
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Genet., 09 May 2023
Sec. Human and Medical Genomics
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1137817
 Pedro Guimarães de Azevedo1Maria de Lourdes Rabelo Guimarães1
Pedro Guimarães de Azevedo1Maria de Lourdes Rabelo Guimarães1 Anna Luiza Braga Albuquerque1
Anna Luiza Braga Albuquerque1 Rayane Benfica Alves1
Rayane Benfica Alves1 Bianca Gomes Fernandes1Flavia Marques de Melo1Raony Guimaraes Corrêa Do Carmo Lisboa Cardenas1Eitan Friedman2Luiz De Marco1,3
Bianca Gomes Fernandes1Flavia Marques de Melo1Raony Guimaraes Corrêa Do Carmo Lisboa Cardenas1Eitan Friedman2Luiz De Marco1,3 Luciana Bastos-Rodrigues1,4*
Luciana Bastos-Rodrigues1,4*Background: Obstructive sleep apnea syndrome (OSAS) (OMIM #107650) is characterized by complete or partial obstruction of the upper airways, resulting in periods of sleep associated apnea. OSAS increases morbidity and mortality risk from cardiovascular and cerebrovascular diseases. While heritability of OSAS is estimated at ∼40%, the precise underlying genes remain elusive. Brazilian families with OSAS that follows as seemingly autosomal dominant inheritance pattern were recruited.
Methods: The study included nine individuals from two Brazilian families displaying a seemingly autosomal dominant inheritance pattern of OSAS. Whole exome sequencing of germline DNA were analyzed using Mendel, MD software. Variants selected were analyzed using Varstation® with subsequent analyses that included validation by Sanger sequencing, pathogenic score assessment by ACMG criteria, co-segregation analyses (when possible) allele frequency, tissue expression patterns, pathway analyses, effect on protein folding modeling using Swiss-Model and RaptorX.
Results: Two families (six affected patients and three unaffected controls) were analyzed. A comprehensive multistep analysis yielded variants in COX20 (rs946982087) (family A), PTPDC1 (rs61743388) and TMOD4 (rs141507115) (family B) that seemed to be strong candidate genes for being OSAS associated genes in these families.
Conclusion: Sequence variants in COX20, PTPDC1 and TMOD4 seemingly are associated with OSAS phenotype in these families. Further studies in more, ethnically diverse families and non-familial OSAS cases are needed to better define the role of these variants as contributors to OSAS phenotype.
Obstructive sleep apnea syndrome (OSAS) (OMIM #107650) affects 936 million adults worldwide (Benjafield et al., 2019) and is hallmarked by the interruption of or a significant decrease in airflow in the presence of respiratory effort during sleep, leading to increased sleepiness and significantly increases cardiovascular morbidity and mortality risks (Lévy et al., 2015). While the precise etiological factors for OSAS are not clear, disturbances in ventilatory control, craniofacial anatomy, and being overweight, as well as genetic factors are major contributors to OSAS (Peppard and Hagen, 2018).
Familial clusters of OSAS have been reported and in fact heritability and genetic factors have been estimated to contribute ∼40% to OSAS phenotype (Redline et al., 1995; Patel et al., 2008). Over the past few years, sequence variants in candidate genes seemingly associated with OSAS (as objectively quantified by AHI [(apnea–hypopnea index (AHI), defined as the number of apnea and hypopnea events per hour of sleep (American Academy of Sleep Medicine, 2014) (International Classification of Sleep Disorders, 2014) have been reported (Larkin et al., 2010), as well as variants identified in the course of admixture mapping (Wang et al., 2019) and genome wide association studies (GWAS) (Cade et al., 2021). However, few genes have been reported and validated as “OSAS genes” using these approaches (Mohit, Shrivastava, Chand, 2021). The aim of the current study was to identify pathogenic germline sequence variants that could be associated with predisposition and/or development of OSAS using whole-exome DNA sequencing of individuals from two Brazilian OSAS families.
