespread insensitivity to pain (Moss et al., 2018). Thus, the two syndromes caused by PRDM12 easily cause appearance damage to patients, which seriously affects the quality of life.
Because the clinical manifestations caused by PRDM12 mutation are various, a lack of gene analysis as a routine clinical examination makes it difficult to diagnose this disease. Therefore, syndromes caused by a PRDM12 mutation are prone to be missed. Without early detection and early diagnosis, early treatment and early prevention are impossible. At the same time, due to the diversity of clinical manifestations of such diseases, including corneal ulcers, oral ulcers and infection, multidisciplinary collaborative management is needed, which increases the difficulty of treatment.
Our study hopes to summarize the clinical symptoms and treatment plans of two patients with PRDM12 mutations admitted to our hospital and 20 patients with PRDM12 mutations reported in the literature, analyze the impact of related symptoms on patients, formulate corresponding clinical management measures, and systematically elaborate the comprehensive prevention and treatment plan for patients with a PRDM12 mutation. It provides new ideas and comprehensive knowledge for the diagnosis and treatment of this disease.
Two patients with PRDM12 mutations confirmed in Beijing Children’s Hospital affiliated with Capital Medical University were included in this study. All the children’s guardians were informed and signed written informed consent. The terms “PRDM12,” “Hereditary sensory and autonomic neuropathy-VIII,” “Congenital insensitivity to pain 3,” and “Midface toddler excoriation syndrome” were searched in the National Library of Medicine of the United States (PubMed) from the time of establishment to October 2022. Inclusion criteria were the discovery of associated pathogenic genes.
Genomic DNA was extracted from the peripheral blood of patients to construct a genomic library, and then the exon and adjacent intron regions (50bp) of all human genes were captured by probe hybridization and enriched. The enriched target gene fragments were sequenced by a next-generation high throughput sequencer (Illumina). NextGene V2.3.4 software was used to compare the sequencing data with the human genome hg19 reference sequence provided by the UCSC database, and the coverage of target regions and sequencing quality were evaluated.
An 11-month female infant presented with recurrent corneal and oral ulcers that had persisted for 6 months. The infant developed corneal ulcers the size of rice grains in both eyes and oral ulcers successively at the age of 5 months. The local hospital treated her for “traumatic ulcers,” but she showed no significant improvement. The sizes of corneal and oral ulcers increased gradually.
The patient’s family had visited several major hospitals for the corneal ulcers. Traditional treatments such as tobramycin, levofloxacin, calf blood deproteinized extract eye gel, ganciclovir eye gel, and interferon eye drops were applied successively with no obvious improvement. The corneal ulcer gradually got worse, as did her oral ulcers. For the oral ulcer, symptomatic treatments were applied with no obvious improvement. The oral bacterial culture results found Staphylococcus aureus and Streptococcus salivarius, which were susceptible to benzoxicillin. The systemic use of linezolid and ertapenem to control the recurrent infection received little response.
When the patient was admitted to our hospital, the ophthalmic examination found conjunctival congestion and corneal ulcers in both eyes. The corneal ulcers infiltrated the deep stromal layer with an unclear borderline (Figure 1A). The chronic oral ulcer had caused profound defects of tongue and lip (Figure 1E). The medical history showed that the patient had no perspiration after birth, insensitivity to pain, developmental delay, and no obvious joint deformity. Based on the systemic clinical manifestations, we considered that the child had HSAN, so we conducted genetic testing on the child. The report showed a mutation of PRDM12. The patient carried pathogenic compound heterozygous variations of c.682+1G > A and c.502C > T (p.Arg168Cys), which were inherited from her father and mother, respectively. The ACMG criteria showed c.682+1G > A is pathogenic, and c.502C > T (p.Arg168Cys) is of uncertain significance. Her parents were not consanguineous in marriage. The diagnosis was confirmed as PRDM12 mutation-related CIP.
