 Wen-Ting Chen
Wen-Ting Chen Min Li
Min Li Shi-Yun Hu
Shi-Yun Hu Su-Hao Wang
Su-Hao Wang Ming-Long Yuan
Ming-Long Yuan- 1State Key Laboratory of Herbage Improvement and Grassland Agro-Ecosystems, Lanzhou University, Lanzhou, Gansu, China
- 2Key Laboratory of Grassland Livestock Industry Innovation, Ministry of Agriculture and Rural Affairs, Lanzhou, Gansu, China
- 3College of Pastoral Agricultural Science and Technology, Lanzhou University, Lanzhou, Gansu, China
- 4National Demonstration Center for Experimental Grassland Science Education, Lanzhou University, Lanzhou, Gansu, China
Harsh environments (e.g., hypoxia and cold temperatures) of the Qinghai–Tibetan Plateau have a substantial influence on adaptive evolution in various species. Some species in Lycaenidae, a large and widely distributed family of butterflies, are adapted to the Qinghai–Tibetan Plateau. Here, we sequenced four mitogenomes of two lycaenid species in the Qinghai–Tibetan Plateau and performed a detailed comparative mitogenomic analysis including nine other lycaenid mitogenomes (nine species) to explore the molecular basis of high-altitude adaptation. Based on mitogenomic data, Bayesian inference, and maximum likelihood methods, we recovered a lycaenid phylogeny of [Curetinae + (Aphnaeinae + (Lycaeninae + (Theclinae + Polyommatinae)))]. The gene content, gene arrangement, base composition, codon usage, and transfer RNA genes (sequence and structure) were highly conserved within Lycaenidae. TrnS1 not only lacked the dihydrouridine arm but also showed anticodon and copy number diversity. The ratios of non-synonymous substitutions to synonymous substitutions of 13 protein-coding genes (PCGs) were less than 1.0, indicating that all PCGs evolved under purifying selection. However, signals of positive selection were detected in cox1 in the two Qinghai–Tibetan Plateau lycaenid species, indicating that this gene may be associated with high-altitude adaptation. Three large non-coding regions, i.e., rrnS-trnM (control region), trnQ-nad2, and trnS2-nad1, were found in the mitogenomes of all lycaenid species. Conserved motifs in three non-coding regions (trnE-trnF, trnS1-trnE, and trnP-nad6) and long sequences in two non-coding regions (nad6-cob and cob-trnS2) were detected in the Qinghai-Tibetan Plateau lycaenid species, suggesting that these non-coding regions were involved in high-altitude adaptation. In addition to the characterization of Lycaenidae mitogenomes, this study highlights the importance of both PCGs and non-coding regions in high-altitude adaptation.
1 Introduction
Lycaenidae (Insecta: Lepidoptera: Papilionoidea), the second largest family of butterflies after Nymphalidae, consists of approximately 6,000 species in seven subfamilies (Pierce et al., 2002). Species in the family are widely distributed worldwide, with high diversity in morphology and ecology (Artem’eva, 2007; Schar et al., 2018). Lycaenidae species mainly inhabit mountains and forests, and some are adapted to high-altitude environments (Hughes, 2000; Balint et al., 2022; Marabuto et al., 2022). Most lycaenid species (about 75%) are associated with ants, forming a mutually beneficial symbiotic relationship (i.e., myrmecophily) (Pierce et al., 2002; Nemet et al., 2016; Riva et al., 2017; Kubik and Schorr, 2018), and this relationship may be related to the geographical distribution (Schmidt and Rice, 2002; Kaminski, 2008). However, little research has focused on the mechanism underlying environmental adaptation in Lycaenidae, and further analyses using molecular data are needed.
Mitochondria are the sites of energy conversion and metabolism in eukaryotes, known as the “energy factory” (Milenkovic et al., 2007; Tripodi et al., 2018; Bottje, 2019). Both nuclear and mitochondrial genomes (mitogenomes) encode essential proteins in the electron transfer chain of mitochondria. Generally, animal mitogenomes consist of 37 genes, i.e., 13 protein-coding genes (PCGs), 22 transfer RNA genes (tRNAs), and two ribosomal RNA unit genes (rRNAs, rrnL and rrnS). In addition, animal mitogenomes usually contain a large non-coding region, known as the control region (CR) or AT-rich region in arthropods, which contains essential regulatory elements for transcription and replication (Boore, 1999; Zardoya and Suadrez, 2008). Mitogenomes have been used in population genetics, phylogeography, and phylogenetic studies of various taxa, e.g., insects (Yuan et al., 2015; Liu et al., 2022a; Zhang et al., 2022), spiders (Tyagi et al., 2020; Li et al., 2021; Li et al., 2022), and centipedes (Hu et al., 2020; Ding et al., 2022). To date, only nine sequenced mitogenomes of nine Lycaenidae species have been stored in GenBank, which is extremely limited given the species richness, restricting our understanding of the phylogeny and evolution of the family.
It is historically believed that mitogenomes evolve neutrally; however, mitochondrial genes have important functional roles in OXPHOS, suggesting that they are targets of natural selection (Blier et al., 2001; Manoli et al., 2007; da Fonseca et al., 2008). Signals of adaptive evolution in several mitochondrial genes have been detected in various animal taxa, e.g., atp6 in wild Tibetan pigs (Li et al., 2016), atp8 in Freyastera benthophila (Mu et al., 2018) and Gobiidae (Shang et al., 2022), and cob in Calyptogena marissinica (Yang et al., 2019a). The Qinghai–Tibetan Plateau (QTP) is the largest plateau in the world, characterized by hypoxia, cold temperatures, and strong ultraviolet radiation. These harsh environmental conditions influence species diversification and adaptive evolution substantially (Zhang et al., 2017; Yuan et al., 2018). Non-neutral evolution in mitochondrial genes has been found in birds (Zhou et al., 2014; Gu et al., 2016), mammals (Yu et al., 2011; Luo et al., 2012; Peng et al., 2012), fish (Li et al., 2013; Wang et al., 2016), and insects (Zhang et al., 2017; Yuan et al., 2018; Balint et al., 2022) inhabiting the QTP. Further mitogenomic analyses of additional QTP insect groups will improve our understanding of adaptation to high-altitude environments. A large number of genetic analyses have shown that mitogenomes can be used to analyze adaptive evolution (Korkmaz et al., 2017; Yuan et al., 2018; Yuan et al., 2020; Bartakova et al., 2021).
In this study, we proposed that high-altitude adaptation in Lycaenidae inhabiting the QTP is associated not only with PCGs but also non-coding regions. We sequenced four complete mitogenomes of two QTP lycaenid species, Polyommatus amorata and Agriades orbitulus. Combined with sequenced mitogenomes of nine lycaenid species available on GenBank, we performed a detailed comparative mitogenomic analysis and constructed a mitogenomic phylogeny of Lycaenidae. We focused on the importance of PCGs and non-coding regions in the environmental adaptation of Lycaenidae. In addition to characterizing Lycaenidae mitogenomes, our results provide new insights into the high-altitude adaptation and evolution of Lycaenidae.
2 Materials and methods
2.1 Sampling, DNA extraction, and sequencing
Adult specimens were collected from alpine meadows of the QTP, in Menyuan County of Qinghai Province and Naqu County of the Tibet Autonomous Region, China. Detailed sampling information is provided in Supplementary Table S1. Samples were preserved in 100% ethanol during collection and stored at −80°C after transporting to the laboratory until DNA extraction. Samples were deposited in the State Key Laboratory of Herbage Improvement and Grassland Agro-Ecosystems, College of Pastoral Agricultural Science and Technology, Lanzhou University, Lanzhou, China. Total genomic DNA was extracted from a single specimen using a DNeasy Tissue Kit (Qiagen, Hilden, Germany). The DNA quality was detected by 1.2% agarose gel electrophoresis and spectrophotometry using the NanoDrop ND-1000 (Thermo Fisher Scientific, Waltham, MA, United States). DNA was sequenced in both directions using the Illumina NovaSeq 6000 platform (2 × 150 bp) by Wuhan Benagen Tech Solutions (Wuhan, China).
2.2 Mitochondrial genome assembly, annotation, and analysis
Low-quality reads, including reads with a cutoff Phred quality score of Q20, more than 5% N bases, adapter sequences, or repeated reads introduced by PCR duplicates were removed using SOAPnuke (version: 2.1.0) (Chen et al., 2017). The high-quality reads were assembled by using SPAdes (version 3.13.0) (Bankevich et al., 2012), using the mitogenome of Cupido argiades (NC_023088) as a reference. The assembled mitogenomes were annotated by the MITOS web server (http://mitos2.bioinf.uni-leipzig.de) (Bernt et al., 2013) to locate PCGs, tRNAs, and rRNAs by comparisons with homologous regions in other insect mitogenome sequences. All identified PCGs were corrected by sequence alignment using the published Lycaenidae mitogenome sequences, the start and stop codons were identified, and all genes were manually verified and proofread after annotation (to avoid overlap). The secondary structures of the tRNA genes were verified using tRNAscan-SE (version 1.21) (Lowe and Eddy, 1997). The tandem repeats of control regions were detected using Tandem Repeats Finder (version 4.09) (Benson, 1999). All four newly sequenced mitogenome sequences have been deposited at GenBank (under accession number ON411617-20).