The study encompassed individuals from two Brazilian families displaying OSAS in at least two generations (Figure 1). These families were referred to the School of Medicine, Universidade Federal de Minas Gerais. They underwent the OSAS evaluation protocol as previously described (Kapur et al., 2017). All OSAS diagnoses were based on the established criteria as specified by the American Academy of Sleep Medicine (2014) (International Classification of Sleep Disorders, 2014). Briefly, each participant underwent complete OSAS-focused clinical evaluation and polysomnography. The inclusion criteria were included: diagnosis of OSAS as defined by the American Academy of Sleep Medicine criteria (2014) and absence of craniofacial dysmorphism, genetic syndromes with OSAS as part of the spectrum of manifestations, drug and alcohol abuse, psychiatric disorders, and dementia; age 18–85 years. Participants with upper airway resistance syndrome (UARS) and central sleep apnea were excluded. In addition, dental data were collected such as the presence of self-reported bruxism, pain in masticatory muscles, noise upon movement of the temporomandibular joint (TMJ), tongue size, floor of mouth (i.e., sublingual space) U-shaped region, bordered inferiorly by the mylohyoid muscle, laterally by the gingiva overlying the lingual surface of the mandible, superiorly by the oral tongue, and posteriorly at the insertion of the anterior tonsillar pillar into the tongue. Genomic DNA was extracted from peripheral blood samples of participants using standard protocols (Lahiri and Nurnberger, 1991). The quality and quantity of each DNA sample were tested by NanoDrop ND -2000 UV-Vis Spectrophotometer (Thermofisher, Waltham, MA). The experimental protocols were approved by the Institutional Review Board at the Universidade Federal de Minas Gerais (CEP UFMG 2.980.453). All participants signed an informed consent.
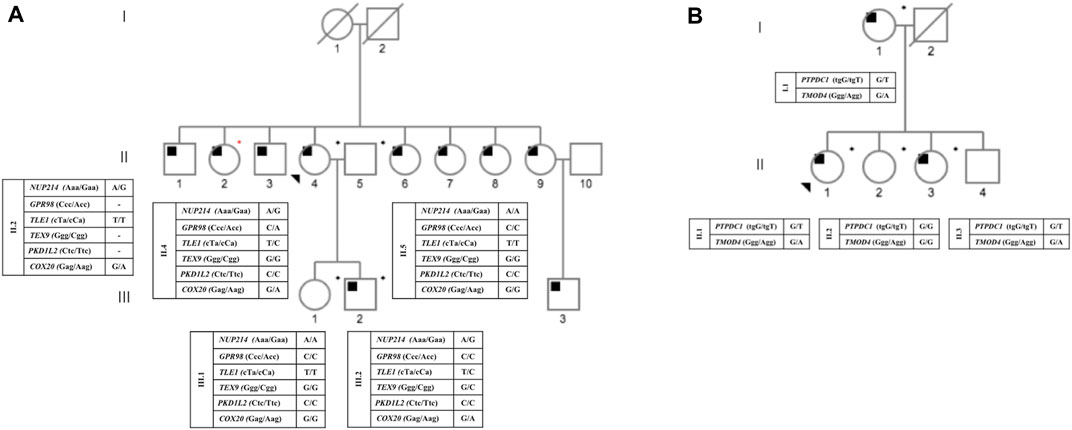
FIGURE 1. Pedigree status from families A and B, with respect to OSA and the genotypes for each identified heterozygous mutation. Circles and squares represent female and male members, respectively. The proband is indicated by a black arrow. Deceased members are shown by diagonal lines. Black squares represent affected individuals; blank eigenforms indicate unaffected individuals; *DNA from individuals submitted to exome analysis; *individual who participated only in the segregation study. (A) pedigree from family A; (B) pedigree from family B.