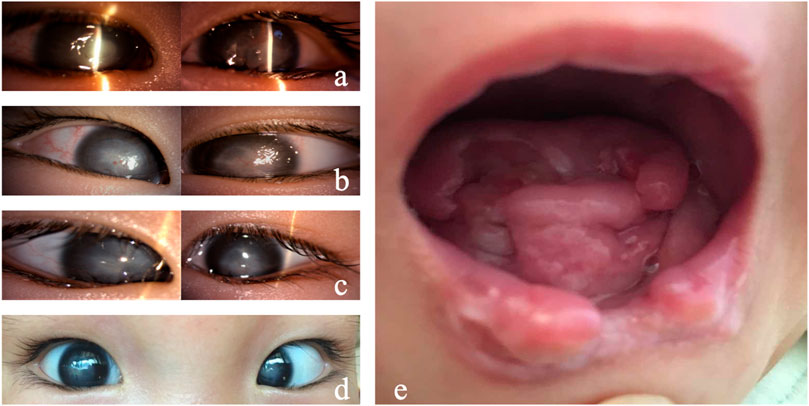
FIGURE 1. Clinical manifestations of Case 1. (A) Both eyes showed conjunctival hyperemia, corneal ulceration, deep stromal infiltration, borderline ambiguity, and opacity. (B) On day 7, the corneal ulcers in both eyes gradually healed, and the conjunctival congestion was reduced. (C) On day 14, the corneal ulcers healed in both eyes. (D) On day 40, conjunctival hyperemia (−) occurred in both eyes, the corneal epithelium was intact, and corneal leukoplakia formed. (E) Tongue and lip defects and oral ulcers.
As for the treatment, considering the infant with unknown etiology, weak general conditions, fever, and no sweat, surgery is not optimal as the anesthesia is risky. In addition, the penetrating keratoplasty requires intense care after surgery, which could be difficult as she perceives no pain. A strong eye rubbing or a delayed blink could lead to graft damage. After a comprehensive systemic status assessment, we used natamycin eye drops and gatifloxacin eye gel for both eyes and continued anti-infection symptomatic treatment for the whole body. The corneal ulcer improved significantly 2 days later. The oral secretion culture results showed Candida guilliermondii infection. After 7 days of ocular antifungal treatment, the corneal ulcer was limited, and conjunctival congestion was relieved. Systemic antifungal treatment with fluconazole injection was added. The patient’s condition gradually stabilized, and the fever was controlled (Figure 1B). After 2 weeks, systemic medication was gradually stopped, and the corneal and oral ulcers gradually healed (Figure 1C). Thereafter, the systemic medical treatment was symptomatic, and the eyes were regularly moisturized with preservative-free, artificial tears (0.3% sodium hyaluronate) eye drops. The cornea remained stable 4 weeks after discharge (Figure 1D).
A 15-month female infant presented with corneal opacity in both eyes for 5 months. She came to the hospital with a body temperature of 38.5 °C. The ophthalmic examination showed that the corneal reflex disappeared, tear secretion decreased, conjunctival congestion was present in both eyes, and both eyes showed corneal opacity (Figure 2A,B). Her tongue and lip were defective (Figure 2C). A purulent mossy attachment was noted around the oral cavity, on the oral mucosa and on the tongue surface. She was born at full term. The parents recalled little blinking after her birth. Eight months after birth, the infant manifested continuously sucking and biting the lower lip with no obvious incentives. Large mucosal ulcers were also noticed in the oral cavity. Oral anti-infection treatment and behavioral interventions such as sticking the lower lip with medical tape or usage of a pacifier to correct the habit of biting the lower lip were not effective. At the age of 10 months, her right eye was noticed to be cloudy, and her motor and intellectual development were delayed. Their parents were not consanguineous in marriage. There was no history of genetic family disease. Confirmation of the PRDM12 mutation in the patient was made through telephone follow-up with her family members. The type of mutation is not available, and co-segregation analysis has not been performed in this family. The diagnosis was confirmed as PRDM12 mutation-related CIP.
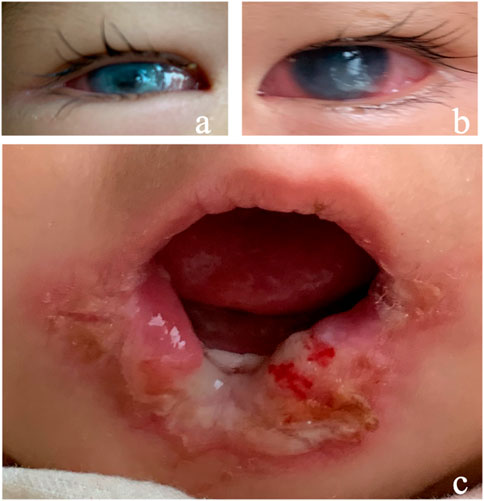
FIGURE 2. Clinical manifestations of Case 2. (A,B) Conjunctival hyperemia, corneal ulcer; (C) tongue and lip defects and oral ulcers.