Nucleotide diversity (π) was calculated for the 13 PCGs of the mitochondrial genome using DnaSP (version 6) with a sliding window of 100 bp and a step size of 25 bp (Rozas et al., 2017). The nucleotide composition and codon usage were analyzed using MEGA (version 10.2) (Kumar et al., 2018). The GC content, GC-skew, and AT-skew were estimated to evaluate the overall nucleotide composition. The GC content (GC %) was defined as the content of G + C, and the formula for compositional skewness was as follows: AT-skew = [A−T]/[A + T], GC-skew = [G−C]/[G + C] (Perna and Kocher, 1995). The effective number of codons (ENCs), codon bias index (CBI), and G + C contents of the first, second, and third codon positions were used to analyze codon usage. The ENC and CBI were determined using DnaSP (version 6) (Rozas et al., 2017), and the G + C contents at the first, second, and third codon positions were determined using MEGA (version 10.2) (Kumar et al., 2018). The correlation between the G + C content of all codons (GCa), G + C content of the third codon position (GC3), ENC, and CBI as well as the relationship between nucleotide composition and codon bias for all PCGs were analyzed using Excel. Secondary structures of sequences were predicted by the minimum free energy model using mfold (http://www.unafold.org/) (Zuker, 2003). The most stable structure (i.e., the structure with the lowest free energy) was selected when there were multiple structures.
2.3 Phylogenetic analysis
Thirteen lycaenid mitogenomes were included in a phylogenetic analysis, including the four newly sequenced mitogenomes and nine lycaenid mitogenomes (nine species) available from GenBank (Supplementary Table S2). Apodemia mormo (NC_024571) and Abisara fylloides (NC_021746) belonging to Riodinidae were used as outgroups. All 13 PCGs were individually aligned by ClustalW (Codons), and two rRNAs (rrnL and rrnS) were aligned by ClustalW, implemented in MEGA (version 10.2) (Kumar et al., 2018). Poorly aligned and divergent sequences were removed using the Gblocks server (http://molevol.cmima.csic.es/castresana/Gblocks_server.html). Three datasets were generated for phylogenetic analyses: 1) the P123 dataset, with nucleotide sequences at all codon positions of 13 PCGs; 2) the P123RNA dataset, with P123 and the nucleotide sequences of two rRNAs; 3) the P123AA dataset, with the inferred amino acid sequences of 13 PCGs. Each dataset was tested for substitution saturation by using DAMBE (version 5.3.74) (Xia, 2013). There was no substantial sequence saturation (Supplementary Table S3), indicating that the datasets can be used in phylogenetic analysis. The best partitioning schemes and corresponding nucleotide substitution models for each dataset were identified using the IQ-TREE web server (http://iqtree.cibiv.univie.ac.at/) (Trifinopoulos et al., 2016), and the results were used for downstream phylogenetic analysis (Supplementary Table S4).
Maximum likelihood (ML) phylogenetic analysis was performed using RAxML-HPC2 (version 8.0.24) (Stamatakis, 2014) with the GTRGAMMA model and 1,000 bootstrap (BS) replicates. Bayesian inference (BI) was performed using MrBayes (version 3.2.7) (Ronquist et al., 2012), with 1 × 108 generations and sampling every 100 generations (Yuan et al., 2015). Stationarity was achieved when the estimated sample size was over 100 and when the potential scale reduction factor approached 1.0, and default settings were used for the remaining parameters (Ronquist et al., 2012).
2.4 Evolutionary rates and selective pressure analysis
The number of synonymous substitutions per synonymous site (dS), the number of non-synonymous substitutions per non-synonymous site (dN), and the ratio of non-synonymous to synonymous substitutions (ω) were calculated for the 13 PCGs in Lycaenidae. Generally, ω (dN/dS) < 1 indicates negative/purifying selection, ω > 1 indicates positive/diversifying selection, and ω = 1 indicates neutral expectation (Anisimova and Kosiol, 2009). All dN and dS values were calculated using MEGA (version 10.2) (Kumar et al., 2018), and ω was calculated using Excel. To analyze the selective pressure for each PCG in Polyommatinae species under branch-specific and branch-site models, the CodeML program in PAML (version 4.7) was used, applying a ML approach (Yang, 2007). Two Polyommatinae species (P. amorata and A. orbitulus) inhabiting the QTP were used as the foreground branch. Positive selection was inferred when ω > 1, and the log-likelihood ratio test (LRT) was significant (p < 0.1) (Yang, 2007). The Bayes empirical Bayes (BEB) method was used to calculate posterior probabilities for site classes to determine which codon positions experienced positive selection (ω > 1) (Zhang et al., 2005).
We also used the Datamonkey web server (http://www.datamonkey.org/) to analyze the evolutionary rate of each PCG, with the fixed-effects likelihood mode (FEL, site-by-site analysis) to detect which codons were under selection (Pond and Frost, 2005).
3 Results
3.1 General features of Lycaenidae mitogenomes
We obtained four complete mitogenomes for two QTP Polyommatinae species (Supplementary Table S1). These newly sequenced mitogenomes were closed circular DNA molecules, with sizes ranging from 15,340 bp (A. orbitulus NQ2) to 15,389 bp (P. amorata), similar to those of other Lycaenidae species (15,162–15,366 bp) (Supplementary Table S2). Thirty-seven typical mitochondrial genes without rearrangements were detected in each mitogenome, comprising 13 PCGs, 22 tRNAs, and two rRNAs. Non-coding regions, consisting of putative control regions (CRs) and 879 bp intergenic nucleotides dispersed among 68 intergenic regions (IGRs) in four newly sequenced mitogenomes, were similar in all lycaenid species.
In Lycaenidae, 13 PCGs began with an ATN codon, except for cox1 (which started with CGA), and terminated with complete (TAA or TAG) or truncated (TA or T) stop codons. Twenty-three genes (nine PCGs and 14 tRNAs) were located on the J-strand, with the remaining 14 genes encoded on the N-strand. The conservation in sequence size was observed in CRs, tRNAs, rrnS, rrnL, and PCGs among lycaenid mitogenomes, with the minimum variation observed in PCGs (27 bp) and the maximum in CRs (139 bp) (Figure 1).
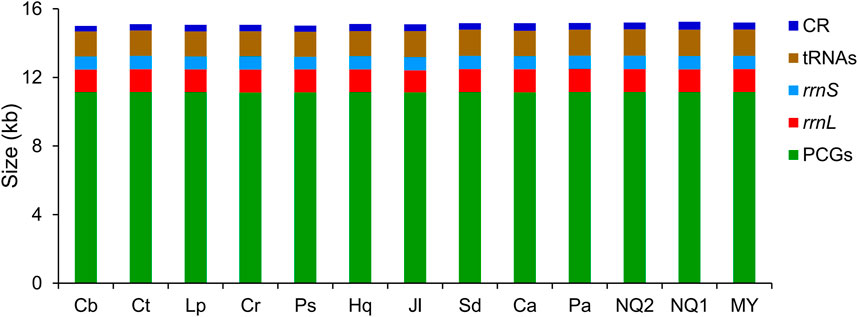
FIGURE 1. Size comparison of protein-coding genes (PCGs), transfer RNA genes (tRNAs), rrnL, rrnS, and control region (CR) among 13 Lycaenidae mitogenomes. Species are abbreviated as follows: Cb, Curetis bulis; Ct, Cigaritis takanonis; Lp, Lycaena phlaeas; Cr, Coreana raphaelis; Ps, Protantigius superans; Hq, Hypaurotis quercus; Jl, Japonica lutea; Sd, Shijimiaeoides divina; Ca, Cupido argiades; Pa, Polyommatus amorata; NQ2, Agriades orbitulus NQ2; NQ1, Agriades orbitulus NQ1; MY, Agriades orbitulus MY.
Nucleotide diversity in 13 lycaenid PCGs differed considerably among taxa and among genes. Nucleotide diversity estimates were significantly lower in QTP species than in other taxa, with atp8 (π = 0.054) exhibiting the highest polymorphism and nad1 (π = 0.032) showing the lowest (Figure 2). In other species, diversity was highest and lowest in nad6 (π = 0.157) and cox2 (π = 0.094), respectively (Figure 2).
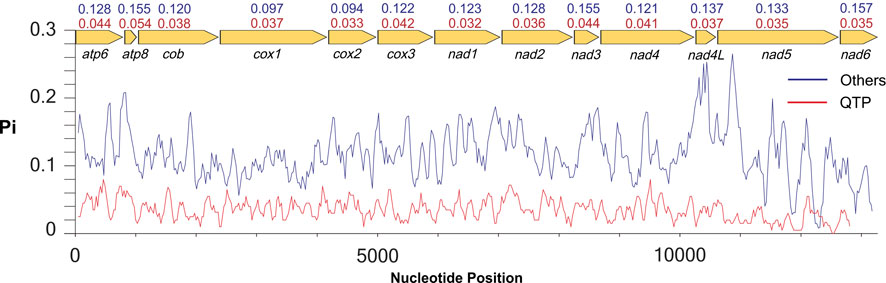
FIGURE 2. Nucleotide diversity (Pi) of 13 PCGs in 13 sequences of Lycaenidae. Lines show the values of Pi in a sliding window analysis. The red line represents the QTP species, and the blue line represents other species. The Pi values for each group and gene are shown on the gene name. Red values represent the QTP species, and blue values represent other species.
3.2 Nucleotide composition and codon usage
All Lycaenidae mitogenomes on the J-strand presented a similar nucleotide composition, characterized by a high A + T content, moderate negative AT skews (−0.0475 to 0.0041), and consistently negative GC skews (−0.1578 to −0.2145). Summaries of A + T% vs. AT-skew and G + C% vs. GC-skew across all available complete mitogenomes of Lycaenidae are shown in Figure 3. Among the 13 mitochondrial sequences, the A + T content ranged from 81.08% (P. amorata) to 82.66% (Coreana raphaelis) and 11 sequences had negative AT-skews, while the other two were positive ( Cigaritis takanonis and P. amorata) (Figure 3). Moreover, a comparative analysis of Lycaenidae mitogenomes indicated that the A + T content was always highest in the CR (90.02%–94.61%) and lowest in PCGs (79.58%–81.46%). Furthermore, it differed substantially among codon positions of PCGs; in particular, the third codon position had a higher A + T content (92.17%–96.87%) than those of the first (74.58%–76.82%) and second (70.35%–71.92%) positions (Figure 4).