DNA was subjected to whole exome capturing and sequencing using the Roche NimbleGen V2 chip (Madison, WI) or Nextera (San Diego, CA) with the Illumina HiSeq2000 sequencing platform (San Diego, CA) (Horn, 2012). Sanger sequencing was used to validate all pathogenic variants identified via WES.
For each studied sample, raw sequence files were prepared using the Genome Analysis Tool Kit (GATK). Each fastq file was aligned against the human hg19/GRCh37 reference genome and a variant call format (VCF) file generated for each sample. PCR duplicates were removed using Picard (http://broadinstitute.github.io/picard/), reads around known and detected indels were realigned, and base quality was recalibrated using GATK.
VCF files were analyzed using two different tools: Mendel, MD (available at https://mendelmd.org/) (Cardenas et al., 2017), and Varstation® (available at https://varstation.com/). Only variants selected by both software and excluding those common to cases and controls were further analyzed according to the ACMG Standards and guidelines (Richards et al., 2015), using published data and the VarSome platform (https://varsome.com). Genes that harbored “pathogenic”, “likely pathogenic” or “uncertain significance”, were queried for potential relevance to OSA pathogenesis by applying the following criteria: i) predicted functional consequences using the Ensembl database; ii) publications relating each gene to cancer, based on PUBMED search of the gene name; iii) pathway annotation, which includes all pathways in which a given gene product has been involved at Genecards, OMIM, and UniProt databases; and iv) minor allele frequency of less than 1% in gnomAD database. After this analyze, seven variants in genes in Family A and in eight variants in Family B, classified as “Likely benign” or “Benign”, were excluded (Supplementary Tables S1, 2). After these steps of filtering and validating candidate pathogenic variants by Sanger sequencing, we generated a final list of candidate genes (Supplementary Figure S1).
In addition, allele frequencies of all candidate variants were queried with data derived from The Genome Aggregation Database (gnomAD; https://gnomad.broadinstitute.org/; accessed 2nd August 2022) as well as the AbraOM: Online Archive of Brazilian Mutations database (https://abraom.ib.usp.br/; accessed 15th August 2022).
Wild-type protein models were established according to protein sequence available at UniProt (https://www.uniprot.org/blast/). To obtain model of proteins predicted to be derived from the specific gene variants, Swiss-Model and RaptorX (http://raptosx.uchicago.edu) were used.
The study included nine individuals from two Brazilian families displaying a seemingly autosomal dominant inheritance pattern of OSAS (Figure 1). The average age and BMI of family A was 55.25 years old and 23.36 kg/m2, respectively, of family B the average age and BMI was 62.75 years old and 30.72 kg/m2. Table 1 summarizes all relevant clinical and laboratory data.
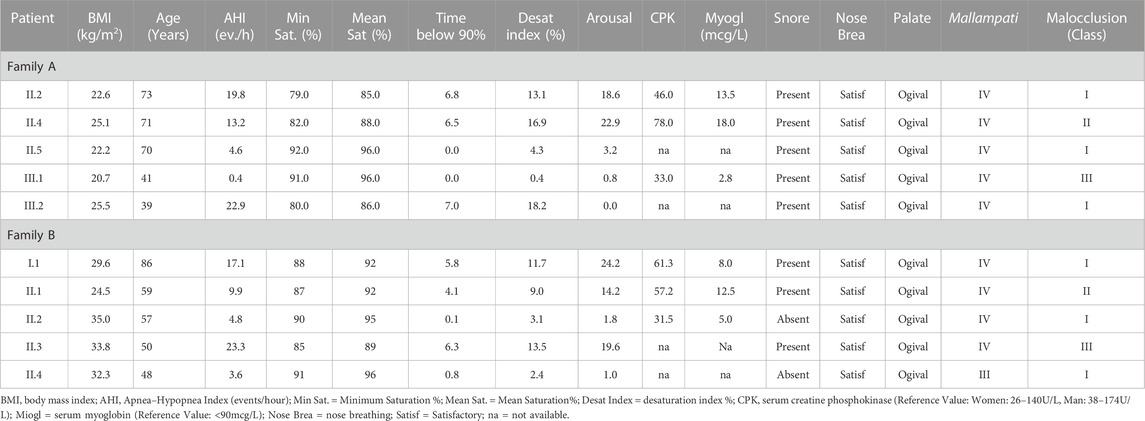
TABLE 1. Clinical and laboratory data of the individuals studied.