A total of 11 articles that had reported the PRDM12 mutation-related disease were retrieved, including 20 cases with detailed case reports. Together with our cases, 22 cases were included. The demographic characteristics and ocular, oral, facial, and skeletal manifestations of the patients were summarized and analyzed. We summarized the clinical manifestations of all patients with PRDM12 mutations reported so far in Table 1 and summarized the patients with detailed disease descriptions in Table 2. All variations of PRDM12 are summarized in Table 3.
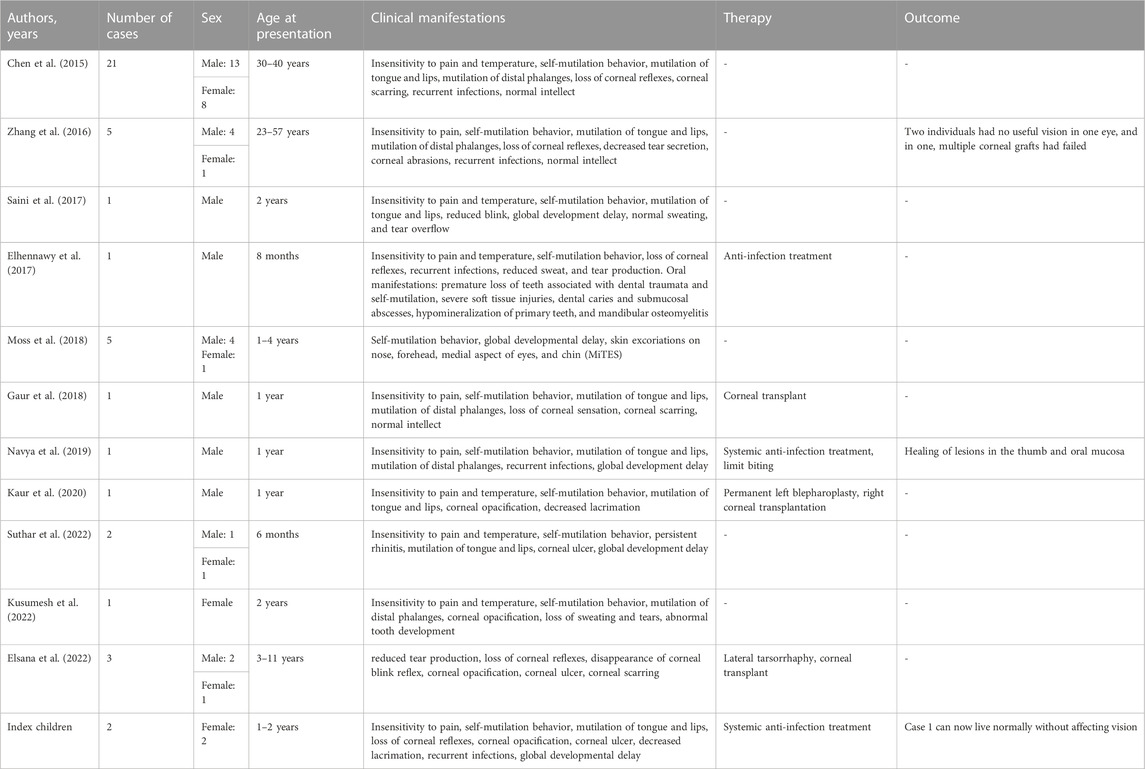
TABLE 1. Review of cases with PRDM12-related pain insensitivity.
