 Xianzhi Zhang
Xianzhi Zhang Xinyi Wang1
Xinyi Wang1 Wencai Wang
Wencai Wang- 1College of Horticulture and Landscape Architecture, Zhongkai University of Agriculture and Engineering, Guangzhou, China
- 2Science and Technology Innovation Center, Guangzhou University of Chinese Medicine, Guangzhou, China
Eucommia ulmoides is a famous rubber-producing and medicinal tree species that produces unisexual flowers on separate individuals from the earliest stage of stamen/pistil primordium formation. To explore the genetic regulation pathway of sex in E. ulmoides, comprehensive genome-wide analyses and tissue-/sex-specific transcriptome comparisons of MADS-box transcription factors were performed for the first time in this work. Quantitative real-time PCR technique was employed to further validate the expression of genes that are assigned to floral organ ABCDE model. A total of 66 non-redundant E. ulmoides MADS-box (EuMADS) genes were identified, they were classified into Type I (M-type, 17 genes) and Type II (MIKC, 49 genes). Complex protein-motif composition, exon-intron structure and phytohormone-response cis-elements were detected in MIKC-EuMADS genes. Furthermore, 24 differentially-expressed EuMADS genes (DEGs) between male and female flowers, and two DEGs between male and female leaves were revealed. Amongst the 14 floral organ ABCDE model-related genes, there were 6 (A/B/C/E-class) and 5 (A/D/E-class) genes displayed male- and female-biased expression respectively. In particular, one B-class gene EuMADS39 and one A-class gene EuMADS65 were almost exclusively expressed in male trees, no matter in flower or leaf tissues. Collectively, these results suggested a critical role of MADS-box transcription factors in sex determination of E. ulmoides, which is conducive to decoding the molecular regulation mechanism of sex in E. ulmoides.
1 Introduction
MADS-box genes encode a class of transcription factors (TFs) that contain a highly conserved MADS domain (∼60 amino acids, aa) and are able to combine CArG-box cis-elements to regulate gene expression at transcriptional level (Theissen et al., 2018). ‘MADS’ is abbreviated from initials of four proteins in different organisms: MCM1 from yeast (M), AGAMOUS from Arabidopsis (A), DEFICIENS from snapdragon (D) and SRF from human (S) (Dong et al., 2021). In plants, the MADS-box gene family is categorized into two subfamilies, i.e., Type-I (M-type) and Type-II (MICK). M-type subfamily generally contains SRF domain and is classified into three lineages, i.e., Mα, Mβ, and Mγ (Wang et al., 2019). Whereas, MIKC subfamily usually possesses SRF or MEF2 domains and is divided into two lineages: MIKC* and MIKCC (superscript C means classic) (Gutierrez et al., 2022). Wherein genes belong to MIKCC are the most well-studied MADS-box genes by far, they contain as many as 13 subgroups: AG/SHP, AGL6, AGL12, AGL15, ANR1, AP1, AP3/PI, FLC, SEP, SOC1, SVP, TM8, and TT16 (Theißen et al., 2016; Alhindi and Al-Abdallat, 2021).
MADS-box TFs play critical roles in various processes of plant development, such as flowering control, floral organogenesis, and fruit ripening (Theissen and Saedler, 2001; Lai et al., 2022). Nearly all the well-known floral homeotic genes of ‘ABCDE model’ belong to the MADS-box family (Theien, 2001; Soltis et al., 2007). According to ‘ABCDE model’, the sepal, petal, stamen and carpel of bisexual flower are specifically regulated by A + E, A + B + E, B + C + E and C + E genes respectively, and ovules by C + D + E (Theissen and Saedler, 2001; Theißen et al., 2016). In the well-studied Arabidopsis there are 12 A/B/C/D/E-class genes, i.e., A-class: APETALA1 (AP1) and APETALA2 (AP2); B-class: APETALA3 (AP3) and PISTILLATA (PI); C-class: AGAMOUS (AG); D-class: SHATTERPROOF1, 2 (SHP1 and SHP2) and SEEDSTICK (STK); and E-class: SEPALLATA1, 2, 3, 4 (SEP1, SEP2, SEP3 and SEP4) (Soltis et al., 2007). All these 12 genes, except for AP2 belong to MADS-box family (Theien, 2001).
Recent studies revealed that plant sex is highly associated with MADS-box genes (Zhang et al., 2022). For example, LcMADS42/46/47/51/75/93/100 are probably involved in the unisexual flower formation of litchi (Litchi chinensis Sonn.) (Guan et al., 2021). One MADS-box gene QsPISTILLATA is also exclusively expressed in male flowers of Quercus suber L., and is proved to be functional for stamen development (Sobral and Costa, 2017). Remarkably, B-class MADS-box TFs are essential for sex determination of several dioecious plants (Zhang et al., 2022). In spinach (Spinacia oleracea L.), B-class genes SpAP3 and SpPI are expressed specifically in the floral primordial of male. Suppression of SpAP3 or SpPI turned male individuals into female (Pfent et al., 2005; Sather et al., 2010). Similarly, in Thalictrum dioicum L., all B-class genes (ThdAP3-1, ThdAP3-2a, ThdAP3-2b, ThdPI-1 and ThdPI-2) are only expressed in floral buds of male but not female at very initial stage. Knocking-down the expression of ThdPI-1 or ThdPI-2 also converted male individuals into female (Di Stilio et al., 2005; Larue et al., 2013).
Eucommia ulmoides Oliv. is known as the hardy rubber tree that can synthesize gutta-rubber (trans-1,4-polyisoprene, TPI) almost in the whole plant, especially in fruits (Wang et al., 2018). It is also the resource of well-known traditional Chinese medicine ‘DuZhong’ (Ouyang et al., 2021). Meanwhile, this species is a strict dioecious plant with stamen primordium or pistil primordium initiating in separate individuals, suggesting that its flowers are unisexual from inception (Wang et al., 2018; Qing et al., 2021). Some MADS-box TFs, especially them involved in ‘ABCDE model’ of flower development, have been suggested to take part in sex determination of dioecious plants with such type of unisexual flowers (Diggle et al., 2011; Zhang et al., 2022). However, the characteristics and roles of MADS-box TFs in sex determination of E. ulmoides remain largely unknown by far.
In this work, MADS-box TFs of E. ulmoides were identified throughout the genome and comprehensively characterized, e.g., their evolutionary relationships, protein motif compositions, gene structures, and phytohormone response cis-elements. Tissue- and sex-specific expression profiling of all the MADS-box genes were investigated based on transcriptome data. Quantitative real-time PCR (qRT-PCR) analyses were further conducted to uncover the sex-biased expression patterns of A/B/C/D/E-class genes involved in flower development. The results obtained herein will on the one hand shed lights on our understanding of the evolution and function of MADS-box TFs in E. ulmoides, on the other, will promote decoding the molecular regulation mechanism of sex in this dioecious plant.
2 Results
2.1 Genome-wide identification and synteny analysis of MADS-Box TFs in E. ulmoides
A total of 66 non-redundant MADS-box TFs were identified in E. ulmoides for the first time according to BLASTP and HMMER analyses, named as EuMADS01-66 (Table 1). Wherein 64 of them were derived from the haploid E. ulmoide genome (Li et al., 2020), the rest two were discovered in the male genome (Wuyun et al., 2018), with 58 members being commonly detected from both the haploid and male genomes. The identified 66 EuMADS TFs were further verified in SMART and CDD databases for the presence of conserved MADS domain. These MADS-box TFs were sized from 95 aa (EuMADS04) to 471 aa (EuMADS62) with an average of 232 aa. Their isoelectric points (pl) varied from 4.7 (EuMADS02) to 11.5 (EuMADS05), molecular weight (MW) were between 10.7 kDa (EuMADS04) and 53.4 kDa (EuMADS62), number of phosphorylation sites (Ser, Tyr and Thr sites) were from 11 (EuMADS04) to 72 (EuMADS62). As expected, subcellular locations of all the 66 EuMADS TFs were predicted to be in the nucleus. Information of the nucleotide and amino-acid sequences of these 66 EuMADS genes were documented in Additional Data1.
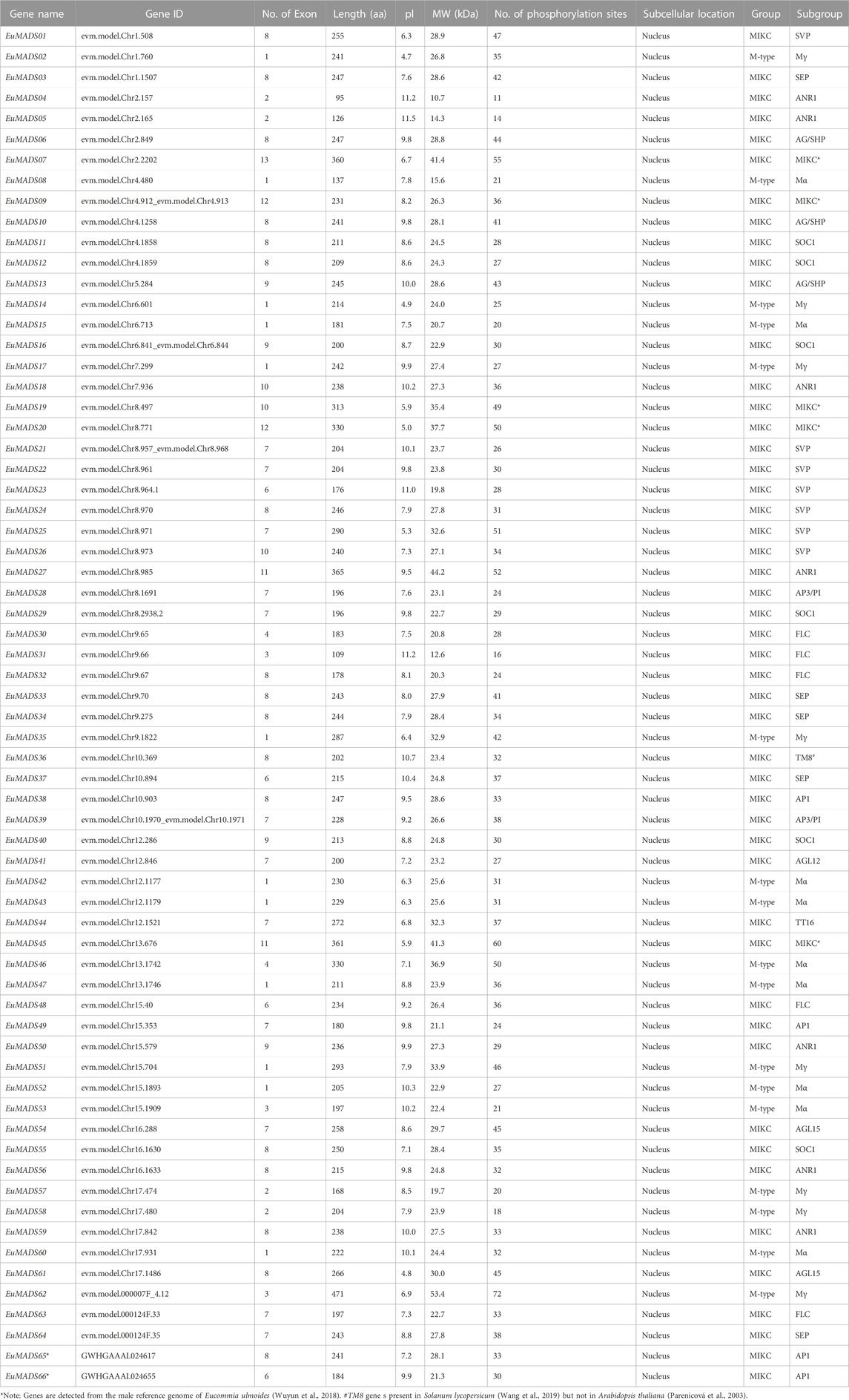
TABLE 1. Detailed information of the EuMADS genes.