A total of 38,612 common variants in 7,462 genes were detected in two affected individuals of family A (II.4 and III.2). After the initial filtering steps (see Methods) a total of 13 heterozygous sequence variants in 13 genes were selected for further analyses. After ACMG pathogenicity classification, Sanger sequence validation, and co-segregation analyses, only one variant remained as a possible OSAS-related genetic variant (COX20, rs946982087) (Supplementary Figure S1A). This variant is not reported in the Brazilian population according to the AbraOM database (https://abraom.ib.usp.br/) or in the GnomAD database (gnomAD; https://gnomad.broadinstitute.org/).
The predicted effect of this sequence variant on protein structure by the Swiss Model/MolProbity (https://swissmodel.expasy.org) is to eliminate the Glu9-Glu11 hydrogen bond [15] thereby destabilizing the protein (Supplementary Figure S2).
A total of 40,318 sequence variants shared by the three affected individuals of family B (I.1, II.1 and II.3) were present in 6,526 genes. After the initial filtering steps (Supplementary Figure S1B), two missense variants in two genes (PTPDC1 and TMOD4) were selected as candidate genes for validation (Supplementary Figure S1B). The reported frequency of these variants in the Brazilian population (https://abraom.ib.usp.br/) was 0.0017 for PTPDC1 and 0.00042 for TMOD4, respectively. In the GnomAD database the reported frequencies are 0.00046 for PTPDC1 and 0.00097 for TMOD4 (https://gnomad.broadinstitute.org/), respectively. No segregation analysis was performed in this family due the lack of available samples.
The pathogenicity of PTPDC1 variant (rs61743388) was investigated. The substitution p.Trp159Cys occurs in the tyrosine-protein phosphatase domain, known to be involved in the control of processes such as cellular division and differentiation and was predicted to be pathogenic according to software that estimates the impact on the biological function of a protein, such as PROVEAN (http://provean.jcvi.org/index.php), MutationTaster and SIFT (https://sift.bii.a-star.edu.sg/) (Supplementary Figure S3).
TMOD4 protein is highly evolutionarily conserved between the amino acids 1–151 and 172–344, encompassing the mutated residue p.Gly323Arg. This missense variant localizes at the beginning of an alpha helix and hence may be deleterious for its function, according to Mutation Taster analysis (Supplementary Figure S4).
In this study, three heterozygous sequence variants that seemingly exert a deleterious effect on protein function in three genes (COX20, PTPDC1, and TMOD4) were identified in two Brazilian OSAS families. COX20 is a nucleus-encoded protein, essential for the assembly of the mitochondrial respiratory chain complex IV (CIV)/cytochrome C oxidase (Szklarczyk et al., 2013). COX20 assists in the stabilization and translocation of newly synthesized COX2 C-terminal into the intermembrane space during CIV assembly and promotes the association between COX2 and metallochaperones SCO1/2, essential for the biogenesis of the cooper core in Complex IV (Lorenzi et al., 2018). Consequently, limiting COX20 levels was associated with unstable, degradable COX2 proteins (Bourens et al., 2014), resulting in impaired complex IV biogenesis and reduced respiratory capacity (Otero et al., 2018). Homozygous COX2 gene pathogenic variants lead to mitochondrial complex IV deficiency (OMIM# 220110), a disorder clinically hallmarked by neuromuscular findings such as hypotonia, dystonia, and ataxia (Szklarczyk et al., 2013; Doss et al., 2014). Polysomnography showed that individuals with central apnea and obstructive sleep apnea, display a decreased ventilatory drive in response to hypoxia and/or hypercapnia (Brunetti et al., 2021).