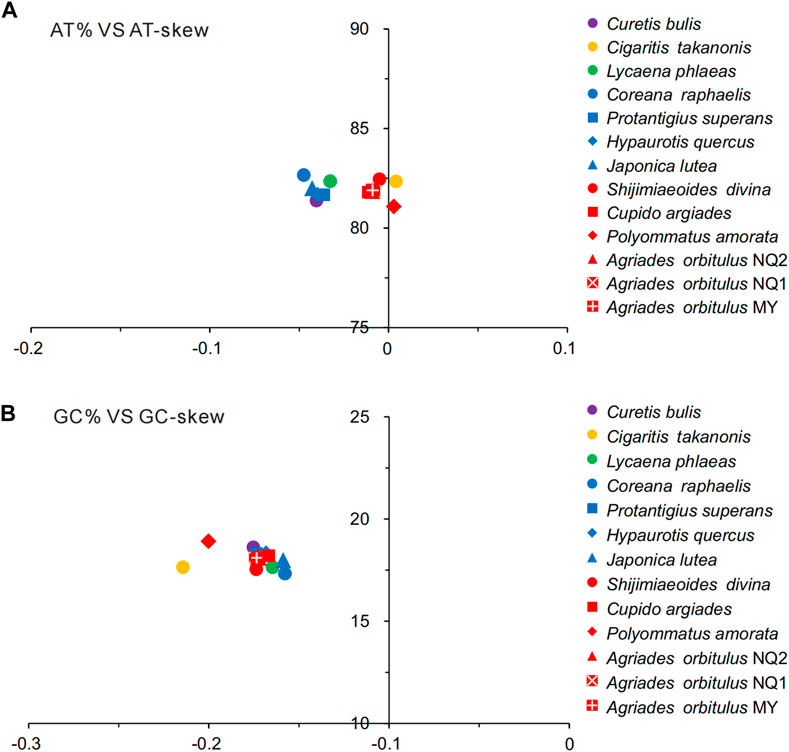
FIGURE 3. AT% vs. AT-skew (A) and GC% vs. GC-skew (B) in the 13 Lycaenidae mitochondrial genomes. Results are shown as the bp percentage (Y-axis) and nucleotide skews (X-axis). Values are calculated for J-strands for full-length mitogenomes.
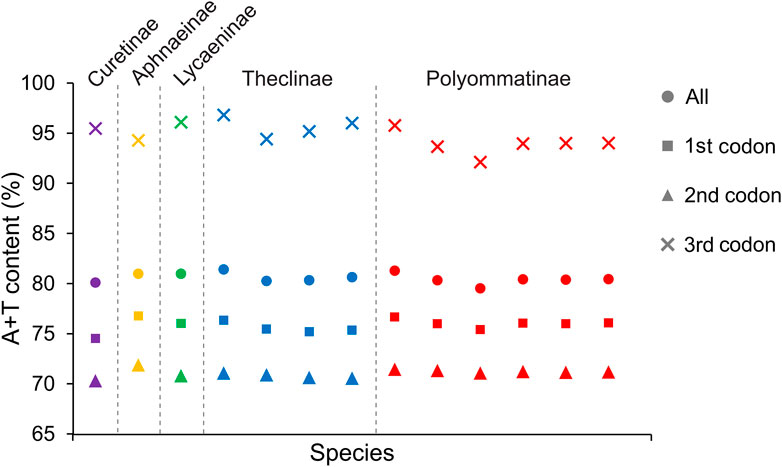
FIGURE 4. A + T contents of mitochondrial protein-coding genes among five subfamilies within Lycaenidae.
There was a small difference in the number of codons but a bias in codon usage (Supplementary Table S5). The number of codons in the 13 sequences ranged from 3,708 (C. raphaelis) to 3,717 (A. orbitulus MY and A. orbitulus NQ2). A relative synonymous codon use (RSCU) analysis showed that the mitogenomes used up to 59 invertebrate mitochondrial codons (in four sequences) and at least 51 codons (C. raphaelis) (Supplementary Table S5). In Lycaenidae, at least one of the five GC-rich codons CCG (P), GCG (A), CGC (R), CGG (R), and GGC (G) was not used, and the five AT-rich codons UUA (L), UUU (F), AUU (I), UAU (Y), and AUA (M) were the most frequently used codons (Supplementary Table S5).
To further investigate codon usage bias among Lycaenidae species, we analyzed the correlation between the ENC, CBI, GCa, and GC3 for the 13 PCGs (Figure 5). The ENC ranged from 29.90 (C. raphaelis) to 32.98 (P. amorata), with an average of 31.21, and the CBI ranged from 0.78 (P. amorata) to 0.86 (C. raphaelis), with an average of 0.82. The ENC was positively correlated with GCa (R2 = 0.48, p < 0.01) (Figure 5A) and GC3 (R2 = 0.90, p < 0.01) (Figure 5B). CBI was negatively correlated with GCa (R2 = 0.66, p < 0.01) (Figure 5C) and GC3 (R2 = 0.96, p < 0.01) (Figure 5D), and the ENC and CBI were significantly negatively correlated (R2 = 0.88, p < 0.01) (Figure 5E). In addition, the ENC and GC3 values were compared with those of the standard curve (ENC*, codon bias only determined by the base composition at the third position), and the actual sample points for all Lycaenidae fell far below the ENC* (Figure 6).
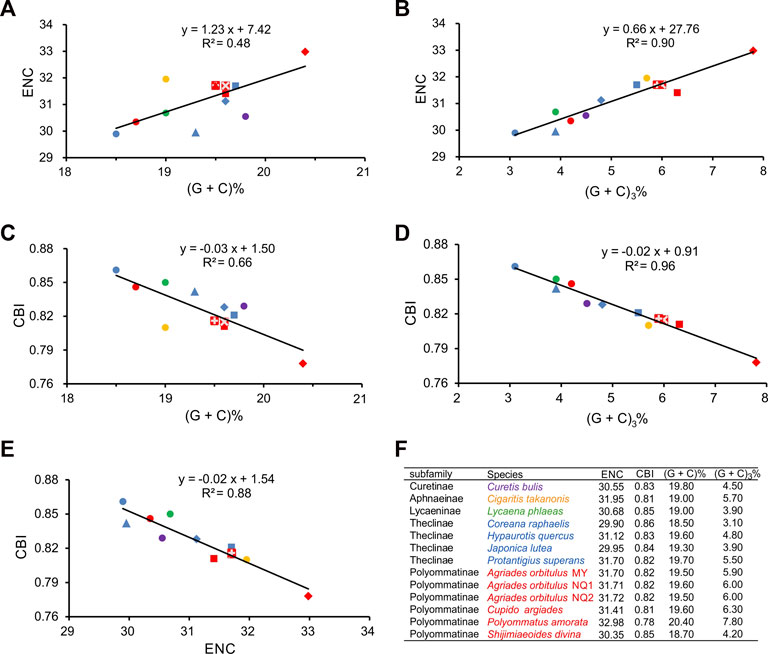
FIGURE 5. Evaluation of codon bias in the 13 Lycaenidae mitogenomes. (G + C)%, G + C content at all codon positions in the 13 protein-coding genes (PCGs). (G + C)3%, G + C content at the third codon positions in 13 PCGs. ENC, effective number of codons. CBI, codon bias index. The colors of symbols match those in Figure 3.

FIGURE 6. Correlation between the effective number of codons (ENC) and G + C content at the third codon position (GC3) in the 13 protein-coding genes (PCGs) for Lycaenidae species. The solid line represents the relationship between the ENC and GC3 content.
3.3 Transfer RNA genes
Twenty-two tRNAs were detected in these four new mitogenomes and were conserved in sequence and structure in lycaenids (Figure 7). High conservation was confirmed at the family level (64.06%–92.86%), subfamily level (71.43%–98.57%), and QTP species level (89.06%–100.00%). A stable canonical cloverleaf secondary structure was forecasted. Variations in trnS1 were detected in the absence of the dihydrouridine arm, the type of anticodon, and copy number, and this variation may be associated with environmental adaptation.
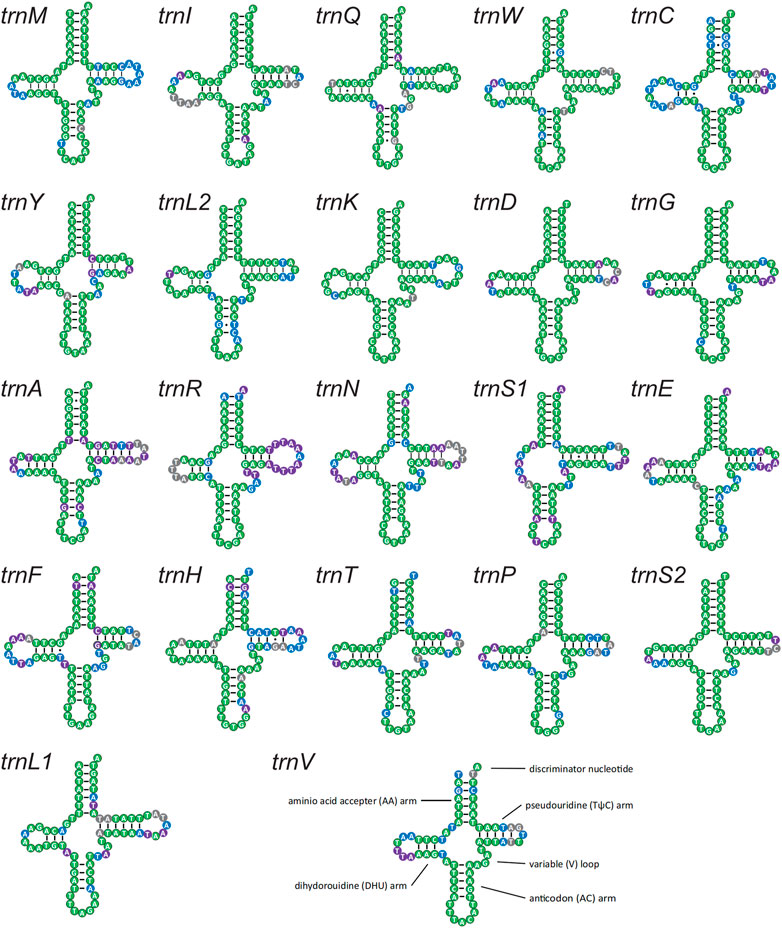
FIGURE 7. Putative secondary structures of the 22 tRNA genes identified in the mitochondrial genome of Lycaenidae. All tRNA genes are shown in the order of occurrence in the mitochondrial genome starting from trnM. The nucleotides showing 100% identity within Lycaenidae, Polyommatinae, and QTP species are marked in green, blue, and purple, respectively. Bars indicate Watson–Crick base pairings, and dots between G and T pairs mark canonical base pairings in tRNA.