The 66 EuMADS genes were then mapped to the assembled 17 chromosomes of haploid E. ulmoides (Li et al., 2020) (Figure 1A). As a result, a highly uneven distribution pattern of EuMADS genes was revealed, with Chromosome 8 (Chr8) harboring the maximum number of EuMADS genes (11, 16.7%), followed by Chr9 and Chr15 (both contain 6, 9.1%), and Chr4 and Chr17 (both contain 5, 7.6%). While only one was localized on Chr5, none was seen on Chr3, Chr11 and Chr14 (Figure 1B). Besides, five EuMADS genes were unable to be assigned to any of the 17 assembled chromosomes (Li et al., 2020). Moreover, intra-genome and inter-genome relations of EuMADS genes were analyzed by MCScanX. The results revealed that 15 out of 66 EuMADS genes locating in 14 pairs of segmental duplicates showed collinearity between chromosomes in E. ulmoides (Figure 2A, Supplementary Table S1). Meanwhile, synteny analyses of MADS-box genes between Eucommia and Arabidopsis/rice showed more collinear regions with Arabidopsis (27) than that with rice (11) (Figure 2B). This may suggest closer evolutionary relationship between the two eudicot plants, i.e., Eucommia and Arabidopsis.
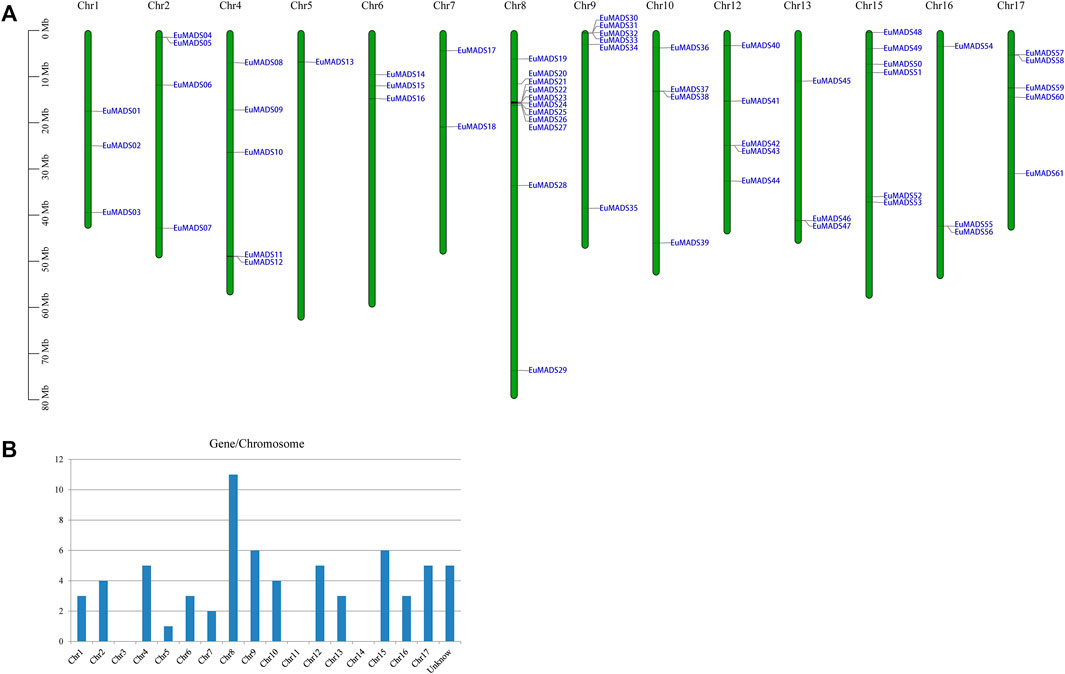
FIGURE 1. Chromosomal distribution of the 66 EuMADS genes. (A) Schematic representation of the chromosomal localization of EuMADS genes. Each vertical bar represents one chromosome indicated by numbers above. Chromosome length is scaled on the left. (B) The density of EuMADS genes on each chromosome. EuMADS genes with unknown chromosomal position are shown as unknown.
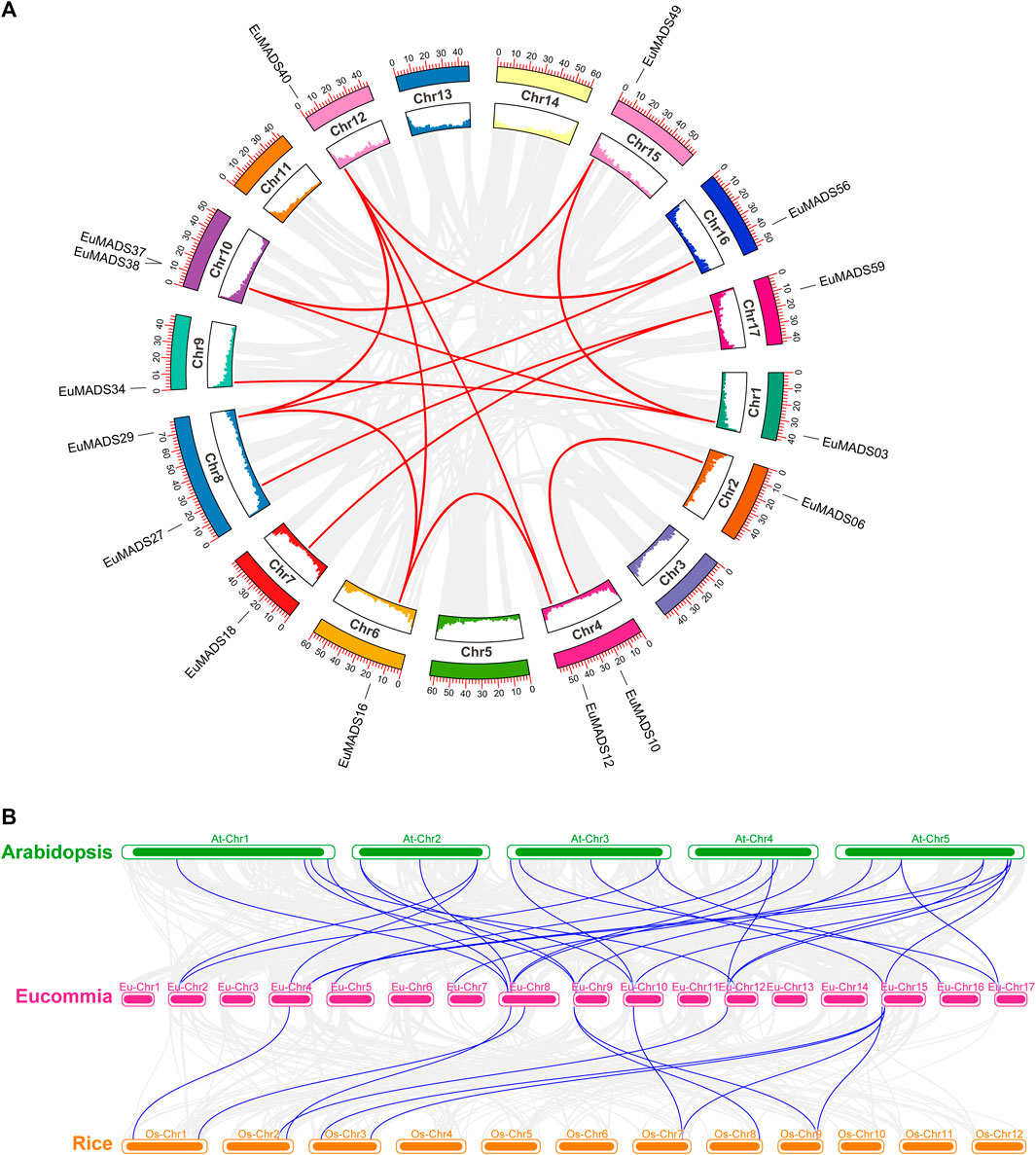
FIGURE 2. Synteny analysis of EuMADS genes within E. ulmoides and between species. (A) Inter-chromosomal relationships of EuMADS genes within E. ulmoides. Gray lines indicate all synteny blocks in E. ulmoides genome, red lines indicate duplicated EuMADS gene pairs. Inside boxes indicate gene density of each corresponding chromosome. (B) Collinear relationships of EuMADS genes between E. ulmoides and the other two species (Arabidopsis and rice). Gray lines indicate the syntenic blocks between Eucommia and Arabidopsis/rice, blue lines highlight the collinear EuMADS gene pairs.
2.2 Phylogenetic relationship and classification of EuMADS TFs
In order to correctly classify the EuMADS genes, phylogenetic relationship among the EuMADS TFs (66) and MADS-box TFs of A. thaliana (AtMADS, 103) were analyzed. Maximum likelihood (ML) tree showed that 17 EuMADS TFs were clustered in Type I (M-type) subfamily, while the rest 49 belonged to Type II (MIKC) (Figure 3). In M-type subfamily, nine EuMADS TFs were clustered in Mα lineage while the remaining eight genes were distributed in Mγ lineage. The Mβ lineage was absent in E. ulmoides.
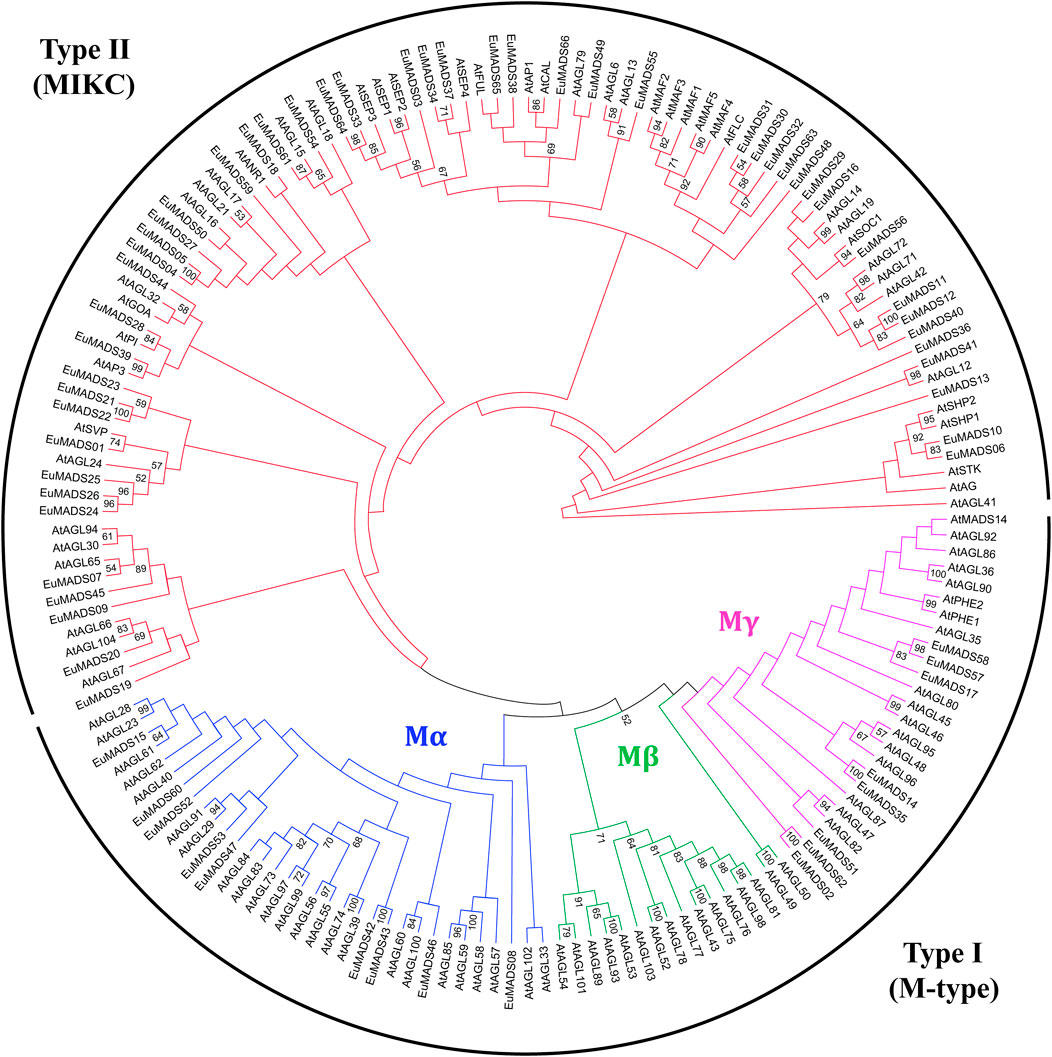
FIGURE 3. Maximum likelihood tree of MADS-box transcription factors in E. ulmoides and A. thaliana. The tree reveals two subfamilies of the MADS-box gene family, i.e., Type I (M-type) and Type II (MIKC). In M-type group, three subgroups, i.e., Mα, Mβ and Mγ are shown in blue, green and purple, respectively. MIKC group is colored in red.