Although not located in the transmembrane domains of COX20, p.Glu9Lys is at a position of considerable importance for the stability and functionality of COX20. This variant is predicted (but shown conclusively) to disrupt protein structure causing the loss of an important hydrogen bond.
In family B, sequence variants in two genes seemingly underlie OSAS pathogenesis. While the precise function of the Protein tyrosine phosphatase domain-containing 1 (PTPDC1) protein is yet to be elucidated, its down regulation has been associated with phenotypes such as obesity (Zhu et al., 2020) and dementia (Wang et al., 2020). Functionally, PTPDC1 protein is involved in cilium assembly and degradation, directing fluid movement (motile cilia), sensing extracellular environment, and controlling signal transduction pathways (primary cilia). Thus, PTPDC1 depletion leads to an increase in cilia length, suggesting that it negatively regulates cilia length via increased anterograde transport (Lai et al., 2011).
The other variant present in family B is TMOD4, one of the four members of the Tropomodulin family, a group of proteins that bind to the pointed ends of actin filaments thereby stabilizing them and inhibiting filament elongation (Fischer & Fowler, 2003). TMOD4 gene is highly expressed in skeletal muscle and heart (Cox & Zoghbi, 2000). The Inactivating biallelic pathogenic sequence variants in the TMOD4 have been associated with myopathy (e.g., nemaline myopathy OMIM #609284) also reported in zebrafish-mutated model (Berger et al., 2014), suggesting that TMOD4 mutations may lead to OSAS relevant myopathy, in the intercostal muscles and/or diaphragm.
An OSAS -like phenotype has previously been reported in cases with nemaline myopathy (Sasaki et al., 1997; Cook & Berkowitz, 2005). Sasaki et al. (1997), reported the results of polysomnography in two nemaline myopathy (NM) cases who had apnea or irregular chest movements with hypercapnia that occurred only during REM sleep. Cook and Berkowitz (2005) also described a milder form of NM in a child who presented at polysomnography, severe obstructive sleep apnea with an oxygen saturation nadir of 55% during REM sleep. Thus, these single reports are in line with the notion that loss of function in TMDO4 may play a role in muscle strength and may contribute to OSAS pathogenesis.
The wild-type amino acids in COX20, PTPDC1 and TMOD4 are highly conserved between species, which demonstrates its evolutionary relevance. Additionally, it is interesting to notice that the variants observed are rare in general population, demonstrating that it could be related to less adaptive subjects.
Noteworthy, no studies have previously identified possible genes related to mendelian forms of OSA and none of the above cited variants or other variants in the three genes reported herein have been identified in previous OSAS-GWAS studies. A search in GWAS catalog (https://www.ebi.ac.uk/gwas/genes) for variants within 150 Kb of the variants reported in the current study led to no results associated with sleep apnea. A possible reason is that SNPs of modest effect are missed as they do not reach a significance threshold (Eichler et al., 2010). Non-etheless, among the 41 OSA-associated GWAS variants, some trace back to genes involved in cytoskeletal structure and dynamic functionality, such as CDH4 (a calcium-dependent cell-cell adhesion glycoprotein that plays a role in muscle development), similarly to the PTPDC1 and TMDO4 genes mutated in Family B, which exhibit mainly structural functions important for muscle cell functionality. Furthermore, many of the GWAS OSA-associated variants display a pattern of participating in neuromuscular physiologic pathways and neurogenesis, such as RMST, CAMK1D and ATP2BA (Cade et al., 2016; Strausz et al., 2021). So far, most of the studies on OSA have focused on the effect of metabolic disorders such as obesity and environmental factors in the pathogenesis of OSA. However, the present finding of a syndromic form of OSA related to three genes with possible important roles in neuromuscular development and functionality, along with the existence of GWAS variants participating in similar pathways suggests that organic alterations and genetic susceptibility may play a pivotal role on the pathogenesis of OSA.