The anticodon of trnS1 was not unique and varied from the most common TCT (10 sequences) to ACT (Japonica lutea and Cupido argiades) or GCT (Curetis bulis). In addition, reports of a second copy of trnS1 with the anticodon ACT in two species (C. raphaelis and Hypaurotis quercus) suggested that multiple functional copies contribute to environmental adaptation.
3.4 Non-coding regions
The largest non-coding region of Lycaenidae with a length of 324 bp (Curetis bulis) to 463 bp (A. orbitulus NQ1) was the CR, located at a conserved position between rrnS and trnM. The CR had special structures that make it the starting point of DNA replication, and the following three characteristics were observed in 13 Lycaenidae sequences. 1) Each sequence had at least one maximal tandem repeat unit ranging from 12 to 24 bp in length, with a maximum length of 24 bp in C. raphaelis. Only two perfect repeats were observed for each tandem repeat unit in 13 sequences, with imperfect repeats in few species (Supplementary Table S6). 2) The longest TATA motif of 56 bp was observed in C. raphaelis, and the motif was lacking in Lycaena phlaeas. 3) A large poly(T/A) motif with a length of 19 or 20 bp and a small (T/A) motif with a length of 7–10 bp were observed in all sequences.
Additionally, many IGRs were detected in Lycaenidae, besides the CR. Thirteen sequences shared two IGRs (trnQ-nad2 and trnS2-nad1) (Figures 8, 9). Special structural domains were found in three IGRs (trnE-trnF, trnS1-trnE, and trnP-nad6) in two QTP species (Supplementary Figure S1), and two relatively large IGRs (nad6-cob and cob-trnS2) were found in two QTP species.

FIGURE 8. Non-coding region between trnQ and nad2 in Lycaenidae mitogenomes. dG, free energy, kcal/mol. The most stable structure was selected.
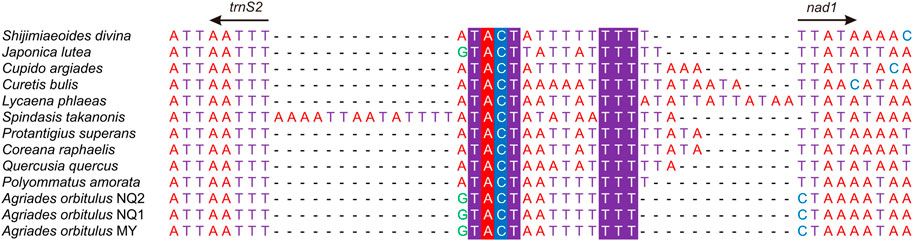
FIGURE 9. Non-coding region between trnS2 and nad1 in the 13 Lycaenidae mitogenomes. Conserved bases in the 13 Lycaenidae are highlighted.
Seven IGRs could be characterized as follows. 1) The secondary structures of trnQ-nad2 (48–60 bp) were predicted for 13 sequences, revealing the typical stem-loop structure (Figure 8). 2) Two conserved motifs were observed at ends, “TACT” near trnS2 and “TTT” near nad1, the predicted binding site of the Drosophila mitochondrial transcription termination factor (DmTTF). The middle parts of the two motifs were composed of A and T in 13 lycaenids at trnS2-nad1 (15–31 bp) (Figure 9). 3) A 27-bp motif of (TAT)2, (TA)8, and “CATAT” was observed in two QTP species but did not exceed 3 bp in non-QTP species at trnE-trnF (29–39 bp) (Supplementary Figure S1A). 4) A 10-bp motif of “TTATATTATT” was observed in QTP species and was not detected in non-QTP species (no more than 11 bp) at trnS1-trnE (12–30 bp) (Supplementary Figure S1B). 5) A 7-bp sequence of “ATTTGAT” was observed in two QTP species, while other species had only 2 bp at trnP-nad6 (Supplementary Figure S1C). 6) A 6-bp sequence was observed in QTP species but was lacking in non-QTP species at nad6-cob. 7) A short sequence (2–10 bp) was observed at cob-trnS2 only in QTP species.
Furthermore, a large IGR was detected in A. orbitulus at cox3-trnG (29 bp); this may be a species-specific sequence and was likely to have a stem-loop structure (Supplementary Figure S2). Other non-coding regions showed substantial variation in size and shared similar characteristics, including a high A + T content and poly(T/A), AT motif, or stem-loop structures.
3.5 Mitochondrial phylogeny
BI and ML methods for phylogenetic reconstruction based on mitogenomes yielded similar tree topologies for Lycaenidae at the species level (Figure 10; Supplementary Figure S3). The only incongruence was the relationship among A. orbitulus NQ1, A. orbitulus NQ2, and A. orbitulus MY (Supplementary Figure S3). Five subfamilies (Curetinae, Aphnaeinae, Lycaeninae, Theclinae, and Polyommatinae) in Lycaenidae were recovered consistently with high support (Figure 10; Supplementary Figure S3). These clades were identified as monophyletic groups. Curetinae was placed at the basal position in Lycaenidae, followed by the divergence of Aphnaeinae and Lycaeninae, and Theclinae clustered with Polyommatinae (Figure 10). Furthermore, the relationship among the genera in Polyommatinae were well-supported by both BI and ML analyses, indicating the basal position of Shijimiaeoides and the following topology: Shijimiaeoides divina + (Cupido argiades + (P. amorata + A. orbitulus)) (Figure 10).
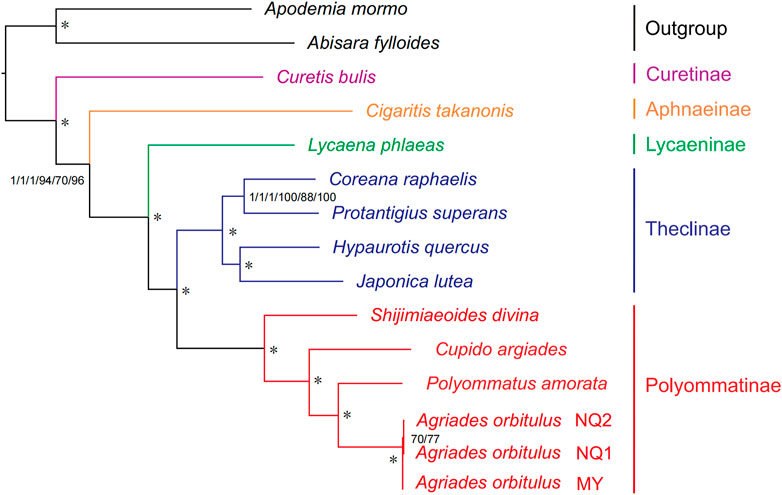
FIGURE 10. Phylogenetic relationships among five subfamilies within Lycaenidae inferred from mitogenomic data. Numbers from left to right are Bayesian posterior probabilities (PP) and ML bootstrap (BS) values for each of the three datasets (P123, P123AA, and P123RNA). Asterisk (*) indicates PP = 1.0 and BS > 90. Full phylogenetic results are provided in Supplementary Figure S3.
3.6 Adaptive evolution of protein-coding genes
The ω values of 13 PCGs were low (ω < 1), suggesting that these genes were under purifying selection (Figure 11B). The lowest ω value was presented in cox1, and atp8 showed the highest ω value (Figure 11B). This was also true for the percentage of negatively selective sites (Figure 11A). In the branch-specific model, only two PCGs (atp8 and nad6) were significantly different between the QTP lycaenid species and non-QTP species (Supplementary Table S7). In the branch-site model analysis, signatures of positive selection were detected at one codon (position 404) of cox1, approaching significance (p = 0.055) (Supplementary Table S8), suggesting that this locus may be associated with high-altitude adaptation. The selected codon changed from CCT (Pro) to AAT (Asn) in the QTP species. We detected no evidence of positive selection in other positions or genes; however, the role of positive selection cannot be excluded.
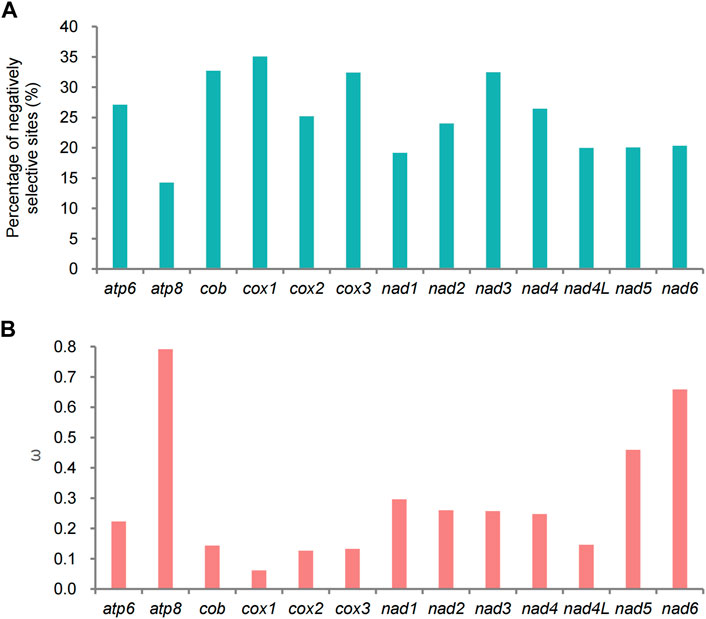
FIGURE 11. Results of selective pressure analyses. (A) Percentage of sites under negative selection in each of the 13 protein-coding genes (PCGs) in Polyommatinae determined by FEL site-by-site analyses. (B) Ratios (ω) of non-synonymous substitutions to synonymous substitutions for each of the 13 PCGs in Polyommatinae.