MIKC MADS-box TFs of tomato (MIKC-SolyMADS, 32), rice (MIKC-OsMADS, 34) and Arabidopsis (MIKC-AtMADS, 42) were further used to classify MIKC-EuMADS TFs (49). The phylogenetic tree was branched into 14 clades, corresponding to the 13 known subgroups identified in MIKCC lineage, plus MIKC* subgroup (Figure 4). There were respectively four A-class genes (EuMADS38, 49, 65, and 66), two B-class genes (EuMADS28 and 39), three C-/D-class genes (EuMADS06, 10 and 13), and five E-class genes (EuMADS03, 04, 33, 37, and 64) detected in E. ulmoides. Some MIKC-EuMADS TFs were scattered across other nine clades of MIKCC. For instance, each of the AGL6, TM8, TT16, AGL12 subgroup harbored one different MIKC-EuMADS member, while AGL15, FLC subgroup contained two and five respectively, both SOC1 and ANR1 subgroup had six, SVP subgroup included seven. Note, there were only five MIKC-EuMADS TFs clustered in MIKC* lineage.
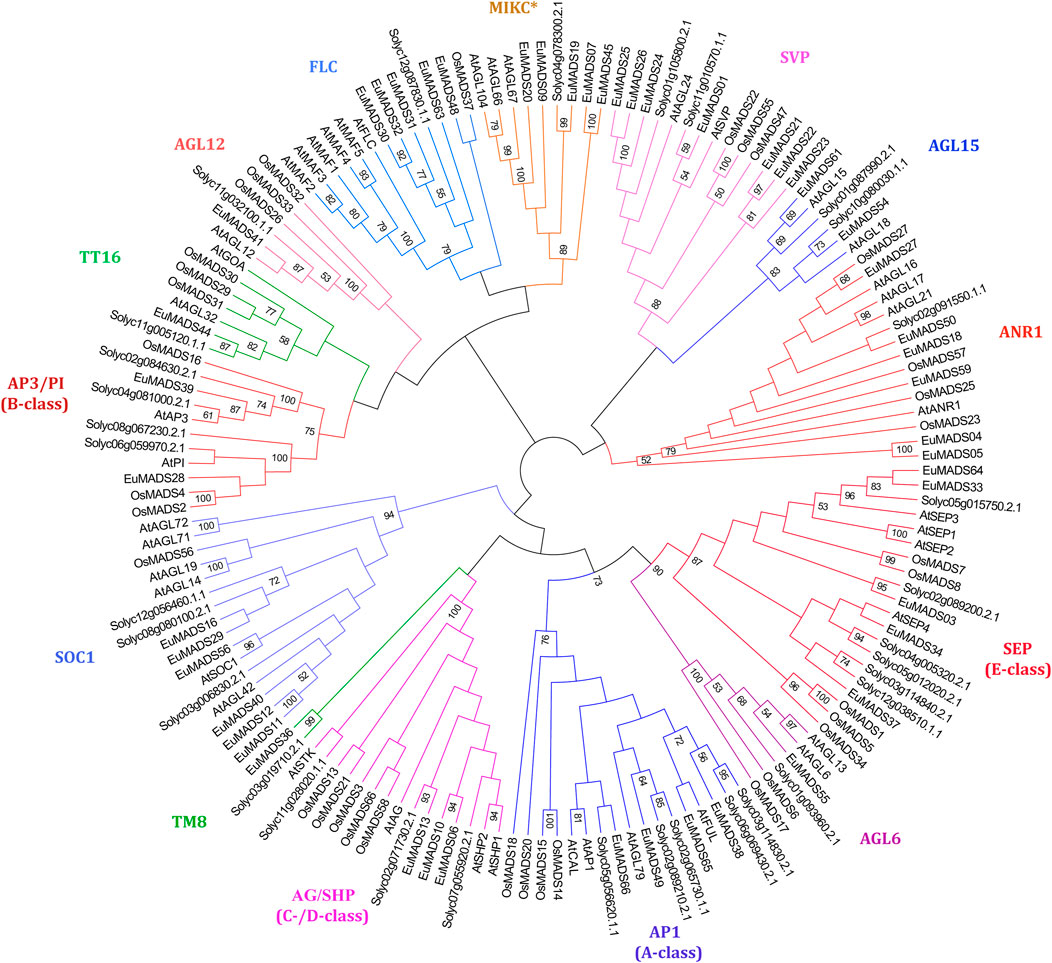
FIGURE 4. Maximum likelihood tree of MIKC MADS-box transcription factors in E. ulmoides, A. thaliana, S. lycopersicum and Oryza sativa. MIKC* clade and 13 known MIKCC subgroups are indicated in different colors.
2.3 Protein domain, gene structure, and promotor cis-element of EuMADS TFs
To obtain more information about functions of EuMADS TFs, the protein domain/motif composition, exon-intron organization, and cis-elements in promotor region were analyzed for each EuMADS gene. The conserved domains of 66 EuMADS TFs were identified using Batch CD-Search in NCBI. In summary, EuMADS TFs contained seven types of conserved domains in two categories, i.e., MADS (MADS-MEF2-like, MADS superfamily, MADS-SRF-like, MADS, SRF-TF) and K-box (K-box, K-box superfamily) (Figure 5B). As expected, all the 66 EuMADS proteins contained conserved MADS domain. Among the 49 Type II (MIKC-EuMADS) TFs, 38 proteins possessed both MADS and K-box domains, whereas the rest 11 proteins lacked K-box domain. Meanwhile, there was only MADS domain but no K-box domain in all the 17 Type I (M-type EuMADS) TFs. These results suggested functional differentiation among the EuMADS members. Moreover, the protein motif composition also varied obviously between the two EuMADS subfamilies (Figure 5C). Motif 1 was the most conserved motif in the EuMADS TF family and distributed in the vast majority of members (60/66, 90.9%). Motif 2 was present in most MIKC-EuMADS members (41/49, 83.7%), but not found in any M-type EuMADS TFs. Interestingly, motif composition of EuMADS TFs from the same phylogenetic clade was similar, indicating possible resemblance in biological functions. For example, protein motifs in the AP3/PI subgroup (B-class) all arranged conservatively as 1-3-2, while the AG/SHP subgroup (C-/D-class) all usually arrayed as 1-3-7-2-4.

FIGURE 5. Protein domain, motif and gene structure of EuMADS transcription factors. (A) Phylogenetic relationship of the EuMADS gene family. Type I (M-type) including 2 clades and Type II (MIKC) including 14 clades are indicated by colored vertical lines. (B) Conserved protein domains of EuMADS transcription factors. (C) Protein motif composition of EuMADS transcription factors. The seven domains and 20 motifs are boxed in different colors, respectively. (D) Exon-intron structure of EuMADS genes. CDS, UTR and intron are represented by green, yellow boxes and black lines, respectively.
Gene structure analysis showed that the composition of exon/introns between the M-type and MIKC subfamily were distinct (Table 1; Figure 5D). M-type EuMADS genes embodied 1-4 exons per gene (averagely c. 1.5), whereas MIKC genes housed 2–13 exons (averagely c. 7.8). Most M-type EuMADS genes (12/17, 70.6%) contained only one exon but none of introns, almost all the MIKC-EuMADS genes (45/49, 91.8%) harbored at least six exons and five introns. More exons in MIKC-EuMADS genes may imply their complexity and/or variation of biological functions. The number of exons in genes from the same phylogenetic clade was usually similar, e.g., seven exons present in EuMADS28 and EuMADS39, both belong to clade AP3/PI (B-class). The size of EuMADS genes varied largely, from 527 bp (EuMADS52, Mα subgroup of M-type) to 78,479 bp (EuMADS25, clade SVP of MIKC).
The 2,000 bp promoter sequences upstream of the start codon (ATG) of each EuMADS gene were obtained from the genome of E. ulmoides (Wuyun et al., 2018; Li et al., 2020) to predict the cis-acting regulatory elements (CAREs) by PlantCARE. A total of 1,140 CAREs belonging to 22 types and 12 functional modules were found (Figure 6A, Supplementary Table S2), suggesting extensive biological functions of EuMADS TFs in the growth and development of E. ulmoides. Among the 12 functional modules, light-response CAREs had the largest number (313), including seven types, i.e., G-Box, Sp1, GT1-motif, 3-AF1 binding site, 4cl-CMA2b, ACE, and AAAC-motif. Meanwhile, EuMADS63 in Type II (MIKC) subfamily contained the most CAREs (36), while EuMADS35 of Type I (M-type) subfamily had the fewest (5). Notably, a total of 568 phytohormone-response CAREs were detected, consisting of 172 abscisic acid (ABA), 51 auxin (IAA), 31 zein, 60 gibberellin (GA), 212 methyl jasmonate (MeJA), and 42 salicylic acid (SA) (Figure 6B).
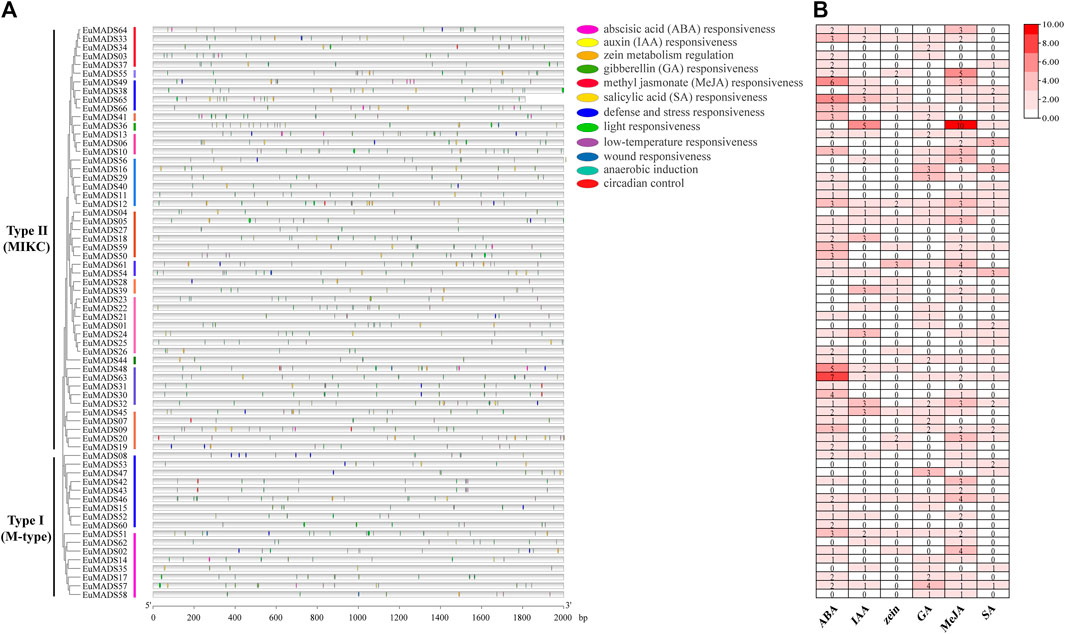
FIGURE 6. Predicted cis-acting regulatory elements for EuMADS genes. (A) The physical locations of cis-elements in the promoter region of each EuMADS gene, arranged by the phylogenetic tree. The 12 functional modules are shown as colored ellipse boxes. (B) Counts of phytohormone (ABA, IAA, zein, GA, MeJA, and SA) response cis-elements in each EuMADS gene.