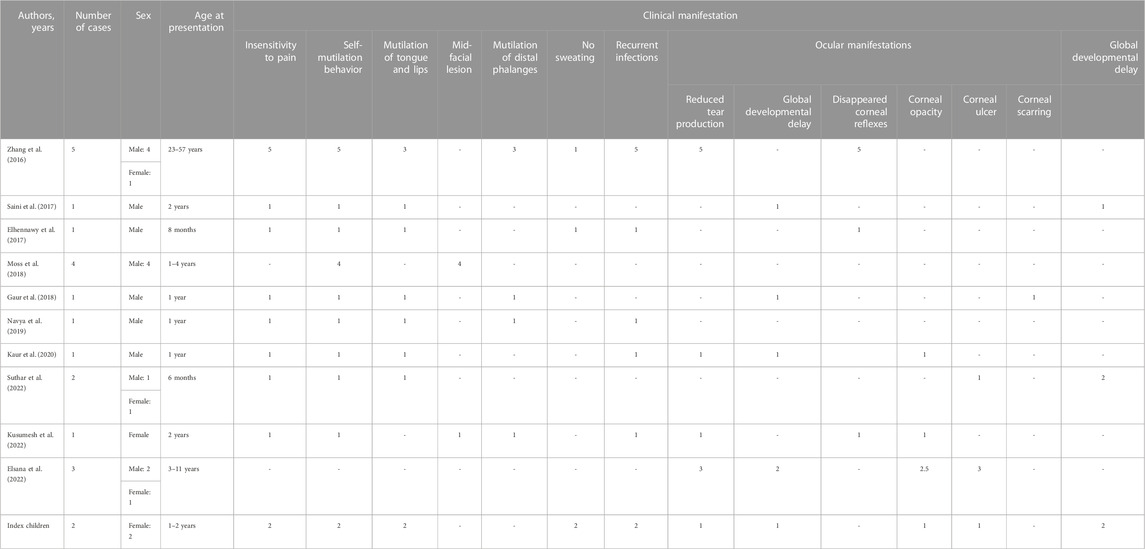
TABLE 2. Clinical features of patients diagnosed with PRDM12 mutations.

TABLE 3. Mutation sites identified in a patient with a PRDM12 gene mutation.
Among the 22 patients, there were 16 males (72.7%) and 6 females (27.3%). The age of onset ranged from 6 months to 57 years. The prevalence of clinic manifestation was 14 cases with insensitivity to pain (63.6%), 19 cases with self-mutilation behaviors (86.4%), 11 cases with tongue and lip defects (50%), 5 cases with a mid-facial lesion (22.7%), 6 cases with distal phalanx injury (27.3%), 11 cases of recurrent infection (50%), 3 cases (13.6%) with anhidrosis, and 5 cases (22.7%) with global developmental delay. The prevalence of ocular symptoms was 11 cases (50%) with reduced tear secretion, 6 cases (27.3%) with decreased corneal sensitivity, 7 cases (31.8%) with disappeared corneal reflexes, 5.5 cases (25%, 0.5 indicated a single eye) with corneal opacity, 5 cases (22.7%) with corneal ulceration, and 1 case (4.5%) with a corneal scar.
CIP3 is a rare inherited pain loss disorder with various clinical manifestations. As the symptoms varied, including insensitivity to pain, self-mutilation behaviors, recurrent infections, and self-injury-induced oral and corneal ulcers, doctors tend to only notice a single symptom and overlook the overall presentation, resulting in misdiagnosis and missed diagnosis.
Genetic analysis, as the only means of genetic disorder diagnosis, can accurately locate the mutated gene, identify the type of disease, and give clues in the prenatal examination. Studies have shown that the number of newborns diagnosed with HSAN-III has decreased significantly over the past decade with the help of prenatal testing (Couzin-Frankel, 2010). Fetuses with PRDM12 mutations may also be identified prenatally by such means. A comprehensive understanding of the PRDM12 mutation-related disease could be helpful for early diagnosis and treatment (Imhof et al., 2020). No association was found among the PRDM12 mutation sites in the 22 reported patients. However, the mutation may affect the structure of the protein and affect the distribution of pain perception. The aim of this study is to systematically review PRDM12 mutation-related CIP based on our two patients and the 20 cases reported in the literature. The overall findings of our study demonstrated that CIP due to PRDM12 mutations usually resulted in pain insensitivity, facial and limb defects, and recurrent infections, which significantly damaged children’s growth.