4 Discussion
4.1 General features of Lycaenidae mitogenomes are conserved
The mitogenomes of animals are generally conserved, reflecting their indispensable functions. However, adaptive evolution may alter mitogenomic characteristics (Li et al., 2016; Yuan et al., 2020), resulting in variation in the size, number, and arrangement of genes (Adams and Palmer, 2003; Wang et al., 2014a; Gan et al., 2016; Chen et al., 2020c). In Lycaenidae, mitogenomes are highly conserved in size, structure, and function (Kim et al., 2011; Zhang et al., 2013; Jeong et al., 2017), as supported by analyses of gene content, gene arrangement, base composition, codon usage, and transfer RNAs. No rearrangements were detected (Boore, 1999).
The nucleotide composition affects codon usage, and different codon usage patterns may lead to differences in biological functions (Foroughmand-Araabi et al., 2015; Ma et al., 2015; Zhou et al., 2021). In Lycaenidae, a strong bias in nucleotide composition (toward A and T) was found, and this pattern is common in insects (Meng et al., 2022; Qi et al., 2022; Sun et al., 2022). Moreover, this codon bias can be explained by broad patterns in both natural selection and mutation.
Transfer RNA genes were highly conserved and exhibited a typical secondary cloverleaf structure. TrnS1 exhibited variation in the presence of the dihydrouridine arm, type of anticodon, and copy number, considered an important indicator of adaptive evolution. In insects, the lack of the dihydrouridine arm in tRNA is not rare (Yang et al., 2019c; Wei et al., 2021; Wu et al., 2022), and isoacceptor tRNAs (which have a different anticodon but charge the same amino acid) may be related to codon usage patterns and selection (Oliveira et al., 2008; Behura et al., 2010). Moreover, observations of more than 22 tRNAs had been reported in only a few insect mitogenomes. In Lycaenidae, two trnS1 genes, one copy containing the anticodon TCT and another copy containing the anticodon ACT, have been observed in C. raphaelis and H. quercus (Kim et al., 2006). Therefore, the observation of multiple copies of trnS1 in P. amorata was reasonable, and the second copy may be functional (Behura et al., 2010; Behura and Severson, 2011; Pang et al., 2014; Zhou et al., 2020). The anticodon of the second copy of trnS1 was ACT in P. amorata and was expected to have a clover structure without the DHU arm. The second copy showed 77.78% sequence identity to the first copy (determined using BLAST as the ratio of matched bases to the total number of aligned bases) (Supplementary Figure S4). These features were important indicators of adaptive evolution in Lycaenidae tRNA, though we cannot exclude the role of random ancestral mutations. Further research including additional lycaenid species and individuals are needed to clarify the strength and role of adaptive evolution.
4.2 Mitogenomic data can provide valid phylogenetic signals for Lycaenidae
Mitogenomes are relatively stable and have excellent characteristics for evolutionary analyses (Yuan et al., 2015; Li et al., 2019; Li et al., 2022), as demonstrated in many lepidopteran groups (Liu et al., 2016; Shi et al., 2020; Sun et al., 2020). Subfamilies in Lycaenidae formed monophyletic groups in the phylogeny. The classification of some species in the family is controversial, mainly stemming from disagreements in analyses based on morphological characteristics (Ugelvig et al., 2011). Despite variation in phylogenetic relationships inferred from morphology, analyses based on molecular data yield highly consistent results. Therefore, scholars have suggested using a combination of morphological and molecular data to confirm the relationships among groups in Lycaenidae (Wahlberg et al., 2005; Stekolnikov et al., 2013; Talavera et al., 2013). A subfamily-level phylogeny of Lycaenidae is clearly defined as [Curetinae + (Aphnaeinae + (Lycaeninae + (Theclinae + Polyommatinae)))], providing an updated view of relationships within this family. The phylogenetic analysis of Lycaenidae is consistent with previous research supported by molecular data, showing that mitogenomes provide useful data for resolving phylogenetic relationships (Jeong et al., 2017; Zhou et al., 2020). Considering the limited taxon sampling in the present study, additional mitogenomes covering more subfamilies/tribes (especially Theclinae and Polyommatinae) will be necessary to improve our understanding of mitogenomic phylogeny in Lycaenidae.
4.3 Adaptive evolution of Lycaenidae mitogenomes
Generally, mitochondrial genes show variation in evolutionary rates, reflecting differences in selective pressure (Chen et al., 2018; Sarvani et al., 2018; Sarkar et al., 2020). Low dN/dS (ω < 1) ratios for 13 PCGs implied that the mitogenomes underwent purifying or stabilizing selection. Actually, a harsh high-altitude environment would shrink the effective population size, resulting in decreased selection on mutations but increased effects of genetic drift (Gomaa et al., 2011). Therefore, the net effect of genetic drift can lose genetic diversity. In this case, the reduced dN/dS ratios might not be directly associated with high-altitude adaptation.
Additionally, it is generally believed that the functional importance of a gene dictates its level of conservation. This can explain the high conservation of cox1, a subunit of cytochrome oxidase, which is the fourth central enzyme complex of the respiratory electron transport chain and plays a crucial role in metabolism. The evolutionary rate of cox1 was the slowest among genes in this study, suggesting that purifying selection played a crucial role in its evolution, consistent with results for other insect species (Singh et al., 2017; Chen et al., 2020b; Yuan et al., 2020; Liu et al., 2022b). The harsh environment of the QTP may lead to strong selection pressure. In particular, mutations in genes related to oxygen use may be favored in the QTP. We detected a relatively significant signature of selection (p = 0.0550) in cox1 and found that non-synonymous substitutions affected the protein structure. The amino acid at residue 404 changed from proline (Pro, P) to asparagine (Asn, N), classified as a missense mutation. Missense mutations can cause the polypeptide chain to lose its original function and lead to protein abnormalities (Henderson et al., 2010; Beer et al., 2013; Caporali et al., 2018). Nevertheless, these were identified as beneficial mutations in this study and may be the key to high-altitude adaptation. The evolutionary process and the role of this beneficial missense mutation need to be verified by experiments.
Moreover, cox1 is used as a DNA barcode for species identification (Hebert et al., 2003). DNA barcoding is widely used in taxonomies (Bakhoum et al., 2018; Choudhary et al., 2018; DeSalle and Goldstein, 2019; Bianchi and Goncalves, 2021) and has been applied to many groups in Lepidoptera (Janzen et al., 2005; Song et al., 2014; Efetov et al., 2019). Therefore, the high conservation of cox1 in our study suggests that it can also be used for DNA barcoding in Lycaenidae. However, the accuracy of cox1 for species identification in Lycaenidae still needs more evaluation using experimental data, and its utility in phylogeny, population genetics, and phylogeography needs to be further verified.
In addition to PCGs, our research suggested that non-coding regions also underwent positive selection in the high-altitude environment. The largest non-coding region was the CR in mitogenomes, considered the start of transcription and showed typical structural characteristics similar to those in other animal mitogenomes (Yu et al., 2015; Sun et al., 2016; Yang et al., 2019b). IGR in trnQ-nad2 has been observed not only in Lycaenidae but also in most Lepidopteran species (Kim and Kim, 2016; Laemmermann et al., 2016; Kim et al., 2017; Sivasankaran et al., 2017; Wang et al., 2022b), and the stem-loop structure may also be widespread in Lepidoptera. IGR in trnS2-nad1 was observed in Lycaenidae, considered DmTTF, found in other insects (Beckenbach, 2012; Wang et al., 2014b). Intergenic motifs significantly longer than those in other species were found in some species of Lycaenidae, and these may be specific fragments, e.g., a 64-bp sequence in trnK-trnD of L. phlaeas (Zhang et al., 2013), a 26-bp sequence in trnR-trnN of Curetis bulis (Zhang et al., 2013), and a 29-bp sequence in cox3-trnG of A. orbitulus. These findings provide evidence for positive selection in non-coding regions.
Non-coding regions may also experience selection in the high-altitude QTP, as evidenced by the extension of non-coding fragments in QTP species and emergence of special structures. Analyses of all particular IGRs provide insight into evolutionary processes, which can be divided into four stages. In the first stage, the non-coding region and nad6-cob and cob-trnS2 IGRs arose. IGRs of 2–10 bp size arose in QTP species, while no or several nucleotides overlapped in other Lycaenidae. In the second stage, non-coding regions were extended. The size of trnP-nad6 IGR increased from 2 bp to 7 bp. In the third stage, secondary structures began to appear; however, the structures were not stable. Small units of “TA” and “TTA” and possible structural features appeared in QTP species at the cox3-trnG IGR, and a 10-bp motif and possible structure appeared in QTP species at the trnS1-trnE IGR. In the final stage, stable structures appeared. Between trnE and trnF, the secondary structure of the sequence could be predicted. The large non-coding region in trnE-trnF was considered DmTTF and had a similar structure to that of other insect species (Roberti et al., 2003; Beckenbach, 2012; Yuan et al., 2016; Wang et al., 2022a). Other insect groups also had the trnE-trnF IGR and other structural features consistent with repeat regions (Wu et al., 2012; Zhang et al., 2014; Chen et al., 2020a).
Research on IGRs has focused on large fragments (CR, trnE-trnF IGR, and trnS2-nad1 IGR) and taxa-specific fragments, with relatively little research on the evolution of small or medium-sized IGRs. These IGRs could provide important evidence for understanding the contribution of non-coding regions to adaptive evolution. The results of this study provide new insight into the adaptability of mitogenomes in extreme environments, like the QTP.