2.4 Tissue- and sex-specific expression profiling and protein-interaction analysis of EuMADS TFs
To understand the biological functions of EuMADS TFs, expression patterns of all the 66 EuMADS genes were investigated in both reproductive (flower) and vegetative (leaf) tissues of male and female, via comparative transcriptome analysis. FPKM values were calculated to measure the expression level of each EuMADS gene. Overall, more EuMADS genes were expressed in flowers (59/66, 89.4%) than that in leaves (25/66, 37.9%, Figure 7). All the MIKC-EuMADS genes, excluding EuMADS19 were actively expressed in flowers. On the contrary, more members of FLC (3/5, EuMADS32, 48, and 63), SVP (6/7, EuMADS01, 21, 22, 23, 24, and 26), and SOC1 (4/6, EuMADS12, 16, 40, and 56) were preferred to express in leaves than in flowers.
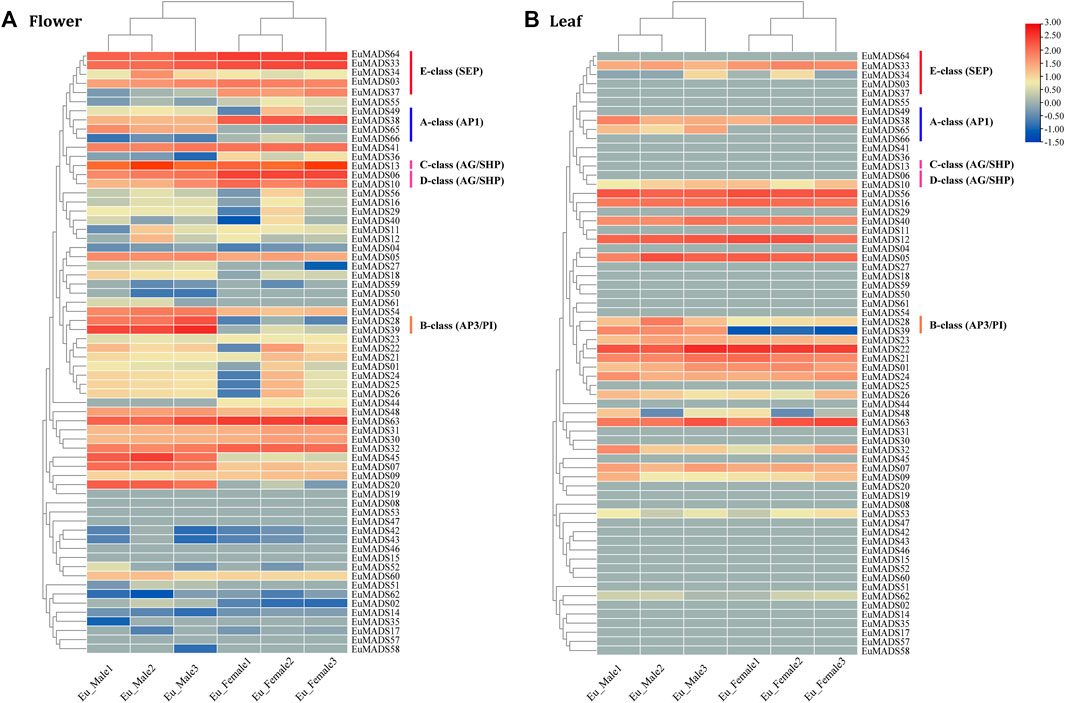
FIGURE 7. Expression profiling of EuMADS genes in tissues of (A) flower and (B) leaf. FPKM values of 66 EuMADS genes were calculated using transcriptome data with normalization. Log2(FPKM value) is displayed from low (blue) to high (red), indicating expression levels. A/B/C/D/E-class genes involved in flower development are highlighted by vertical lines in different colors.
Based on the criteria of a minimal two-fold difference in expression, i.e., log2 (fold change value) ≥1 or ≤ −1, and an adjusted p-value (padj) < 0.05, we found that 24 EuMADS genes were differentially expressed genes (DEGs) between the male and female flowers (Figure 7A, Supplementary Table S3). Among the 24 DEGs, 16 members (EuMADS02, 07, 13, 18, 20, 27, 28, 34, 39, 45, 48, 49, 51, 54, 61, and 65) displayed a male-biased expression pattern, while the rest eight (EuMADS06, 10, 36, 37, 38, 44, 55, and 66) were preferentially expressed in females. In contrast, only two genes, i.e., EuMADS39 (B-class gene) and EuMADS65 (A-class gene) were differentially expressed between the male and female leaves, both appeared male-biased expression (Figure 7B, Supplementary Table S3). Notably, the vast majority of DEGs (19/24, 79.2%) belonged to the MIKCC lineage of MIKC subfamily. This probably implied more important roles of MIKCC-EuMADS genes in floral sex differentiation of E. ulmoides.
Given that some EuMADS TFs may participate together in the same pathway, e.g., floral organ development, protein-interactive analysis was performed by STRING. The results showed that 30 out of 66 EuMADS proteins could interact with each other (Figure 8A, Supplementary Table S4). Among these putatively interacting proteins, 10 were involved in the floral organ ‘ABCDE model’ (Figure 8B). The two B-class genes (EuMADS28 and EuMADS39) were shown to interact with each other, and both of them could interact with the C-class gene (EuMADS13). Some E-class genes, e.g., EuMADS64 and EuMADS37 could interact with all the members of B/C/D-class (EuMADS06, 10, 13, 28, and 39), and two A-class genes (EuMADS65 and EuMADS66).
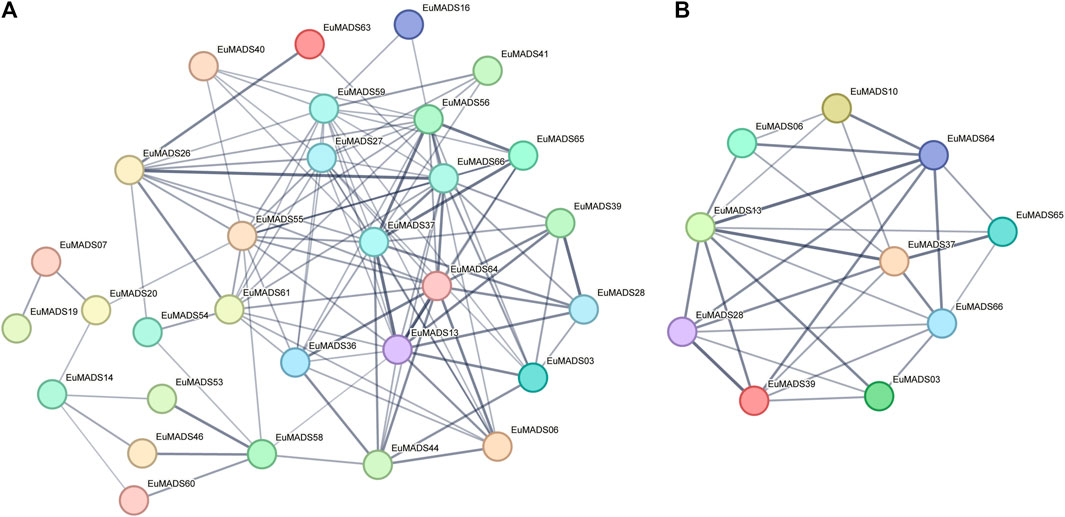
FIGURE 8. Putative interactions between EuMADS proteins. (A) Prediction of the protein interaction network among 66 EuMADS. (B) Prediction of the protein interaction network among 14 EuMADS involved in the floral organ ‘ABCDE model’.
2.5 Sex-biased expression pattern of floral organ ABCDE model-related genes
All the floral organ ABCDE model-related genes that were revealed active in flowers by transcriptome data (Figure 7) were further selected for qRT-PCR analysis. The result revealed most of A/B/C/D/E-class genes (11/14, 78.6%) were DEGs between the male and female flowers (Figure 9), in line with the above results of transcriptome analysis (Figure 7A, Supplementary Table S3). Remarkably, the expression patterns of most A/B/C/D/E-class genes between the male and female flowers were in accordance with their identities in determining floral organs. For example, B-class genes, i.e., EuMADS28 and EuMADS39 both highly expressed in the male flowers, but hardly expressed in the female flowers. Two D-class genes, EuMADS06 and EuMADS10 both showed female-biased expression patterns. One A-class gene (EuMADS66) and two E-class genes (EuMADS03 and EuMADS33) displayed similar expression level between the male and female flowers. Nevertheless, there were some exceptions, for instance, EuMADS49 and EuMADS65 of A-class were highly expressed in male flowers, while their closely relative EuMADS38 expressed preferably in female. C-class gene EuMADS13 was expressed in both male and female flowers, but with a higher expression level in male. In E-class, EuMADS34 showed male-biased expression, while EuMADS37 and EuMADS64 had male-biased expressions.
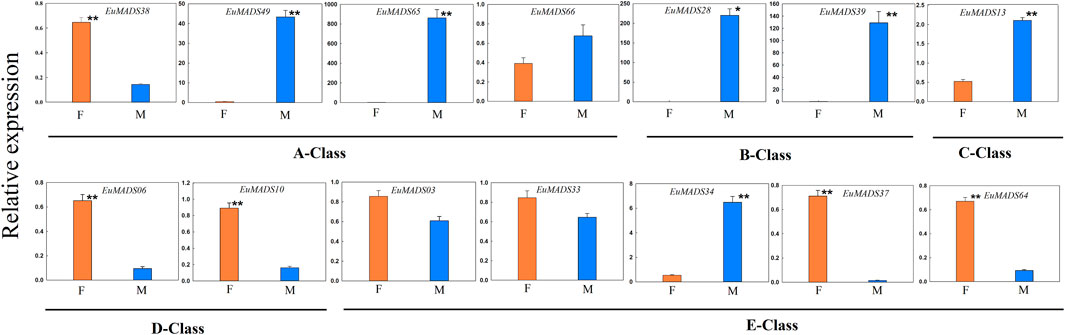
FIGURE 9. Quantitative real-time PCR (qRT-PCR) analyses of A/B/C/D/E-class EuMADS transcription factors in female (F) and male (M) flowers of Eucommia ulmoides. A-class genes (EuMADS38, EuMADS49, EuMADS65 and EuMADS66), B-class genes (EuMADS28 and EuMADS39), C-class gene (EuMADS13), D-class genes (EuMADS06 and EuMADS10) and E-class genes (EuMADS03, EuMADS33, EuMADS34, EuMADS37 and EuMADS64) are all underlined. The error bars were calculated from five biological replicates and two technical replicates. Significant differences between sexes are indicated with *(p < 0.05) and **(p < 0.01).
In addition, two B-class genes (EuMADS28 and EuMADS39) were also actively transcribed in the male leaves (Figure 6B). EuMADS39 that was homologous to AP3 of Arabidopsis and DEF of snapdragon (Antirrhinum majus L.) (Table 2) showed a male-biased expression pattern consistently in both flowers and leaves (Supplementary Tables S1; Figures 6, 7). Notably, EuMADS65 of A-class that shared homology with gene FUL in Arabidopsis and gene DEFH28 in snapdragon was also expressed specifically in both flower and leaf of male individuals. These two genes were therefore most probably involved in sex determination of E. ulmoides.