Pain insensitivity is one of the most distinctive features of all diseases caused by PRDM12 mutations. The insensitivity to pain caused by PRDM12 mutation leads to the defect of the nociceptors during embryonic development (Chen et al., 2015; Landy et al., 2021; Rienzo et al., 2021). With the inability to feel pain, patients often unconsciously show some self-mutilation behaviors. Self-inflicted injuries are more vulnerable to infection and decreased immunity. Recurring infections are the outcome. This causes great difficulties for parents in looking after their children. Self-mutilation behaviors cause defects in various body parts at an early age, which can have lifelong effects on life and appearance. Treatment was limited to plastic surgery, such as functional alginate dressings (Jones et al., 2006), autologous skin grafts (Hu et al., 2015), and cell-based wound healing therapy (Rodrigues et al., 2019). Another difficulty in treating CIP3 is recurrent infection. Because the patient cannot perceive the injury, the healing process may be accompanied by new wounds.
Patients with PRDM12 mutations present with reduced tear secretion, corneal abrasions, and loss of corneal reflexes, resulting in keratitis and corneal scarring (Chen et al., 2015; Zhang et al., 2016). We found that almost every patient had some degree of corneal injury, indicating that the corneal symptoms deserve attention.
There is no clear report in the literature on why patients with PRDM12 mutations have different degrees of eye damage, and we expect to explore this next. Because HSAN autosomal recessive patients often develop the disease at an early age, before irreversible damage is caused, eye symptoms tend to give us a better warning (Schwartzlow and Kazamel, 2019). For example, patients with dry eye signs on the ocular surface, corneal opacity or decreased corneal sensitivity, if further aggravated, such as corneal opacity or increased secretions, should seek medical attention in time to minimize the damage.
As shown in Table 1, when selecting the treatment plan, except for the two patients reported by us, conservative treatment was adopted, and most of the patients with detailed reports of their disease were controlled through corneal transplantation and other operations. In Case 1, penetrating keratoplasty was considered to repair corneal ulcers during a visit to another hospital. Yagev et al. (1999) reported that a child with binocular corneal ulcers caused by painless syndrome was treated with penetrating keratoplasty under good overall condition, but the postoperative effect was poor. Corneal transplantation is a relatively complicated ophthalmic operation that imposes high requirements on patients’ general condition and postoperative nursing (Tan et al., 2012). CIP patients tend to have poor systemic status due to recurrent infections. Moreover, uncontrolled eye rubbing and eye damage will inevitably occur after surgery, resulting in artificial transplant failure. Therefore, after carefully considering the circumstances and full communication with the parents, we adopted conservative treatment. The patient’s vision recovered well, and her daily life was not affected. For Case 1, controlling fever and infection in the early stage is the focus of treatment. When the physician could not find the cause, we started from the local symptoms of the eye and adopted antifungal treatment, which not only avoided the corneal perforation but also effectively controlled the systemic condition of the child.
CIP3 can also lead to global developmental delays. Although the proportion is relatively low, it has a great impact on the growth of children. If the doctor suspects this disease when treating the patients, parents should be reminded to assess the intelligence of their child and avoid missed diagnoses.
MiTES, meanwhile, could be an early warning sign of PRDM12-CIP. MiTES patients often present with pathological itching in the mid-face, an inability to manage the damaged area due to pain insensitivity, and mid-facial lesions after persistent scratching (Moss et al., 2018). Although there is no other evidence of damage to PRDM12-CIP in the disease profile of MiTES patients, four of five had mutations in PRDM12. MiTES, therefore, should be considered if a child is observed unconsciously scratching the midface area. Medical attention is needed to avoid the possibility of MiTES’ progression to PRDM12-CIP. Facial defects often cause the appearance of patients with terror, and their normal growth and integration into society cause great difficulties. Attention should also be paid to the mental health status of this group (McCarron et al., 2021).
Patients with a PRDM12 gene mutation benefit from early detection and diagnosis. Early intervention can greatly control the progression of the disease so that the appearance and vision of the patients will not be affected to the greatest extent. Secondly, different treatment measures should be implemented according to age stages and the severity of the disease, and symptomatic treatment against infection should always be maintained. Finally, the patient’s family should attend to the patient’s psychological problems.
The original contributions presented in the study are included in the article/Supplementary material; further inquiries can be directed to the corresponding authors.