Mitochondrial hypoxia-related adaptive evolution can result in metabolic alterations that promote survival, to some extent. These adaptive changes, e.g., changes in the structure and function of proteins associated with oxidative phosphorylation (Yang et al., 2019a; Bartakova et al., 2021), physiological variation (Scott et al., 2018; Facultative myrmecophily (hymenopteraDawson and Scott, 2022), and metabolite changes (Bacchiocchi and Principato, 2000), are regulated by hypoxia-inducible factor and gene expression (Taylor, 2008; Goda and Kanai, 2012; Scott et al., 2018). The underlying mechanisms differ between groups (Mahalingam et al., 2017). PCGs and non-coding regions involved in mitogenome evolution in high-altitude conditions in Lycaenidae exhibit these changes in protein structure and function and are directly regulated by DmTTF. The shared characteristics in the two QTP species could be associated with high-altitude adaptation; however, elaborating on how the role of ancestral polymorphisms needs to be tested in future studies by using intensive taxon sampling and population genetic analysis.
5 Conclusion
In this study, we sequenced four mitogenomes of two Lycaenidae species inhabiting the QTP. The gene content, gene arrangement, base composition, codon usage, and transfer RNA genes in the sequence and structure showed high conservation within Lycaenidae. Mitogenomic data could provide effective phylogenetic signals for Lycaenidae. Both PCGs and non-coding regions were associated with high-altitude adaptation. Future studies including more mitogenomes and functional analyses of genes under positive selection and non-coding regions associated with environmental adaptation will improve our understanding of Lycaenidae evolution.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, ON411617-20.
Author contributions
M-LY conceived and designed the experiments. W-TC, ML, S-YH, and S-HW sampled butterfly specimens. W-TC, ML, and S-YH conducted experiments. W-TC, ML, S-HW, and M-LY performed data analyses. W-TC and ML wrote the manuscript. M-LY revised the manuscript. All authors read and approved the final version of the manuscript.
Funding
This study was funded by the Second Tibetan Plateau Scientific Expedition and Research (STEP) Program (2019QZKK0302) and the National Science and Technology Fundamental Resources Investigation Program of China (2019FY100400/2019FY100404).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1137588/full#supplementary-material
References
Adams, K. L., and Palmer, J. D. (2003). Evolution of mitochondrial gene content: Gene loss and transfer to the nucleus. Mol. Phylogenetics Evol. 29 (3), 380–395. doi:10.1016/s1055-7903(03)00194-5
Anisimova, M., and Kosiol, C. (2009). Investigating protein-coding sequence evolution with probabilistic codon substitution models. Mol. Biol. Evol. 26 (2), 255–271. doi:10.1093/molbev/msn232
Artem'eva, E. A. (2007). Phenotypic diversity in populations of the common blue butterfly Polyommatus icarus Rott. As a trend in the ecocenotic strategy of the species. Russ. J. Ecol. 38 (1), 58–67. doi:10.1134/s1067413607010109
Bacchiocchi, S., and Principato, G. (2000). Mitochondrial contribution to metabolic changes in the digestive gland of Mytilus galloprovincialis during anaerobiosis. J. Exp. Zoology 286 (2), 107–113. doi:10.1002/(sici)1097-010x(20000201)286:2<107:aid-jez1>3.0.co;2-8
Bakhoum, M. T., Sarr, M., Fall, A. G., Huber, K., Fall, M., Sembene, M., et al. (2018). DNA barcoding and molecular identification of field-collected Culicoides larvae in the Niayes area of Senegal. Parasites Vectors 11 (1), 615. doi:10.1186/s13071-018-3176-y
Balint, Z., Boyer, P., Farfan, J., Cerdena, J., and Pyrcz, T. W. (2022). A new high-altitude species of penaincisalia johnson, 1990 (Lepidoptera, Lycaenidae) from the Peruvian andes. Zootaxa 5154 (1), 49–59. doi:10.11646/zootaxa.5154.1.2
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19 (5), 455–477. doi:10.1089/cmb.2012.0021
Bartakova, V., Bryjova, A., Nicolas, V., Lavrenchenko, L. A., and Bryja, J. (2021). Mitogenomics of the endemic Ethiopian rats: Looking for footprints of adaptive evolution in sky islands. Mitochondrion 57 (8), 182–191. doi:10.1016/j.mito.2020.12.015
Beckenbach, A. T. (2012). Mitochondrial genome sequences of nematocera (lower Diptera): Evidence of rearrangement following a complete genome duplication in a winter crane fly. Genome Biol. Evol. 4 (2), 89–101. doi:10.1093/gbe/evr131
Beer, S., Zhou, J., Szabo, A., Keiles, S., Chandak, G. R., Witt, H., et al. (2013). Comprehensive functional analysis of chymotrypsin C (CTRC) variants reveals distinct loss-of-function mechanisms associated with pancreatitis risk. Gut 62 (11), 1616–1624. doi:10.1136/gutjnl-2012-303090
Behura, S. K., and Severson, D. W. (2011). Coadaptation of isoacceptor tRNA genes and codon usage bias for translation efficiency in Aedes aegypti and Anopheles gambiae. Insect Mol. Biol. 20 (2), 177–187. doi:10.1111/j.1365-2583.2010.01055.x
Behura, S. K., Stanke, M., Desjardins, C. A., Werren, J. H., and Severson, D. W. (2010). Comparative analysis of nuclear tRNA genes of Nasonia vitripennis and other arthropods, and relationships to codon usage bias. Insect Mol. Biol. 19 (1), 49–58. doi:10.1111/j.1365-2583.2009.00933.x
Benson, G. (1999). Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 27 (2), 573–580. doi:10.1093/nar/27.2.573
Bernt, M., Donath, A., Juehling, F., Externbrink, F., Florentz, C., Fritzsch, G., et al. (2013). Mitos: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenetics Evol. 69 (2), 313–319. doi:10.1016/j.ympev.2012.08.023
Bianchi, F. M., and Goncalves, L. T. (2021). Borrowing the pentatomomorpha tome from the DNA barcode library: Scanning the overall performance of cox1 as a tool. J. Zoological Syst. Evol. Res. 59 (5), 992–1012. doi:10.1111/jzs.12476
Blier, P. U., Dufresne, F., and Burton, R. S. (2001). Natural selection and the evolution of mtDNA-encoded peptides: Evidence for intergenomic co-adaptation. Trends Genet. 17 (7), 400–406. doi:10.1016/s0168-9525(01)02338-1
Boore, J. L. (1999). Animal mitochondrial genomes. Nucleic Acids Res. 27 (8), 1767–1780. doi:10.1093/nar/27.8.1767
Bottje, W. G. (2019). Oxidative metabolism and efficiency: The delicate balancing act of mitochondria. Poult. Sci. 98 (10), 4223–4230. doi:10.3382/ps/pey405
Caporali, L., Iommarini, L., La Morgia, C., Olivieri, A., Achilli, A., Maresca, A., et al. (2018). Peculiar combinations of individually nonpathogenic missense mitochondrial DNA variants cause low penetrance Leber's hereditary optic neuropathy. PLoS Genet. 14 (2), e1007210. doi:10.1371/journal.pgen.1007210
Chen, Y. X., Chen, Y. S., Shi, C. M., Huang, Z. B., Zhang, Y., Li, S. K., et al. (2017). SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. GigaScience 7 (1), 1–6. doi:10.1093/gigascience/gix120
Chen, W., Zhang, C. L., Pan, T., Liu, W., Li, K. X., Hu, C. C., et al. (2018). The mitochondrial genome of the kentish plover Charadrius alexandrinus (charadriiformes: Charadriidae) and phylogenetic analysis of charadrii. Genes. & Genomics. 40 (9), 955–963. doi:10.1007/s13258-018-0703-3
Chen, J., Jiang, L. Y., Zhang, X. L., and Qiao, G. X. (2020a). The complete mitochondrial genome of Schoutedenia ralumensis Rubsaamen, 1905 (Hemiptera: Aphididae: Greenideinae). Mitochondrial DNA Part B 5 (3), 2217–2218. doi:10.1080/23802359.2020.1768938
Chen, L., Wahlberg, N., Liao, C. Q., Wang, C. B., Ma, F. Z., and Huang, G. H. (2020b). Fourteen complete mitochondrial genomes of butterflies from the genus Lethe (Lepidoptera, Nymphalidae, Satyrinae) with mitogenome-based phylogenetic analysis. Genomics 112 (6), 4435–4441. doi:10.1016/j.ygeno.2020.07.042
Chen, Z., Liu, Y. Q., Wu, Y. F., Song, F., Cai, W. Z., and Li, H. (2020c). Novel tRNA gene rearrangements in the mitochondrial genome of Camarochiloides weiweii (Hemiptera: Pachynomidae). Int. J. Biol. Macromol. 165, 1738–1744. doi:10.1016/j.ijbiomac.2020.10.051
Choudhary, J. S., Naaz, N., Lemtur, M., Das, B., Singh, A. K., Bhatt, B. P., et al. (2018). Genetic analysis of Bactrocera zonata (Diptera: Tephritidae) populations from India based on cox1 and nad1 gene sequences. Mitochondrial DNA Part A 29 (5), 727–736. doi:10.1080/24701394.2017.1350952