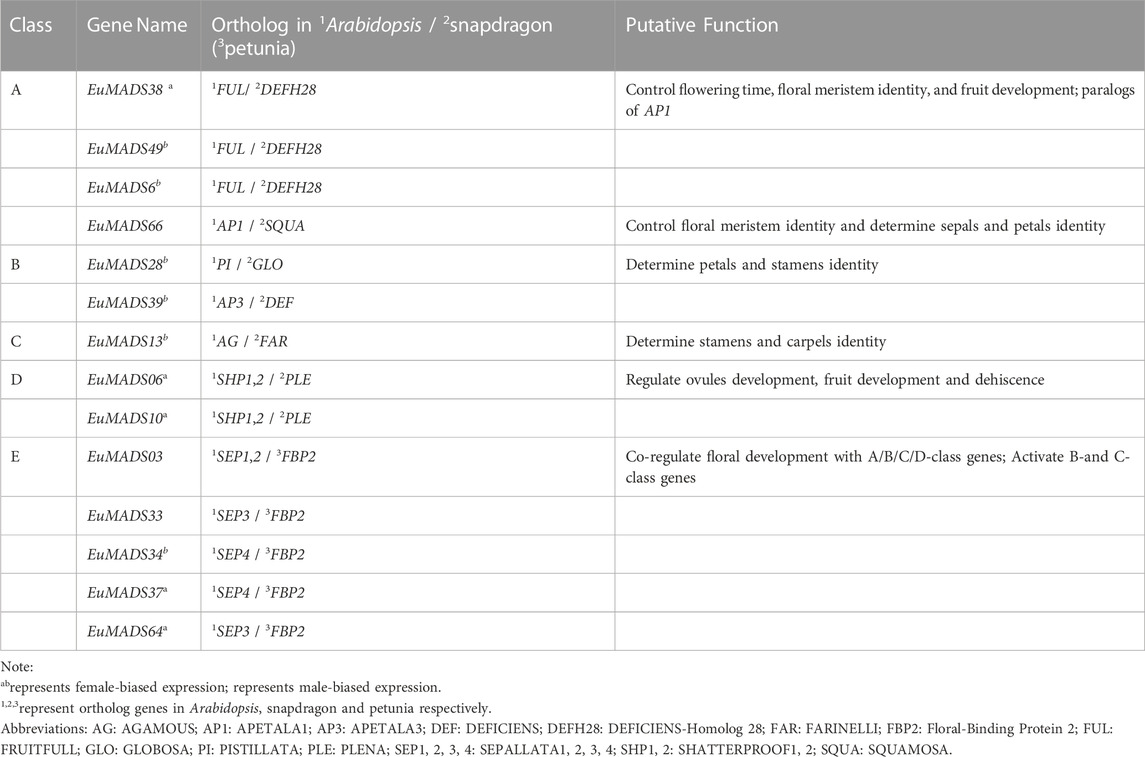
TABLE 2. Function annotation of EuMADS genes involved in ‘ABCDE model’ of flower development.
3 Discussion
3.1 Genome-wide characterization of MADS-box TFs in E. ulmoides
MADS-box TFs play foundational and indispensable roles in floral organogenesis and flowering of plants (Theien, 2001; Theissen et al., 2018). In present work, 66 MADS-box TFs of E. ulmoides were identified (Table 1), they were unevenly distributed across the genome (mainly on 14 chromosomes, Figure 1). The number of MADS-box genes identified in present work (66) is fewer than that in previous study (100) (Wang and Zhang, 2017). This inconsistency may be largely attributed to the alternative splicing of genes (Parenicová et al., 2003; Arora et al., 2007). In E. ulmoides, 49 MADS-box genes of the Type II (MIKC) subfamily averagely contained 7.8 exons per gene (Table 1; Figure 5), which likely would generate >49 transcripts due to different exon combinants during mRNA processing. Under such circumstance, prediction of more putative EuMADS unigenes solely based on transcriptome data is probable. For example, two putative MADS-box unigenes (Cluster-47702.5450 and Cluster-47702.5456) predicted previously (Wang and Zhang, 2017) were practically derived from the same gene locus of EuMADS65 (Supplementary Table S3) in present work. In addition, the quality of data assembling (both transcriptome and genome) and approaches employed are certainly essential for MADS-box family screening (Wang et al., 2019; Guan et al., 2021).
The MADS-box TF family has been studied in certain species with bisexual flowers, e.g., tomato (137 genes) (Wang et al., 2019), foxtail millet (89 genes) (Lai et al., 2022) and American beautyberry (78 genes) (Alhindi and Al-Abdallat, 2021), and a few species with unisexual flowers, such as litchi (L. chinensis, 101 genes) (Guan et al., 2021), hop (H. lupulus L., 65 genes) (Gutierrez et al., 2022) and Populus trichocarpa Torr. & A. Gray ex Hook (105 genes) (Leseberg et al., 2006). Interestingly, the size of MADS-box TF family identified in H. lupulus (65) (Gutierrez et al., 2022) is very close to the numbers of EuMADS TFs we found here. Neither male nor female flowers in E. ulmoides comprise sepal and petal, only have bracts encompassing 8–12 stamens (male) or one pistil (female) (Wang et al., 2020). Likewise, in H. lupulus the female flowers entirely lack sepal and petal, while male flowers comprise five sepals with five stamens inside (Shephard et al., 2000). Their resembling dioecious sexual system and unisexual floral composition may reflect similar family size of MADS-box TFs in E. ulmoides and H. lupulus, or vice versa.
The 66 EuMADS TFs were categorized into two groups, i.e., Type I (M-type) with 17 members and Type II (MIKC) with 49 members (Table 1) according to phylogenetic relationships (Figure 3). Subsequently, the 17 M-type EuMADS TFs were divided into two subgroups (Mα and Mγ), but no Mβ members were found (Figure 3), same as the result from American beautyberry (Callicarpa americana L.) (Alhindi and Al-Abdallat, 2021). Additionally, conserved protein domain analysis confirmed the presence of MADS domain in all the 66 EuMADS TFs and K-box domain in most MIKC-EuMADS TFs (Figure 5), as previously reported in tomato (Wang et al., 2019), physic nut (Tang et al., 2020), and cultivated alfalfa (Dong et al., 2021). Meanwhile, MIKC-EuMADS genes had more exons (7.8 per gene on average) than the M-type members (1.5 per gene on average, Table 1 and Figure 5). Promotor cis-element analysis also predicted more phytohormone-response elements MIKC-EuMADS genes (Figure 6, Supplementary Table S2). All of these results showed more complicated characteristics of MIKC-EuMADS TFs, suggesting more functional diversification of these genes (Ng and Yanofsky, 2001; Bemer et al., 2010; Lin et al., 2020).
The 49 MIKC-EuMADS TFs could be classified into 13 MIKCC subgroups (AG/SHP, AGL6, AGL12, AGL15, ANR1, AP1, AP3/PI, FLC, SEP, SOC1, SVP, TM8, and TT16) that are previously reported (Parenicová et al., 2003; Wang et al., 2019), and one MIKC* subgroup (Table 1; Figure 4). Notably, in A. thaliana, there were six FLC members, i.e., AtFLC and AtMAF1-5 (Figure 4, Supplementary Table S5). The AtFLC gene encodes a floral repressor to inhibit plant flowering (Theissen et al., 2018). When prolonged cold exposure in winter, the expression of AtFLC gene was epigenetically silenced by gene COOLAIR, leading to normal flowering, this is known as the vernalization flowering pathway (Hepworth and Dean, 2015). In the vernalization pathway, AtFLC represses the expression of FT (FLOWERING LOCUS T) and delays flowering (Hepworth and Dean, 2015). In certain species, e.g., hop (Gutierrez et al., 2022), foxtail millet (Lai et al., 2022) and litch (Guan et al., 2021), that require no vernalization for flowering, the AtFLC homologs are dispensable. As expected, the FLC lineage has been completely eliminated from hop (Gutierrez et al., 2022) and foxtail millet (Lai et al., 2022), and litchi only retained far fewer FLC members (1) compared to that (6) of Arabidopsis (Guan et al., 2021). E. ulmoides is a deciduous, temperate zone-growing tree, it would not flower without experiencing freezing winter (Wuyun et al., 2018; Li et al., 2020; Qing et al., 2021). It is thus easy to understand the presence of comparable number (5) of AtFLC homologs (EuMADS30, 31, 32, 48, and 63) in E. ulmoides (Table 1; Figure 4). Further functional analysis of these EuMADS genes would facilitate to understand the flowering regulation of E. ulmoides.
3.2 Essential position of floral organ ABCDE model-related genes in sex determination pathway of E. ulmoides
Sex determination of dioecious plants are usually reflected in floral phenotype, which are related with the expression of floral organ ABCDE model-related genes (Cronk and Muller, 2020; Zhang et al., 2022). In fig (Ficus hispida L.), a MADS-box gene FhAG2 was found locating at the sex determination region of Y chromosome as potential male activator (Zhang et al., 2020). Likewise, studies on cycad (Cycas panzhihuaensis L. Zhou and S. Y. Yang) (Liu et al., 2022), papaya (Carica papaya L.) (Yu et al., 2007), ginkgo (Ginkgo biloba L.) (Liao et al., 2020; Gong and Filatov, 2022), Nepenthes pitcher species (Scharmann et al., 2019), and Silene latifolia Poir. (Matsunaga et al., 2003). also suggested floral MADS-box genes were putative sex determination genes to exhibit sex-linked inheritance. In P. tremula L., ARR17 was a feminizing factor (Müller et al., 2020), CRISPR/Cas9-induced arr17 mutants upregulated B-class gene popPI to turn female individuals into male (Leite Montalvão et al., 2022). Similarly, in Diospyros lotus L., B-class gene DlPI was negatively controlled by the feminizing gene MeGI and a regulation cascade of OGI-MeGI-DlSVP-DlPI was proposed for sex determination in persimmon (Akagi et al., 2014; Yang et al., 2019).
Herein we found that 24 out of the 66 EuMADS genes were DEGs between the male and female flowers, meanwhile two EuMADS genes were also DEGs between the male and female leaves (Figure 7, Supplementary Table S3). It is worth noting that most (11/14, 78.6%) of the floral organ ABCDE model-related genes showed sex-biased expression patterns in E. ulmoides (Figures 7A, 9). Unisexual flowers can be produced when the spatiotemporal expression domain of B- and C-class MADS-box genes are overlapped (Irish, 2017; Jabbour et al., 2022). As expected, two B-class genes we identified in E. ulmoides, i.e., EuMADS28 and EuMADS39 (Table 1; Figure 4) were both highly expressed in the male flowers based on transcriptome analysis and qRT-PCR validation (Figures 7A, 9). Protein interactive analysis also predicted protein-protein interactions among B/C/E-class genes (Figure 8). In particular, EuMADS39 was expressed specifically in the male leaves too (Figure 7B), suggesting its constitutive-expression pattern in male individuals. This gene was previously described as EuAP3 (Wang and Zhang, 2017) by our group or EuDEF (Zhu, 2019) by another team, consistent with its functional annotation here (Table 2). More importantly, EuMADS39 was also physically adjacent to a male-linked molecular marker (MSL4) on Chr10 (Wang et al., 2020) and exhibited a persistently male-biased expression pattern during the whole period of floral bud differentiation (Qing et al., 2021). All these above clues lead us to hypothesize that B-class EuMADS genes, particularly EuMADS39, seem to participate in sex determination pathway of E. ulmoides as (in) direct players. Gene function verification of EuMADS39 is urgently needed in the future.