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Data curation: HY; writing (original draft): HY, JW, and JC; writing (review and editing): all authors.
This work was supported by the Natural Science Foundation of China (82070921).
We thank the families for their help.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Chen, Y. C., Auer-Grumbach, M., Matsukawa, S., Zitzelsberger, M., Themistocleous, A. C., Strom, T. M., et al. (2015). Transcriptional regulator PRDM12 is essential for human pain perception. Nat. Genet. 47, 803–808. doi:10.1038/ng.3308
Couzin-Frankel, J. (2010). Chasing a disease to the vanishing point. Science 328, 298–300. doi:10.1126/science.328.5976.298
Desiderio, S., Vermeiren, S., Van Campenhout, C., Kricha, S., Malki, E., Richts, S., et al. (2019). Prdm12 directs nociceptive sensory neuron development by regulating the expression of the NGF receptor TrkA. Cell Rep. 26, 3522–3536. doi:10.1016/j.celrep.2019.02.097
Di Zazzo, E., De Rosa, C., Abbondanza, C., and Moncharmont, B. (2013). PRDM proteins: Molecular mechanisms in signal transduction and transcriptional regulation. Biology 2, 107–141. doi:10.3390/biology2010107
Drissi, I., Woods, W. A., and Woods, C. G. (2020). Understanding the genetic basis of congenital insensitivity to pain. Br. Med. Bull. 133, 65–78. doi:10.1093/bmb/ldaa003
Elhennawy, K., Reda, S., Finke, C., Graul-Neumann, L., Jost-Brinkmann, P. G., and Bartzela, T. (2017). Oral manifestations, dental management, and a rare homozygous mutation of the PRDM12 gene in a boy with hereditary sensory and autonomic neuropathy type VIII: A case report and review of the literature. J. Med. case Rep. 11, 233. doi:10.1186/s13256-017-1387-z
Elsana, B., Imtirat, A., Yagev, R., Gradstein, L., Majdalani, P., Iny, O., et al. (2022). Ocular manifestations among patients with congenital insensitivity to pain due to variants in PRDM12 and SCN9A genes. Am. J. Med. Genet. Part A 188, 3463–3468. doi:10.1002/ajmg.a.62968
Gaur, N., Meel, R., Anjum, S., and Singh, P. (2018). Hereditary sensory and autonomic neuropathy in a male child: 'The other side of not feeling pain'. BMJ case Rep. 2018, bcr2018226873. doi:10.1136/bcr-2018-226873
Hu, Z. C., Chen, D., Guo, D., Liang, Y. Y., Zhang, J., Zhu, J. Y., et al. (2015). Randomized clinical trial of autologous skin cell suspension combined with skin grafting for chronic wounds. Br. J. Surg. 102, e117–e123. doi:10.1002/bjs.9688
Imhof, S., Kokotović, T., and Nagy, V. (2020). PRDM12: New opportunity in pain research. Trends Mol. Med. 26 (10), 895–897. doi:10.1016/j.molmed.2020.07.007
Jones, V., Grey, J. E., and Harding, K. G. (2006). Wound dressings. BMJ 332, 777–780. doi:10.1136/bmj.332.7544.777
Kaur, J., Singanamalla, B., Suresh, R. G., and Saini, A. G. (2020). Insensitivity to pain, self-mutilation, and neuropathy associated with PRDM12. Pediatr. Neurol. 110, 95–96. doi:10.1016/j.pediatrneurol.2020.03.007
Kusumesh, R., Ambastha, A., Singh, V., and Singh, A. (2022). Hereditary sensory and autonomic neuropathy type VIII: Congenital insensitivity to pain with anhidrosis. Indian dermatology online J. 13, 257–258. doi:10.4103/idoj.idoj_427_21
Landy, M. A., Goyal, M., Casey, K. M., Liu, C., and Lai, H. C. (2021). Loss of Prdm12 during development, but not in mature nociceptors, causes defects in pain sensation. Cell Rep. 34, 108913. doi:10.1016/j.celrep.2021.108913