da Fonseca, R. R., Johnson, W. E., O'Brien, S. J., Ramos, M. J., and Antunes, A. (2008). The adaptive evolution of the mammalian mitochondrial genome. BMC Genomics 9 (9), 119. doi:10.1186/1471-2164-9-119
Dawson, N. J., and Scott, G. R. (2022). Adaptive increases in respiratory capacity and O2 affinity of subsarcolemmal mitochondria from skeletal muscle of high-altitude deer mice. FASEB J. 36 (7), e22391. doi:10.1096/fj.202200219R
DeSalle, R., and Goldstein, P. (2019). Review and interpretation of trends in DNA barcoding. Front. Ecol. Evol. 7, 302. doi:10.3389/fevo.2019.00302
Ding, J. Y., Lan, H., Xu, W., Chen, Y. N., Wu, H., Jiang, H. M., et al. (2022). Two complete mitochondrial genomes in Scolopendra and a comparative analysis of tRNA rearrangements in centipedes. Mol. Biol. Rep. 49 (7), 6173–6180. doi:10.1007/s11033-022-07409-x
Efetov, K. A., Kirsanova, A. V., Lazareva, Z. S., Parshkova, E. V., Tarmann, G. M., Rougerie, R., et al. (2019). DNA barcoding of zygaenidae (Lepidoptera): Results and perspectives. Nota Lepidopterol. 42 (2), 137–150. doi:10.3897/nl.42.33190
Foroughmand-Araabi, M. H., Goliaei, B., Alishahi, K., Sadeghi, M., and Goliaei, S. (2015). Codon usage and protein sequence pattern dependency in different organisms: A bioinformatics approach. J. Bioinforma. Comput. Biol. 13 (2), 1550002. doi:10.1142/S021972001550002X
Gan, H. M., Tan, M. H., Gan, H. Y., Lee, Y. P., and Austin, C. M. (2016). The complete mitogenome of the Norway lobster Nephrops norvegicus (linnaeus, 1758) (Crustacea: Decapoda: Nephropidae). Mitochondrial DNA Part A 27 (5), 3179–3180. doi:10.3109/19401736.2015.1007325
Goda, N., and Kanai, M. (2012). Hypoxia-inducible factors and their roles in energy metabolism. Int. J. Hematol. 95 (5), 457–463. doi:10.1007/s12185-012-1069-y
Gomaa, N. H., Montesinos-Navarro, A., Alonso-Blanco, C., and Pico, F. X. (2011). Temporal variation in genetic diversity and effective population size of Mediterranean and subalpine Arabidopsis thaliana populations. Mol. Ecol. 20 (17), 3540–3554. doi:10.1111/j.1365-294X.2011.05193.x
Gu, P., Liu, W., Yao, Y. F., Ni, Q. Y., Zhang, M. W., Li, D. Y., et al. (2016). Evidence of adaptive evolution of alpine pheasants to high-altitude environment from mitogenomic perspective. Mitochondrial DNA Part A 27 (1), 455–462. doi:10.3109/19401736.2014.900667
Hebert, P. D. N., Cywinska, A., Ball, S. L., and DeWaard, J. R. (2003). Biological identifications through DNA barcodes. Proc. R. Soc. B Biol. Sci. 270 (1512), 313–321. doi:10.1098/rspb.2002.2218
Henderson, D. M., Lee, A., and Ervasti, J. M. (2010). Disease-causing missense mutations in actin binding domain 1 of dystrophin induce thermodynamic instability and protein aggregation. Proc. Natl. Acad. Sci. U. S. A. 107 (21), 9632–9637. doi:10.1073/pnas.1001517107
Hu, C. Y., Wang, S. B., Huang, B. S., Liu, H. G., Xu, L., Hu, Z. G., et al. (2020). The complete mitochondrial genome sequence of Scolopendra mutilans L. Koch, 1878 (Scolopendromorpha, Scolopendridae), with a comparative analysis of other centipede genomes. ZooKeys 925 (1), 73–88. doi:10.3897/zookeys.925.47820
Hughes, J. B. (2000). The scale of resource specialization and the distribution and abundance of lycaenid butterflies. Oecologia 123 (3), 375–383. doi:10.1007/s004420051024
Janzen, D. H., Hajibabaei, M., Burns, J. M., Hallwachs, W., Remigio, E., and Hebert, P. D. N. (2005). Wedding biodiversity inventory of a large and complex Lepidoptera fauna with DNA barcoding. Philosophical Trans. R. Soc. B Biol. Sci. 360 (1462), 1835–1845. doi:10.1098/rstb.2005.1715
Jeong, S. Y., Kim, M. J., Kim, S. S., and Kim, I. (2017). Complete mitochondrial genome of the endangered Lycaenid butterfly Shijimiaeoides divina (Lepidoptera: Lycaenidae). Mitochondrial DNA Part A 28 (2), 242–243. doi:10.3109/19401736.2015.1115860
Kaminski, L. A. (2008). Polyphagy and obligate myrmecophily in the butterfly Hallonympha paucipuncta (Lepidoptera: Riodinidae) in the neotropical Cerrado Savanna. Biotropica 40 (3), 390–394. doi:10.1111/j.1744-7429.2007.00379.x
Kim, M. J., and Kim, I. (2016). Complete mitochondrial genome of the Mormon metalmark butterfly, Apodemia mormo (Lepidoptera: Riodinidae). Mitochondrial DNA Part A 27 (2), 789–791. doi:10.3109/19401736.2014.915539
Kim, I., Lee, E. M., Seol, K. Y., Yun, E. Y., Lee, Y. B., Hwang, J. S., et al. (2006). The mitochondrial genome of the Korean hairstreak, Coreana raphaelis (Lepidoptera: Lycaenidae). Insect Mol. Biol. 15 (2), 217–225. doi:10.1111/j.1365-2583.2006.00630.x
Kim, M. J., Kang, A. R., Jeong, H. C., Kim, K. G., and Kim, I. (2011). Reconstructing intraordinal relationships in Lepidoptera using mitochondrial genome data with the description of two newly sequenced lycaenids, Spindasis takanonis and Protantigius superans (Lepidoptera: Lycaenidae). Mol. Phylogenetics Evol. 61 (2), 436–445. doi:10.1016/j.ympev.2011.07.013
Kim, S. R., Kim, J. S., Kim, K. Y., Kim, M. J., Jeong, J. S., and Kim, I. (2017). Complete mitochondrial genome of the wild silkmoth, Saturnia boisduvalii (Lepidoptera: Saturniidae). Entomological Res. 47 (6), 344–351. doi:10.1111/1748-5967.12256
Korkmaz, E. M., Aydemir, H. B., Temel, B., Budak, M., and Basibuyuk, H. H. (2017). Mitogenome evolution in cephini (hymenoptera: Cephidae): Evidence for parallel adaptive evolution. Biochem. Syst. Ecol. 71, 137–146. doi:10.1016/j.bse.2017.02.004
Kubik, T. D., and Schorr, R. A. (2018). Facultative myrmecophily (Hymenoptera: Formicidae) in the hops blue butterfly, Celastrina humulus (lepidoptera: Lycaenidae). Entomol. News 127 (5), 490–498. doi:10.3157/021.127.0514
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35 (6), 1547–1549. doi:10.1093/molbev/msy096
Laemmermann, K., Vogel, H., and Traut, W. (2016). The mitochondrial genome of the mediterranean flour moth, Ephestia kuehniella (Lepidoptera: Pyralidae), and identification of invading mitochondrial sequences (numts) in the W chromosome. Eur. J. Entomology 113 (1), 482–488. doi:10.14411/eje.2016.063
Li, Y. L., Ren, Z. M., Shedlock, A. M., Wu, J. Q., Sang, L., Tersing, T., et al. (2013). High altitude adaptation of the schizothoracine fishes (Cyprinidae) revealed by the mitochondrial genome analyses. Gene 517 (2), 169–178. doi:10.1016/j.gene.2012.12.096
Li, M. Z., Jin, L., Ma, J. D., Tian, S. L., Li, R. Q., and Li, X. W. (2016). Detecting mitochondrial signatures of selection in wild Tibetan pigs and domesticated pigs. Mitochondrial DNA Part A 27 (1), 747–752. doi:10.3109/19401736.2014.913169
Li, Q., Wang, Q. F., Jin, X., Chen, Z. Q., Xiong, C., Li, P., et al. (2019). Characterization and comparative analysis of six complete mitochondrial genomes from ectomycorrhizal fungi of the Lactarius genus and phylogenetic analysis of the Agaricomycetes. Int. J. Biol. Macromol. 121 (2), 249–260. doi:10.1016/j.ijbiomac.2018.10.029
Li, F., Lv, Y. Y., Wen, Z. Y., Bian, C., Zhang, X. H., Guo, S. T., et al. (2021). The complete mitochondrial genome of the intertidal spider (Desis jiaxiangi) provides novel insights into the adaptive evolution of the mitogenome and the evolution of spiders. BMC Ecol. Evol. 21 (1), 72. doi:10.1186/s12862-021-01803-y
Li, M., Chen, W. T., Zhang, Q. L., Liu, M., Xing, C. W., Cao, Y., et al. (2022). Mitochondrial phylogenomics provides insights into the phylogeny and evolution of spiders (Arthropoda: Araneae). Zoological Res. 43 (4), 566–584. doi:10.24272/j.issn.2095-8137.2021.418
Liu, Q. N., Xin, Z. Z., Bian, D. D., Chai, X. Y., Zhou, C. L., and Tang, B. P. (2016). The first complete mitochondrial genome for the subfamily Limacodidae and implications for the higher phylogeny of Lepidoptera. Sci. Rep. 6 (1), 35878. doi:10.1038/srep35878
Liu, D., Basso, A., Babbucci, M., Patarnello, T., and Negrisolo, E. (2022a). Macrostructural evolution of the mitogenome of butterflies (Lepidoptera, Papilionoidea). Insects 13 (4), 358. doi:10.3390/insects13040358
Liu, N., Fang, L. J., and Zhang, Y. L. (2022b). The complete mitochondrial genomes of four species in the subfamily Limenitidinae (Lepidoptera, Nymphalidae) and a phylogenetic analysis. Insects 13 (1), 16. doi:10.3390/insects13010016
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25 (5), 955–964. doi:10.1093/nar/25.5.955
Luo, Y. J., Chen, Y., Liu, F. Y., and Gao, Y. Q. (2012). Mitochondrial genome of Tibetan wild ass (Equus kiang) reveals substitutions in NADH which may reflect evolutionary adaptation to cold and hypoxic conditions. Asia Life Sci. 21 (1), 1–11.