According to the classical ‘ABCDE model’, we know that A/C/D/E-class MADS-box genes direct the organogenesis of (sepal and petal)/(stamen and carpel)/ovule/the whole floral organ, respectively (Theien, 2001; Theissen and Saedler, 2001; Soltis et al., 2007). As expected, D-class genes (EuMADS06 and EuMADS10) both showed female-biased expression pattern (Figures 7A, 9), in line with its role in ovary development. However, expression patterns of certain A/C/E-class EuMADS genes unfit their identity as described in ‘ABCDE model’. For example, EuMADS65 (A-class gene), homologous to FUL in Arabidopsis and DEFH28 in snapdragon (Table 2), was specifically expressed in male individuals, both in the flowers and leaves (Figures 7, 9). In A. thaliana, FUL is paralog of AP1 and plays multiply functions in controlling flowering time, meristem differentiation, and flower development, etc. (Ng and Yanofsky, 2001; Parenicová et al., 2003; Soltis et al., 2007). There are different flower arrangements between male and female individuals of E. ulmoides, with the male flowers forming a capitulum inflorescence while the female flowers being solitary on branch (Wang et al., 2020; Qing et al., 2021), similar to that in persimmon (Akagi et al., 2014; Yang et al., 2019) and kiwifruit (Akagi et al., 2018; Varkonyi-Gasic et al., 2021). It is thus possible for A-class EuMADS65 functioning in determining the male flower inflorescence.
Notably, neither sepals nor petals were present in E. ulmoides female flowers (Wang et al., 2020; Qing et al., 2021). Given that A + B + E genes determine the petal identity (Theien, 2001; Theissen and Saedler, 2001; Soltis et al., 2007), the silence of B-class genes (EuMADS28 and EuMADS39) in E. ulmoides female flowers (Figures 7A, 9) may partly contribute to the absence of petals. Meanwhile, the four A-class genes (EuMADS38, EuMADS49, EuMADS65 and EuMADS66) showed varied expression patterns between the male and female flowers of E. ulmoides (Figures 7A, 9) and different protein-protein interactions (Figure 8), suggesting their functional differentiation during evolution. This may lead to the loss of ancestral function of A-class genes in determining sepal and petal identity, resuling in the absence of both sepals and petals in E. ulmoides female flowers. Additionally, the number of stamens (8–12) in each male flower is much larger than that of carpel (only 1) in each female flower of E. ulmoides (Zhu, 2019; Wang et al., 2020), as in poplar (Xue et al., 2020) and willow (Wang et al., 2022). This probably leades to higher expression level of C-class gene (EuMADS13) in male flowers than that in female flowers (Figures 7A, 9). Further functional analysis of A-class genes (e.g., EuMADS65) and the C-class gene (EuMADS13) is required to understand their roles in sex determination pathway of E. ulmoides.
4 Materials and methods
4.1 Identification of MADS-Box TFs, genome mapping and synteny analysis
The high-quality haploid genome of E. ulmoides (Li et al., 2020) was downloaded from NCBI (https://www.ncbi.nlm.nih.gov/data-hub/genome/GCA_016647705.1/), together with a male reference genome (Wuyun et al., 2018) from Genome Warehouse (https://ngdc.cncb.ac.cn/gwh/Assembly/13/show). The MADS-box protein sequences of A. thaliana (https://www.arabidopsis.org/) and rice (Oryza sativa L., http://rice.plantbiology.msu.edu/) were separately downloaded on 13 June 2022 (Additional Data1). Two methods were independently used to identify MADS-box TFs in E. ulmoides. In one approach, all the annotated E. ulmoides proteins were reciprocally BLASTed (Camacho et al., 2009) with AtMADS and OsMADS TFs, the best hits (score value ≥100 and e-value ≤ 1e−10) yielded were selected as candidate MADS-box TFs. In the other method, conserved SRF (PF00319) or MEF2 (PF09047) domains were retrieved from Pfam (Mistry et al., 2021) and used to identify the candidate MADS-box TFs in E. ulmoides by Hidden Markov Model (HMM) search in HMMER v. 3.0 (Potter et al., 2018). The shared sequences resulted from the above two methods were further validated by Conserved Domain Database of NCBI (CDD, http://www.ncbi.nlm.nih.gov/cdd/) and Simple Modular Architecture Research Tool (SMART, http://smart.embl-heidelberg.de/) (Letunic et al., 2012). Eventually, 66 non-redundant MADS-box TFs were obtained and named as EuMADS01 to 66. The online tool ExPasy (https://web.expasy.org/compute_pi/) was employed to determine their physicochemical properties, including length of amino acid (aa), isoelectric points (PI) and molecular weight (MW, kDa). Phosphorylation sites, i.e., Ser, Tyr and Thr sites, of EuMADS TFs were counted in Geneious V9.0 (Kearse et al., 2012). Subcellular localizations of EuMADS TFs were further predicted by Plant-mPLoc (http://www.csbio.sjtu.edu.cn/bioinf/plant-multi/) (Chou and Shen, 2010).
Based on GFF3 file of genome annotation (https://www.ncbi.nlm.nih.gov/data-hub/genome/GCA_016647705.1/), the physical positions of 66 EuMADS genes were mapped against the assembled 17 chromosomes of E. ulmoides (Li et al., 2020). TBtools software (Chen et al., 2020) was used for visualization of genes on chromosomes. The number of EuMADS genes per chromosome was then counted and plotted. Moreover, the information of length and gene-density of 17 chromosomes of E. ulmoides were obtained from the reference genome (Li et al., 2020). The multiple collinearity scan toolkit X (MCScanX) program (Wang et al., 2012) was performed with default parameters for collinearity analysis among the 66 EuMADS genes. Then the circos map was drawn using TBtools software (Chen et al., 2020). Similarly, the homology of these EuMADS genes among E. ulmoides, A. thaliana, and O. sativa were also analyzed using MCScanX (Wang et al., 2012). The synteny genome blocks between species were then visualized in TBtools (Chen et al., 2020).
4.2 Phylogenetic analysis and classification of EuMADS TFs
MADS-box TFs from A. thaliana (Parenicová et al., 2003), rice (O. sativa) (Arora et al., 2007) and tomato (Solanum lycopersicum L.) (Wang et al., 2019) were used for the classification of MADS-box family in E. ulmoides. Full-length protein sequences of AtMADS and EuMADS (Additional Data1, Supplementary Table S5) were aligned by using MUSCLE program (Edgar, 2004) in Geneious V9.0 (Kearse et al., 2012) with default parameters. An unrooted maximum likelihood (ML) phylogenetic tree was constructed by MEGA-X software (Kumar et al., 2018), with 1,000 bootstrap replicates under JTT model. The gaps and missing data were manually checked and processed by partial deletion (Wang and Zhang, 2017; Wang et al., 2018). Type I (M-type) and Type II (MIKC) MADS-box TFs of E. ulmoides were determined based on the phylogenetic tree of AtMADS and EuMADS.
Subsequently, to better categorize the EuMADS TFs of MIKC subfamily (MIKC-EuMADS), MIKC protein sequences of tomato (MIKC-SolyMADS), Arabidopsis (MIKC-AtMADS) and rice (MIKC-OsMADS) were all utilized. MIKC-SolyMADS were downloaded from PlantTFDB v5.0 database (Jin et al., 2014) and aligned with MIKC-AtMADS, MIKC-OsMADS and MIKC-EuMADS (Additional Data1, Supplementary Table S5) using the MUSCLE tool (Edgar, 2004). MEGA-X software (Kumar et al., 2018) was applied to infer the ML tree under JTT model with a bootstrap value of 1,000. The resulted ML tree was visualized in FigTree viewer (http://tree.bio.ed.ac.uk/software/figtree/). The MIKC-EuMADS TFs were then classified into different subgroups following the phylogenetic relationships with MIKC-AtMADS, MIKC-OsMADS and MIKC-SlMADS. The subgroups were named following available classification systems in Arabidopsis and tomato (Parenicová et al., 2003; Arora et al., 2007; Wang et al., 2019).
4.3 Protein domain, gene structure, and cis-element analyses of EuMADS TFs
Conserved protein domains of the 66 EuMADS TFs were blasted in the NCBI-CDD website (https://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi) (Marchler-Bauer et al., 2015). The online Multiple Em for Motif Elicitation (MEME) program (http://meme-suite.org/tools/meme) (Bailey et al., 2009) was also employed to analyze the protein motifs of EuMADS TFs. Parameters for MEME search were set as follows: maximum number of motifs = 20, motif width = 6–60, number of repetitions = 0/1, according to our knowledge of plant MADS-box proteins (Wang et al., 2019; Alhindi and Al-Abdallat, 2021; Dong et al., 2021; Gutierrez et al., 2022). Moreover, exon-intron structures of the EuMADS genes were constructed based on the alignment between the full-length coding sequences (CDS) and the corresponding genomic sequences (Wuyun et al., 2018; Li et al., 2020). The annotation information for each EuMADS gene was also extracted from the GFF3 files (PRJNA599775 and PRJCA000677) of the genome data to verify the constructed gene structure. The protein domains, motif composition and gene structure of EuMADS TFs were then visualized using TBtools (Chen et al., 2020).
The 2,000 bp promotor sequences upstream the initiation codon (ATG) of each EuMADS gene were extracted from the genome data (Wuyun et al., 2018; Li et al., 2020) in TBtools (Chen et al., 2020). The online PlantCARE tool (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) was then performed for the cis-acting regulatory element (CARE) prediction. The cis-elements related to phytohormone responsiveness, defense and stress responsiveness, light responsiveness, low-temperature responsiveness, wound responsiveness, anaerobic induction, and circadian control were analyzed. In the phytohormone-response cis-elements, ABRE was involved in ABA-responsiveness, AuxRR-core and TGA-element were involved in IAA-responsiveness, O2-site was involved in zein-metabolism, GARE-motif, TATC-box and P-box were involved in GA-responsiveness, TGACG-motif and CGTCA-motif were involved in MeJA-responsiveness, and TCA-element was involved in SA-responsiveness (Alhindi and Al-Abdallat, 2021; Ye et al., 2022) The location of cis-elements in promoter region of each EuMADS gene was visualized in TBtools (Chen et al., 2020). Cis-elements associated with phytohormones (ABA, IAA, zein, GA, MeJA, and SA) were also counted and plotted in TBtools (Chen et al., 2020).
4.4 Tissue- and sex-specific expression analysis of EuMADS TFs by RNA-seq and protein-interaction prediction
To reveal the expression patterns of MADS-box genes in both vegetative and reproductive tissues of E. ulmoides, leaves and flowers from multiple individuals of male and female were sampled for transcriptome analysis. The RNA-seq data of male and female leaves with three biological replicates were from our previous work (Wang and Zhang, 2017). Male and female flowers from the same corresponding trees that offered leaf tissues were collected in April 2021. Flower samples were named as Eu_Male1, Eu_Male2, Eu_Male3 and Eu_Female1, Eu_Female2, Eu_Female3. Total RNA was extracted using RNeasy Plant Mini Kit (74904, Qiagen, German). mRNA library construction and Illumina sequencing were conducted according to manufacturer’s procedures at BGI Technologies Corporation (Shenzhen, China).
About 6 Gb clean reads were generated for each sample after quality control. These reads were then used to quantify the expression level of each EuMADS gene using RSEM software (Li and Dewey, 2011). FPKM (fragments per kilobase of transcript per million mapped) values of EuMADS genes were calculated from the uniquely mapped reads (Zhao et al., 2021). For each tissue, i.e., flower and leaf, reads from male and female individuals were batched into one dataset, respectively for differential expression analysis with DESeq R package v1.10.1 (Anders, 2010). Benjamini and Hochberg’s approach was employed to adjust the resulting p-values for false discovery rate (FDR) control (Benjamini and Hochberg, 1995). Genes with log2 (fold change value) ≥ 1 or ≤ −1, and padj < 0.05 were considered as differentially expressed genes (DEGs) (Wang and Zhang, 2017; Guan et al., 2021), i.e., male- or female-biased expression. Flower RNA-seq data newly generated in this study were deposited in SRA database (accession number: PRJNA399774).