Lischka, A., Lassuthova, P., Çakar, A., Record, C. J., Van Lent, J., Baets, J., et al. (2022). Genetic pain loss disorders. Nat. Rev. Dis. Prim. 8 (1), 41. doi:10.1038/s41572-022-00365-7
McCarron, R. M., Shapiro, B., Rawles, J., and Luo, J. (2021). Depression. Ann. Intern. Med. 174, ITC65–ITC80. doi:10.7326/AITC202105180
Moss, C., Srinivas, S. M., Sarveswaran, N., Nahorski, M., Gowda, V. K., Browne, F. M., et al. (2018). Midface toddler excoriation syndrome (MiTES) can be caused by autosomal recessive biallelic mutations in a gene for congenital insensitivity to pain, PRDM12. Br. J. dermatology 179, 1135–1140. doi:10.1111/bjd.16893
Nahorski, M. S., Chen, Y. C., and Woods, C. G. (2015). New mendelian disorders of painlessness. Trends Neurosci. 38, 712–724. doi:10.1016/j.tins.2015.08.010
Navya, M. K., Pramod, G. V., Sujatha, G. P., and Ashok, L. (2019). Congenital insensitivity to pain in a 1-year-old boy. J. Indian Soc. Pedod. Prev. Dent. 37, 308–310. doi:10.4103/JISPPD.JISPPD_340_18
Rienzo, M., Di Zazzo, E., Casamassimi, A., Gazzerro, P., Perini, G., Bifulco, M., et al. (2021). PRDM12 in health and diseases. Int. J. Mol. Sci. 22, 12030. doi:10.3390/ijms222112030
Rodrigues, M., Kosaric, N., Bonham, C. A., and Gurtner, G. C. (2019). Wound healing: A cellular perspective. Physiol. Rev. 99, 665–706. doi:10.1152/physrev.00067.2017
Rotthier, A., Baets, J., Timmerman, V., and Janssens, K. (2012). Mechanisms of disease in hereditary sensory and autonomic neuropathies. Nat. Rev. Neurol. 8, 73–85. doi:10.1038/nrneurol.2011.227
Saini, A. G., Padmanabh, H., Sahu, J. K., Kurth, I., Voigt, M., and Singhi, P. (2017). Hereditary sensory polyneuropathy, pain insensitivity and global developmental delay due to novel mutation in PRDM12 gene. Indian J. Pediatr. 84, 332–333. doi:10.1007/s12098-016-2284-y
Schwartzlow, C., and Kazamel, M. (2019). Hereditary sensory and autonomic neuropathies: Adding more to the classification. Curr. neurology Neurosci. Rep. 19, 52. doi:10.1007/s11910-019-0974-3
Suthar, R., Sharawat, I. K., Eggermann, K., Padmanabha, H., Saini, A. G., Bharti, B., et al. (2022). Hereditary sensory and autonomic neuropathy: A case series of six children. Neurol. India 70, 231–237. doi:10.4103/0028-3886.338691
Tan, D. T., Dart, J. K., Holland, E. J., and Kinoshita, S. (2012). Corneal transplantation. Lancet (London, Engl.) 379 (9827), 1749–1761. doi:10.1016/S0140-6736(12)60437-1
Yagev, R., Levy, J., Shorer, Z., and Lifshitz, T. (1999). Congenital insensitivity to pain with anhidrosis: Ocular and systemic manifestations. Am. J. Ophthalmol. 127, 322–326. doi:10.1016/s0002-9394(98)00370-5
Keywords: Prdm12, insensitivity to pain, corneal disease, self-mutilation behavior, HSAN, hereditary and sensory autonomic neuropathy
Citation: Yu H, Wu J, Cong J, Chen M, Huang Y, Yu J and Wang L (2023) Congenital insensitivity to pain associated with PRDM12 mutation: Two case reports and a literature review. Front. Genet. 14:1139161. doi: 10.3389/fgene.2023.1139161
Received: 06 January 2023; Accepted: 27 February 2023;
Published: 20 March 2023.
Edited by:
Jeremy Guggenheim, Cardiff University, United KingdomReviewed by:
Diego Maria Michele Fornasari, University of Milan, ItalyCopyright © 2023 Yu, Wu, Cong, Chen, Huang, Yu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liqiang Wang, bGlxaWFuZ3czMDFAMTYzLmNvbQ==; Jifeng Yu, amVmZmVybnl1QDEyNi5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.