The limitations of our study should be acknowledged, such as lack of functional data, and limitations of exome sequencing for identification of non-coding and structural variants. Furthermore, the number of analyzed individuals from two families is limited and no segregation analysis could be performed in one of the two families, and the lack of replication in a second OSA family with variants in the same gene. Added to this, the results reported herein may only pertain to OSAS cases from Brazil.
In conclusion, the present study suggests that sequence variants in the COX20, PTPDC1, and TMDO4 genes could be associated with OSAS phenotype in Brazilian families. The putative involvement of these genes in OSAS phenotype needs to be validated and expanded using more families of diverse ethnicity.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by CEP/UFMG 2.980.453. The patients/participants provided their written informed consent to participate in this study.
PA, LM, and LB-R designed the study. FM and RG analyzed and BG, RA, MG, and AA performed the experiments. LM, EF, and LB-R supervised the study. PA, LM, EF, LB-R, and AA interpreted the data and wrote the manuscript. All authors vouch for the data and analysis, approve the final version, and agree to publish the manuscript. All authors contributed to the article and approved the submitted version
This work was partially funded by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq# 405053/2013-4), CAPES and Fundação de Amparo à Pesquisa de Minas Gerais (FAPEMIG# CDS-RED-00019-16). The sponsors had no role in the design or conduct of this research.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1137817/full#supplementary-material
American Academy of Sleep Medicine (2014). International classification of sleep disorders. 3rd ed. American Academy of Sleep Medicine.
Benjafield, A. V., Ayas, N. T., Eastwood, P. R., Heinzer, R., Ip, M. S. M., Morrell, M. J., et al. (2019). Estimation of the global prevalence and burden of obstructive sleep apnoea: A literature-based analysis. Lancet Respir. Med. 7 (8), 687–698.
Berger, J., Tarakci, H., Berger, S., Li, M., Hall, T. E., Arner, A., et al. (2014). Loss of Tropomodulin4 in the zebrafish mutant träge causes cytoplasmic rod formation and muscle weakness reminiscent of nemaline myopathy. Dis. Model Mech. 7 (12), 1407–1415. doi:10.1242/dmm.017376
Bourens, M., Boulet, A., Leary, S. C., and Barrientos, A. (2014). Human COX20 cooperates with SCO1 and SCO2 to mature COX2 and promote the assembly of cytochrome c oxidase. Hum. Mol. Genet. 23 (11), 2901–2913. doi:10.1093/hmg/ddu003
Brunetti, V., Della Marca, G., Servidei, S., and Primiano, G. (2021). Sleep disorders in mitochondrial diseases. Curr. Neurol. Neurosci. Rep. 21 (7), 30. doi:10.1007/s11910-021-01121-2
Cade, B. E., Chen, H., Stilp, A. M., Gleason, K. J., Sofer, T., Ancoli-Israel, S., et al. (2016). Genetic associations with obstructive sleep apnea traits in hispanic/latino Americans. Am. J. Respir. Crit. Care Med. 194 (7), 886–897. doi:10.1164/rccm.201512-2431OC
Cade, B. E., Lee, J., Sofer, T., Kurniansyah, N., He, K. Y., Lee, J., et al. (2021). Whole-genome association analyses of sleep-disordered breathing phenotypes in the NHLBI TOPMed program. Genome Med. 13 (1), 136. doi:10.1186/s13073-021-00917-8