Given that MADS-box proteins often form dimers or polymers with each other to play roles in plant growth and development (Theissen and Saedler, 2001; Theißen et al., 2016; Ruelens et al., 2017). The protein sequences of 66 EuMADS TFs were submitted to the STRING website (https://cn.string-db.org/) (Szklarczyk et al., 2015) for protein interactions prediction. Then the protein-protein interaction network was exported and beautified by the Adobe Illustrator tool (McLean, 2002). The protein interactions of the identified 14 A/B/C/D/E-class EuMADS genes was also predicted in the same way.
4.5 Sex-biased expression analysis of floral organ ABCDE model-related genes by qRT-PCR
Ten flower samples were respectively collected from five male and five female E. ulmoides individual trees growing on the campus of Northwest A&F University, Yangling, China (34°16′56″N, 108°04′27″E) in April 2021 for quantitative Real-Time PCR (qRT-PCR) analysis. The collected male and female flowers was about to open. Because there are no sepal and petal in either male or female flowers (Wang and Zhang, 2017; Wuyun et al., 2018; Zhu, 2019), only stamens and pistils were isolated and immediately immersed into liquid nitrogen before stored at −80°C until use. Total RNA extraction and cDNA synthesization were performed using RNeasy Plant Mini Kit (74904, Qiagen, German) and SuperScript™ IV VILO™ Master Mix (Thermo Fisher, United States of) respectively following the manufacturer’s instructions.
qRT-PCR analysis was conducted according to the manuals of Hieff qRT-PCR SYBR Green Master Mix (YEASEN, Shanghai, China) on a LightCycler 480 II Real-Time PCR Platform (Roche, Germany). The reaction system (20 μL) consisted of 10.0 μL of qRT-PCR Mix, 1.0 μL cDNA, 0.4 μL forward primer, 0.4 μL reverse primer, and 8.2 μL of ddH2O. EuGAPDH gene was used as internal control for data normalization (Liu et al., 2018). 2−ΔΔCT method was applied to calculate the relative gene expression of 14 A/B/C/D/E-class EuMADS genes (Livak and Schmittgen, 2001). Primers used for EuMADS and EuGAPDH genes (Supplementary Table S6) were designed using the online program Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/primer3/). Five biological replicates and two technical replicates were carried on for each gene. Differences of gene expression level between male and female flowers were analyzed via ANOVA followed by Student’s t-test in SPSS software (v.24, IBM).
The identified 14 A/B/C/D/E-class EuMADS genes were also functionally annotated by the online BLAST in NCBI database (https://blast.ncbi.nlm.nih.gov/Blast.cgi). The most similar genes in model plants (Arabidopsis, snapdragon/petunia) of flower development studies (Theißen et al., 2016; Irish, 2017; Ruelens et al., 2017) were screened as orthologous genes to predict gene function.
5 Conclusion
In this study, genome-wide comprehensive analyses of the MADS-box TF family in E. ulmoides were performed for the first time. A total of 66 EuMADS TFs were identified, including 49 Type II (MIKC) members and 17 Type I (M-type) members. All the previously recorded 13 MIKCC subgroups were detected in E. ulmoides, but the Mβ lineage was missing. More complicated protein-motif composition, exon-intron architecture, and phytohormone-response cis-elements were found in MICK-EuMADS TFs, suggesting more diverse biological functions in these genes. Furthermore, tissue- and sex-specific transcriptome analyses revealed 24 DEGs between male and female flowers, and two DEGs between male and female leaves. qRT-PCR validation of the 14 EuMADS genes involved in the floral organ ‘ABCDE model’ showed six male-biased A/B/C/E-class genes, five female-biased A/D/E-class genes and three non-biased A/E-class genes in E. ulmoides flowers. Notably, the B-class gene EuMADS39 (ortholog of AtAP3) and the A-class gene EuMADS65 (ortholog of AtFUL) were significantly expressed in the male individuals, no matter in flower or leaf tissues. In short, these results suggested essential roles of floral organ ABCDE model-related MADS-box TFs in sex determination of E. ulmoides, as demonstrated in dioecious poplar (Leite Montalvão et al., 2022), persimmon (Yang et al., 2019) and kiwifruit (Ye et al., 2022) recently.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
XZ: Data analysis and original draft; XW, LP, WG, and YL: sample collection and data analysis; WW: Inputs and revision. All authors read and approved the submitted version.
Funding
Rural Revitalization Project of Guangdong Province (KTP20210181) and Innovation Project for Forestry Science and Technology in Guangdong (2021KJCX015).
Acknowledgments
We would like to thank Dr. Yun Li and Prof. Xiangyang Kang from Beijing Forestry University for the help in genome data collection.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1138703/full#supplementary-material
References
Akagi, T., Henry, I. M., Ohtani, H., Morimoto, T., Beppu, K., Kataoka, I., et al. (2018). A Y-encoded suppressor of feminization arose via lineage-specific duplication of a cytokinin response regulator in kiwifruit. Plant Cell 30, 780–795. doi:10.1105/tpc.17.00787
Akagi, T., Henry, I. M., Tao, R., and Comai, L. A. (2014). Plant genetics. A Y-chromosome-encoded small RNA acts as a sex determinant in persimmons. Science 346, 646–650. doi:10.1126/science.1257225
Alhindi, T., and Al-Abdallat, A. M. (2021). Genome-wide identification and analysis of the MADS-box gene family in American beautyberry (Callicarpa americana). Plants 10, 1805. doi:10.3390/plants10091805
Arora, R., Agarwal, P., Ray, S., Singh, A. K., Singh, V. P., Tyagi, A. K., et al. (2007). MADS-Box gene family in rice: Genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genom 8, 242. doi:10.1186/1471-2164-8-242
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., et al. (2009). Meme suite: Tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208. doi:10.1093/nar/gkp335
Bemer, M., Heijmans, K., Airoldi, C., Davies, B., and Angenent, G. C. (2010). An atlas of type I MADS box gene expression during female gametophyte and seed development in Arabidopsis. Plant Physiol. 154, 287–300. doi:10.1104/pp.110.160770
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57, 289–300. doi:10.1111/j.2517-6161.1995.tb02031.x
Camacho, C. G., Coulouris, G., Avagyan, V., Ma, N., and Papadopoulos, J. (2009). Blast plus: Architecture and applications. BMC Bioinform 10, 1–9.
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Xia, R., He, Y., et al. (2020). TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi:10.1016/j.molp.2020.06.009
Chou, K., and Shen, H. (2010). Plant-mPLoc: A top-down strategy to augment the power for predicting plant protein subcellular localization. PLoS ONE 5, e11335. doi:10.1371/journal.pone.0011335
Cronk, Q., and Muller, N. A. (2020). Default sex and single gene sex determination in dioecious plants. Front. Plant Sci. 11, 1162. doi:10.3389/fpls.2020.01162
Di Stilio, V. S., Kramer, E. M., and Baum, D. A. (2005). Floral MADS box genes and homeotic gender dimorphism in Thalictrum dioicum (Ranunculaceae)-a new model for the study of dioecy. Plant J. 41, 755–766. doi:10.1111/j.1365-313X.2005.02336.x
Diggle, P. K., Stilio, D., Gschwend, V. S., Golenberg, A. R., Moore, E. M., Russell, J. R. W., et al. (2011). Multiple developmental processes underlie sex differentiation in angiosperms. Trends Genet 27, 368–376. doi:10.1016/j.tig.2011.05.003
Dong, X., Deng, H., Ma, W., Zhou, Q., and Liu, Z. (2021). Genome-wide identification of the MADS-box transcription factor family in autotetraploid cultivated alfalfa (Medicago sativa L.) and expression analysis under abiotic stress. BMC Genom 22, 603. doi:10.1186/s12864-021-07911-9
Edgar, R. C. (2004). Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi:10.1093/nar/gkh340
Gong, W., and Filatov, D. A. (2022). Evolution of the sex-determining region in Ginkgo biloba. Ginkgo Biloba. Philos. Trans. R. Soc. B 377, 20210229. doi:10.1098/rstb.2021.0229
Guan, H., Wang, H., Huang, J., Liu, M., Chen, T., Shan, X., et al. (2021). Genome-wide identification and expression analysis of MADS-box family genes in litchi (Litchi chinensis Sonn.) and their involvement in floral sex determination. Plants 10, 2142. doi:10.3390/plants10102142
Gutierrez, R. M., Ribeiro, T. H. C., de Oliveira, R. R., Benedito, V. A., and Chalfun-Junior, A. (2022). Genome-wide analyses of MADS-box genes in Humulus lupulus L. reveal potential participation in plant development, floral architecture, and lupulin gland metabolism. Plants 11, 1237. doi:10.3390/plants11091237
Hepworth, J., and Dean, C. (2015). Flowering locus C’s lessons: Conserved chromatin switches underpinning developmental timing and adaptation. Plant Physiol. 168, 1237–1245. doi:10.1104/pp.15.00496
Irish, V. (2017). The ABC model of floral development. Curr. Biol. 27, R887–R890. doi:10.1016/j.cub.2017.03.045
Jabbour, F., Espinosa, F., Dejonghe, Q., and Le Péchon, T. (2022). Development and evolution of unisexual flowers: A review. Plants 11, 155. doi:10.3390/plants11020155
Jin, J., Zhang, H., Kong, L., Gao, G., and Luo, J. (2014). PlantTFDB 3.0: A portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 42, D1182–D1187. doi:10.1093/nar/gkt1016
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi:10.1093/bioinformatics/bts199
Kumar, S., Stecher, G., Li, M., Knyaz, C., Tamura, K., Battistuzzi, F. U., et al. (2018). Mega X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi:10.1093/molbev/msy096
Lai, D., Yan, J., He, A., Xue, G., Yang, H., Feng, L., et al. (2022). Genome-wide identification, phylogenetic and expression pattern analysis of MADS-box family genes in foxtail millet (Setaria italica). Sci. Rep. 12, 4979. doi:10.1038/s41598-022-07103-9
Larue, N. C., Sullivan, A. M., and Di Stilio, V. S. (2013). Functional recapitulation of transitions in sexual systems by homeosis during the evolution of dioecy in Thalictrum. Thalictrum. Front. Plant Sci. 4, 487. doi:10.3389/fpls.2013.00487
Leite Montalvão, A. P., Kersten, B., Kim, G., Fladung, M., and Müller, N. A. (2022). ARR17 controls dioecy in Populus by repressing B-class MADS-box gene expression. Philos. Trans. R. Soc. B 377, 20210217. doi:10.1098/rstb.2021.0217
Leseberg, C. H., Li, A., Kang, H., Duvall, M., and Mao, L. (2006). Genome-wide analysis of the MADS-box gene family in Populus trichocarpa. Popul. Trichocarpa. Gene 378, 84–94. doi:10.1016/j.gene.2006.05.022