Cardenas, R., D Linhares, N., Ferreira, R., and PenaMendel, S. D. J. M. D. (2017). Mendel,MD: A user-friendly open-source web tool for analyzing WES and wgs in the diagnosis of patients with mendelian disorders. PLOS Comput. Biol. 13 (6), e1005520. doi:10.1371/journal.pcbi.1005520
Cook, B. J., and Berkowitz, R. G. (2005). Tracheostomy in children with nemaline core myopathy. Int. J. Pediatr. Otorhinolaryngol. 69 (2), 263–266. doi:10.1016/j.ijporl.2004.08.020
Cox, P. R., and Zoghbi, H. Y. (2000). Sequencing, expression analysis, and mapping of three unique human tropomodulin genes and their mouse orthologs. Genomics 63 (1), 97–107. doi:10.1006/geno.1999.6061
Doss, S., Lohmann, K., Seibler, P., Arns, B., Klopstock, T., Zühlke, C., et al. (2014). Recessive dystonia-ataxia syndrome in a Turkish family caused by a COX20 (FAM36A) mutation. J. Neuro.l 261 (1), 207–212. doi:10.1007/s00415-013-7177-7
Eichler, E. E., Flint, J., Gibson, G., Kong, A., Leal, S. M., Moore, J. H., et al. (2010). Missing heritability and strategies for finding the underlying causes of complex disease. Nat. Rev. Genet. 11 (6), 446–450. doi:10.1038/nrg2809
Fischer, R. S., and Fowler, V. M. (2003). Tropomodulins: Life at the slow end. Trends Cell Biol. 13 (11), 593–601. doi:10.1016/j.tcb.2003.09.007
Horn, S. (2012). Target enrichment via DNA hybridization capture. Methods Mol. Biol. 840, 177–188. doi:10.1007/978-1-61779-516-9_21
Kapur, V. K., Auckley, D. H., Chowdhuri, S., Kuhlmann, D. C., Mehra, R., Ramar, K., et al. (2017). Clinical practice guideline for diagnostic testing for adult obstructive sleep apnea: An American Academy of Sleep Medicine Clinical Practice Guideline. J. Clin. Sleep. Med. 13 (3), 479–504. doi:10.5664/jcsm.6506
Lahiri, D. K., and Nurnberger, J. I. (1991). A rapid non-enzymatic method for the preparation of HMW DNA from blood for RFLP studies. Nucleic Acids Res. 19 (19), 5444. doi:10.1093/nar/19.19.5444
Lai, C. K., Gupta, N., Wen, X., Rangell, L., Chih, B., Peterson, A. S., et al. (2011). Functional characterization of putative cilia genes by high-content analysis. Mol. Biol. Cell 22 (7), 1104–1119. doi:10.1091/mbc.E10-07-0596
Larkin, E. K., Patel, S. R., Goodloe, R. J., Li, Y., Zhu, X., Gray-McGuire, C., et al. (2010). A candidate gene study of obstructive sleep apnea in European Americans and African Americans. Am. J. Respir. Crit. Care Med. 182 (7), 947–953. doi:10.1164/rccm.201002-0192OC
Lévy, P., Kohler, M., McNicholas, W. T., Barbé, F., McEvoy, R. D., Somers, V. K., et al. (2015). Obstructive sleep apnoea syndrome. Nat. Rev. Dis. Primer 1, 15015. doi:10.1038/nrdp.2015.15
Lorenzi, I., Oeljeklaus, S., Aich, A., Ronsör, C., Callegari, S., Dudek, J., et al. (2018). The mitochondrial TMEM177 associates with COX20 during COX2 biogenesis. Biochim. Biophys. Acta Mol. Cell Res. 1865 (2), 323–333. doi:10.1016/j.bbamcr.2017.11.010
Mohit, , Shrivastava, A., and Chand, P. (2021). Molecular determinants of obstructive sleep apnea. Sleep. Med. 80, 105–112. doi:10.1016/j.sleep.2021.01.032
Otero, M. G., Tiongson, E., Diaz, F., Haude, K., Panzer, K., Collier, A., et al. (2018). Novel pathogenic COX20 variants causing dysarthria, ataxia, and sensory neuropathy. Ann. Clin. Transl. Neurol. 6 (1), 154–160. doi:10.1002/acn3.661
Patel, S. R., Larkin, E. K., and Redline, S. (2008). Shared genetic basis for obstructive sleep apnea and adiposity measures. Int. J. Obes. 32 (5), 795–800. doi:10.1038/sj.ijo.0803803
Peppard, P. E., and Hagen, E. W. (2018). The last 25 years of obstructive sleep apnea epidemiology—And the next 25? Am. J. Respir. Crit. Care Med. 197 (3), 310–312. doi:10.1164/rccm.201708-1614PP
Redline, S., Tishler, P. V., Tosteson, T. D., Williamson, J., Kump, K., Browner, I., et al. (1995). The familial aggregation of obstructive sleep apnea. Am. J. Respir. Crit. Care Med. 151 (3), 682–687. doi:10.1164/ajrccm/151.3_Pt_1.682
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and Genomics and the association for molecular pathology. Genet. Med. 175 (5), 405–424. doi:10.1038/gim.2015.30
Sasaki, M., Takeda, M., Kobayashi, K., and Nonaka, I. (1997). Respiratory failure in nemaline myopathy. Pediatr. Neurol. 16 (4), 344–346. doi:10.1016/s0887-8994(97)00032-5
Strausz, S., Ruotsalainen, S., Ollila, H. M., Karjalainen, J., Kiiskinen, T., Reeve, M., et al. (2021). Genetic analysis of obstructive sleep apnoea discovers a strong association with cardiometabolic health. Eur. Respir. J. 57 (5), 2003091. doi:10.1183/13993003.03091-2020
Szklarczyk, R., Wanschers, B. F., Nijtmans, L. G., Rodenburg, R. J., Zschocke, J., Dikow, N., et al. (2013). A mutation in the FAM36A gene, the human ortholog of COX20, impairs cytochrome c oxidase assembly and is associated with ataxia and muscle hypotonia. Hum. Mol. Genet. 22 (4), 656–667. doi:10.1093/hmg/dds473
Wang, H., Cade, B. E., Sofer, T., Sands, S. A., Chen, H., Browning, S. R., et al. (2019). Admixture mapping identifies novel loci for obstructive sleep apnea in Hispanic/Latino Americans. Hum. Mol. Genet. 28 (4), 675–687. doi:10.1093/hmg/ddy387
Wang, H., Yang, J., Schneider, J. A., De Jager, P. L., Bennett, D. A., and Zhang, H. Y. (2020). Genome-wide interaction analysis of pathological hallmarks in Alzheimer's disease. Neurobiol. Aging 93, 61–68. doi:10.1016/j.neurobiolaging.2020.04.025
Keywords: whole exome, sequencing, obstructive sleep apnea syndrome, OSAS, candidate genes
Citation: de Azevedo PG, Guimarães MdLR, Albuquerque ALB, Alves RB, Gomes Fernandes B, Marques de Melo F, Guimaraes Corrêa Do Carmo Lisboa Cardenas R, Friedman E, De Marco L and Bastos-Rodrigues L (2023) Whole-exome identifies germline variants in families with obstructive sleep apnea syndrome. Front. Genet. 14:1137817. doi: 10.3389/fgene.2023.1137817
Received: 04 January 2023; Accepted: 26 April 2023;
Published: 09 May 2023.
Edited by:
Jared C. Roach, Institute for Systems Biology (ISB), United StatesReviewed by:
Sunil Naik, Penn State Hershey Children’s Hospital, United StatesCopyright © 2023 de Azevedo, Guimarães, Albuquerque, Alves, Gomes Fernandes, Marques de Melo, Guimaraes Corrêa Do Carmo Lisboa Cardenas, Friedman, De Marco and Bastos-Rodrigues. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luciana Bastos-Rodrigues, bHUuYmFzdG9zckBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.