Letunic, I., Doerks, T., and Bork, P. (2012). Smart 7: Recent updates to the protein domain annotation resource. Nucleic Acids Res. 40, D302–D305. doi:10.1093/nar/gkr931
Li, B., and Dewey, C. N. (2011). Rsem: Accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinform 12, 323–399. doi:10.1186/1471-2105-12-323
Li, Y., Wei, H., Yang, J., Du, K., Li, J., Zhang, Y., et al. (2020). High-quality de novo assembly of the Eucommia ulmoides haploid genome provides new insights into evolution and rubber biosynthesis. Hortic. Res. 7, 183. doi:10.1038/s41438-020-00406-w
Liao, Q., Du, R., Gou, J., Guo, L., Shen, H., Liu, H. L., et al. (2020). The genomic architecture of the sex-determining region and sex-related metabolic variation in Ginkgobiloba. Ginkgo biloba Plant J. 104, 1399–1409. doi:10.1111/tpj.15009
Lin, Z., Gao, D., Damaris, R. N., and Yang, P. (2020). Genome-wide identification of MADS-box gene family in sacred lotus (Nelumbo nucifera) identifies a SEPALLATA homolog gene involved in floral development. BMC Plant Biol. 20, 497. doi:10.1186/s12870-020-02712-w
Liu, H., Lu, Y., Wang, J., and Wuyun, T. (2018). Genome-wide screening of long non-coding RNAs involved in rubber biosynthesis in Eucommia ulmoides. J. Integr. Plant. Biol. 60, 1070–1082. doi:10.1111/jipb.12693
Liu, Y., Wang, S., Li, L., Yang, T., Dong, S., Wei, T., et al. (2022). The Cycas genome and the early evolution of seed plants. Nat. Plants 8, 389–401. doi:10.1038/s41477-022-01129-7
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25, 402–408. doi:10.1006/meth.2001.1262
Marchler-Bauer, A., Derbyshire, M. K., Gonzales, N. R., Lu, S., Farideh, C., Geer, L. Y., et al. (2015). Cdd: NCBI’s conserved domain database. Nucleic Acids Res. 43, D222–D226. doi:10.1093/nar/gku1221
Matsunaga, S., Isono, E., Kejnovsky, E., Vyskot, B., Dolezel, J., Kawano, S., et al. (2003). Duplicative transfer of a MADS box gene to a plant Y chromosome. Mol. Biol. Evol. 20, 1062–1069. doi:10.1093/molbev/msg114
McLean, D. (2002). Adobe photoshop and illustrator techniques. J. Audiov. Media Med. 25, 79–81. doi:10.1080/01405110220140865
Mistry, J., Chuguransky, S., Williams, L., Qureshi, M., Salazar, G. A., Sonnhammer, E. L. L., et al. (2021). Pfam: The protein families database in 2021. Nucleic Acids Res. 49, D412–D419. doi:10.1093/nar/gkaa913
Müller, N. A., Kersten, B., Leite Montalvão, A. P., Mähler, N., Bernhardsson, C., Bräutigam, K., et al. (2020). A single gene underlies the dynamic evolution of poplar sex determination. Nat. Plants 6, 630–637. doi:10.1038/s41477-020-0672-9
Ng, M., and Yanofsky, M. F. (2001). Function and evolution of the plant MADS-box gene family. Nat. Rev. Genet. 2, 186–195. doi:10.1038/35056041
Ouyang, D., Wang, L., Tang, T., and Feng, H. (2021). Genomic-wide identification and characterization of the uridine diphosphate glycosyltransferase family in Eucommia ulmoides Oliver. Plants 10, 1934. doi:10.3390/plants10091934
Parenicová, L., De, F. S., Kieffer, M., Horner, D. S., Favalli, C., Busscher, J., et al. (2003). Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: New openings to the MADS world. Plant Cell 15, 1538–1551. doi:10.1105/tpc.011544
Pfent, C., Pobursky, K. J., Sather, D. N., and Golenberg, E. M. (2005). Characterization of SpAPETALA3 and SpPISTILLATA, B class floral identity genes in Spinacia oleracea, and their relationship to sexual dimorphism. Dev. Genes Evol. 215, 132–142. doi:10.1007/s00427-004-0459-4
Potter, S. C., Luciani, A., Eddy, S. R., Park, Y., Lopez, R., and Finn, R. D. (2018). HMMER web server: 2018 update. Nucleic Acids Res. 46, W200–W204. doi:10.1093/nar/gky448
Qing, J., Zhu, L., and Du, H. (2021). Flower bud differentiation and dynamic changes of endogenous hormone in Eucommia ulmoides. J. Central South Univ. For. Technol. 41, 58–65. (in Chinese).
Ruelens, P., Zhang, Z., Mourik, H. V., Maere, S., Kaufmann, K., and Geuten, K. (2017). The origin of floral organ identity quartets. Plant Cell 29, 229–242. doi:10.1105/tpc.16.00366
Sather, D. N., Jovanovic, M., and Golenberg, E. M. (2010). Functional analysis of B and C class floral organ genes in spinach demonstrates their role in sexual dimorphism. BMC Plant Biol. 10, 46–14. doi:10.1186/1471-2229-10-46
Scharmann, M., Grafe, T. U., Metali, F., and Widmer, A. (2019). Sex is determined by XY chromosomes across the radiation of dioecious Nepenthes pitcher plants. Evol. Lett. 3, 586–597. doi:10.1002/evl3.142
Shephard, H. L., Parker, J. S., Darby, P., and Ainsworth, C. C. (2000). Sexual development and sex chromosomes in hop. New Phytol. 148, 397–411. doi:10.1046/j.1469-8137.2000.00771.x
Sobral, R., and Costa, M. M. R. (2017). Role of floral organ identity genes in the development of unisexual flowers of Quercus suber L. Sci. Rep. 7, 10368–10415. doi:10.1038/s41598-017-10732-0
Soltis, D. E., Ma, H., Frohlich, M. W., Soltis, P. S., Albert, V. A., Oppenheimer, D. G., et al. (2007). The floral genome: An evolutionary history of gene duplication and shifting patterns of gene expression. Trends Plant Sci. 12, 358–367. doi:10.1016/j.tplants.2007.06.012
Szklarczyk, D., Franceschini, A., Wyder, S., Forslund, K., Heller, D., Huerta-Cepas, J., et al. (2015). STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452. doi:10.1093/nar/gku1003
Tang, Y., Wang, J., Bao, X., Wu, Q., Yang, T., Li, H., et al. (2020). Genome-wide analysis of Jatropha curcas MADS-box gene family and functional characterization of the JcMADS40 gene in transgenic rice. BMC Genom 21, 325. doi:10.1186/s12864-020-6741-7
Theien, G. (2001). Development of floral organ identity: Stories from the MADS house. Curr. Opin. Plant Biol. 4, 75–85. doi:10.1016/s1369-5266(00)00139-4
Theissen, G., Rumpler, F., and Gramzow, L. (2018). Array of MADS-box genes: Facilitator for rapid adaptation? Trends Plant Sci. 23, 563–576. doi:10.1016/j.tplants.2018.04.008
Theissen, G., and Saedler, H. (2001). Plant biology. Floral quartets. Nature 409, 469–471. doi:10.1038/35054172
Theißen, G., Melzer, R., and Rümpler, F. (2016). MADS-Domain transcription factors and the floral quartet model of flower development: Linking plant development and evolution. Development 143, 3259–3271. doi:10.1242/dev.134080
Varkonyi-Gasic, E., Wang, T., Cooney, J., Jeon, S., Voogd, C., Douglas, M. J., et al. (2021). Shy Girl, a kiwifruit suppressor of feminization, restricts gynoecium development via regulation of cytokinin metabolism and signalling. New Phytol. 230, 1461–1475. doi:10.1111/nph.17234
Wang, D., Li, M., Li, Y., Yang, W., Ma, X., Zhang, L., et al. (2022). Repeated turnovers keep sex chromosomes young in willows. Genome Biol. 23, 200. doi:10.1186/s13059-022-02769-w
Wang, W., Chen, S., and Zhang, X. (2018). Whole-genome comparison reveals heterogeneous divergence and mutation hotspots in chloroplast genome of Eucommia ulmoides Oliver. Int. J. Mol. Sci. 19, 1037. doi:10.3390/ijms19041037
Wang, W., Yang, G., Deng, X., Shao, F., Li, Y., Guo, W., et al. (2020). Molecular sex identification in the hardy rubber tree (Eucommia ulmoides Oliver) via ddRAD markers. Int. J. Genomics 2020, 2420976. doi:10.1155/2020/2420976
Wang, W., and Zhang, X. (2017). Identification of the sex-biased gene expression and putative sex-associated genes in Eucommia ulmoides Oliver using comparative transcriptome analyses. Molecules 22, 2255. doi:10.3390/molecules22122255
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49. doi:10.1093/nar/gkr1293
Wang, Y., Zhang, J., Hu, Z., Guo, X., Tian, S., and Chen, G. (2019). Genome-wide analysis of the MADS-box transcription factor family in Solanum lycopersicum. Solanum Lycopersicum. Int. J. Mol. Sci. 20, 2961. doi:10.3390/ijms20122961
Wuyun, T. N., Wang, L., Liu, H., Wang, X., Zhang, L., Bennetzen, J. L., et al. (2018). The hardy rubber tree genome provides insights into the evolution of polyisoprene biosynthesis. Mol. Plant 11, 429–442. doi:10.1016/j.molp.2017.11.014
Xue, L., Wu, H., Chen, Y., Li, X., Hou, J., Lu, J., et al. (2020). Evidences for a role of two Y-specific genes in sex determination in Populus deltoides. Nat. Commun. 11, 5893–5912. doi:10.1038/s41467-020-19559-2
Yang, H. W., Akagi, T., Kawakatsu, T., and Tao, R. (2019). Gene networks orchestrated by MeGI: A single-factor mechanism underlying sex determination in persimmon. Plant J. 98, 97–111. doi:10.1111/tpj.14202
Ye, L., Luo, M. M., Wang, Z., Bai, F., Luo, X., Gao, L., et al. (2022). Genome-wide analysis of MADS-box gene family in kiwifruit (Actinidia chinensis var. chinensis) and their potential role in floral sex differentiation. Front. Genet. 13, 1043178. doi:10.3389/fgene.2022.1043178
Yu, Q., Steiger, D., Kramer, E. M., Moore, P. H., and Ming, R. (2007). Floral MADS-box genes in trioecious papaya: Characterization of AG and AP1 subfamily genes revealed a sex-type-specific gene. Trop. Plant Biol. 1, 97–107. doi:10.1007/s12042-007-9000-z
Zhang, X., Pan, L., Guo, W., Li, Y., and Wang, W. (2022). A convergent mechanism of sex determination in dioecious plants: Distinct sex-determining genes display converged regulation on floral B-class genes. Front. Plant Sci. 13, 953445. doi:10.3389/fpls.2022.953445
Zhang, X., Wang, G., Zhang, S., Chen, S., Wang, Y., Wen, P., et al. (2020). Genomes of the Banyan tree and pollinator wasp provide insights into fig-wasp coevolution. Cell 183, 875–889.e17. doi:10.1016/j.cell.2020.09.043
Zhao, Y., Li, M. C., Konaté, M. M., Chen, L., Das, B., Karlovich, C., et al. (2021). Or normalized counts? A comparative study of quantification measures for the analysis of RNA-seq data from the NCI patient-derived models repository. J. Transl. Med. 19, 1–15.
Keywords: Eucommia ulmoides, MADS-box transcription factors, sex determination, floral organ ABCDE model-related genes, genome-wide identification, expression analysis
Citation: Zhang X, Wang X, Pan L, Guo W, Li Y and Wang W (2023) Genome-wide identification and expression analysis of MADS-box transcription factors reveal their involvement in sex determination of hardy rubber tree (Eucommia ulmoides oliv.). Front. Genet. 14:1138703. doi: 10.3389/fgene.2023.1138703
Received: 06 January 2023; Accepted: 06 February 2023;
Published: 21 February 2023.
Edited by:
Madhav P. Nepal, South Dakota State University, United StatesReviewed by:
Jianling Zhang, Chongqing University, ChinaTareq Alhindi, The University of Jordan, Jordan
Copyright © 2023 Zhang, Wang, Pan, Guo, Li and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wencai Wang, d2VuY2Fpd2FuZ0BnenVjbS5lZHUuY24=