Ma, X. X., Feng, Y. P., Bai, J. L., Zhang, D. R., Lin, X. S., and Ma, Z. R. (2015). Nucleotide composition bias and codon usage trends of gene populations in Mycoplasma capricolum subsp capricolum and M. agalactiae. J. Genet. 94 (2), 251–260. doi:10.1007/s12041-015-0512-2
Mahalingam, S., McClelland, G. B., and Scott, G. R. (2017). Evolved changes in the intracellular distribution and physiology of muscle mitochondria in high-altitude native deer mice. J. Physiology 595 (14), 4785–4801. doi:10.1113/JP274130
Manoli, I., Alesci, S., Blackman, M. R., Su, Y. A., Rennert, O. M., and Chrousos, G. P. (2007). Mitochondria as key components of the stress response. Trends Endocrinol. Metabolism 18 (5), 190–198. doi:10.1016/j.tem.2007.04.004
Marabuto, E., Pires, P., Romao, F., Lemos, P., and Merckx, T. (2022). A review of the distribution and ecology of the elusive Brown Hairstreak butterfly Thecla betulae (Lepidoptera, Lycaenidae) in the Iberian Peninsula. Nota Lepidopterol. 45 (161), 101–118. doi:10.3897/nl.45.76222
Meng, Y. F., Chen, C. F., Huang, Y. X., Wang, X., and Zhang, B. (2022). Phylogenetic relationship and characterization of the complete mitochondrial genome sequence of Smerinthus planus (Lepidoptera: Sphingidae). Mitochondrial DNA Part B 7 (6), 941–943. doi:10.1080/23802359.2022.2080014
Milenkovic, D., Mueller, J., Stojanovski, D., Pfanner, N., and Chacinska, A. (2007). Diverse mechanisms and machineries for import of mitochondrial proteins. Biol. Chem. 388 (9), 891–897. doi:10.1515/BC.2007.097
Mu, W. D., Liu, J., and Zhang, H. B. (2018). The first complete mitochondrial genome of the Mariana Trench Freyastera benthophila (Asteroidea: Brisingida: Brisingidae) allows insights into the deep-sea adaptive evolution of Brisingida. Ecol. Evol. 8 (22), 10673–10686. doi:10.1002/ece3.4427
Nemet, E., Czekes, Z., Marko, B., and Rakosy, L. (2016). Host plant preference in the protected myrmecophilous Transylvanian Blue (Pseudophilotes bavius hungarica) butterfly (Lepidoptera: Lycaenidae) and its relationship with potential ant partners. J. Insect Conservation 20 (5), 765–772. doi:10.1007/s10841-016-9907-5
Oliveira, M. T., Barau, J. G., Martins Junqueira, A. C., Feijao, P. C., da Rosa, A. C., Abreu, C. F., et al. (2008). Structure and evolution of the mitochondrial genomes of Haematobia irritans and Stomoxys calcitrans: The muscidae (Diptera: Calyptratae) perspective. Mol. Phylogenetics Evol. 48 (3), 850–857. doi:10.1016/j.ympev.2008.05.022
Pang, Y. L. J., Abo, R., Levine, S. S., and Dedon, P. C. (2014). Diverse cell stresses induce unique patterns of tRNA up- and down-regulation: tRNA-Seq for quantifying changes in tRNA copy number. Nucleic Acids Res. 42 (22), e170. doi:10.1093/nar/gku945
Peng, Q. K., Tang, L., Tan, S., Li, Z. G., Wang, J. F., and Zou, F. D. (2012). Mitogenomic analysis of the genus pseudois: Evidence of adaptive evolution of morphological variation in the ATP synthase genes. Mitochondrion 12 (5), 500–505. doi:10.1016/j.mito.2012.07.107
Perna, N. T., and Kocher, T. D. (1995). Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 41 (3), 353–358. doi:10.1007/BF00186547
Pierce, N. E., Braby, M. F., Heath, A., Lohman, D. J., Mathew, J., Rand, D. B., et al. (2002). The ecology and evolution of ant association in the Lycaenidae (Lepidoptera). Annu. Rev. Entomology 47 (1), 733–771. doi:10.1146/annurev.ento.47.091201.145257
Pond, S. L. K., and Frost, S. D. W. (2005). Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 22 (5), 1208–1222. doi:10.1093/molbev/msi105
Qi, L. Q., Zhang, H., Wang, X., and Huang, Y. X. (2022). The complete mitochondrial genome sequence of the hawkmoth, Dahira obliquifascia (Lepidoptera: Sphingidae) and phylogenetic analysis. Mitochondrial DNA Part B 7 (2), 339–340. doi:10.1080/23802359.2022.2032436
Riva, F., Barbero, F., Bonelli, S., Balletto, E., and Casacci, L. P. (2017). The acoustic repertoire of lycaenid butterfly larvae. Bioacoustics 26 (1), 77–90. doi:10.1080/09524622.2016.1197151
Roberti, M., Polosa, P. L., Bruni, F., Musicco, C., Gadaleta, M. N., and Cantatore, P. (2003). DmTTF, a novel mitochondrial transcription termination factor that recognises two sequences of Drosophila melanogaster mitochondrial DNA. Nucleic Acids Res. 31 (6), 1597–1604. doi:10.1093/nar/gkg272
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Hohna, S., et al. (2012). MrBayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61 (3), 539–542. doi:10.1093/sysbio/sys029
Rozas, J., Ferrer-Mata, A., Sanchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of largedata sets. Mol. Biol. Evol. 34 (12), 3299–3302. doi:10.1093/molbev/msx248
Sarkar, I., Dey, P., Sharma, S. K., Ray, S. D., Kochiganti, V. H. S., Singh, R., et al. (2020). Turdoides affinis mitogenome reveals the translational efficiency and importance of NADH dehydrogenase complex-I in the Leiothrichidae family. Sci. Rep. 10 (1), 16202. doi:10.1038/s41598-020-72674-4
Sarvani, R. K., Parmar, D. R., Tabasum, W., Thota, N., Sreenivas, A., and Gaur, A. (2018). Characterization of the complete mitogenome of Indian mouse deer, Moschiola indica (Artiodactyla: Tragulidae) and its evolutionary significance. Sci. Rep. 8 (1), 2697. doi:10.1038/s41598-018-20946-5
Schar, S., Eastwood, R., Arnaldi, K. G., Talavera, G., Kaliszewska, Z. A., Boyle, J. H., et al. (2018). Ecological specialization is associated with genetic structure in the ant-associated butterfly family Lycaenidae. Proc. R. Soc. B Biol. Sci. 285 (1886), 20181158. doi:10.1098/rspb.2018.1158
Schmidt, D. J., and Rice, S. J. (2002). Association of ants with juvenile Ogyris amaryllis amaryllis Hewitson (Lepidoptera: Lycaenidae) in south-eastern Queensland. Aust. J. Entomology 41 (2), 164–169. doi:10.1046/j.1440-6055.2002.00278.x
Scott, G. R., Guo, K. H., and Dawson, N. J. (2018). The mitochondrial basis for adaptive variation in aerobic performance in high-altitude deer mice. Integr. Comp. Biol. 58 (3), 506–518. doi:10.1093/icb/icy056
Shang, Y. Q., Wang, X. B., Liu, G., Wu, X. Y., Wei, Q. G., Sun, G. L., et al. (2022). Adaptability and evolution of Gobiidae: A genetic exploration. Animals 12 (14), 1741. doi:10.3390/ani12141741
Shi, Q. H., Sun, G., Fang, Y., Zhang, L. H., and Zhang, J. C. (2020). The complete mitochondrial genome of Punchinello butterfly, Zemeros flegyas (Lepidoptera: Riodinidae) and its phylogenetic implications. Mitochondrial DNA Part B 5 (2), 1567–1569. doi:10.1080/23802359.2020.1742604
Singh, D., Kabiraj, D., Sharma, P., Chetia, H., Mosahari, P. V., Neog, K., et al. (2017). The mitochondrial genome of Muga silkworm (Antheraea assamensis) and its comparative analysis with other lepidopteran insects. PLoS One 12 (11), e0188077. doi:10.1371/journal.pone.0188077
Sivasankaran, K., Mathew, P., Anand, S., Ceasar, S. A., Mariapackiam, S., and Ignacimuthu, S. (2017). Complete mitochondrial genome sequence of fruit-piercing moth Eudocima phalonia (Linnaeus, 1763) (Lepidoptera: Noctuoidea). Genomics data 14, 66–81. doi:10.1016/j.gdata.2017.09.004
Song, S. B., Shi, Z. Y., Jin, Q., Han, H. L., Liu, X. F., Hao, M. D., et al. (2014). Species identification of noctuidae moths (Insecta: Lepidoptera) from baoding and langfang, hebei, China with DNA barcoding and establishment of a local DNA barcoding library. Chin. J. Appl. Entomology 51 (1), 156–168.
Stamatakis, A. (2014). RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30 (9), 1312–1313. doi:10.1093/bioinformatics/btu033
Stekolnikov, A. A., Lukhtanov, V. A., and Korzeev, A. I. (2013). Congruence between comparative morphology and molecular phylogenies: Evolution of the male genitalia skeletal/muscular system in the subtribe Polyommatina (Lepidoptera, Lycaenidae). Entomol. Rev. 92 (3), 517–536. doi:10.1134/s0013873814020031
Sun, L. Y., Zhang, D. C., Guo, H. Y., Jiang, S. G., and Zhu, C. Y. (2016). Complete mitochondrial genome sequence of golden pompano Trachinotus ovatus. Mitochondrial DNA Part A 27 (2), 871–872. doi:10.3109/19401736.2014.919482
Sun, Y. X., Zhu, Y. S., Chen, C., Zhu, Q. S., Zhu, Q. Q., Zhou, Y. Y., et al. (2020). The complete mitochondrial genome of Dysgonia stuposa (Lepidoptera: Erebidae) and phylogenetic relationships within Noctuoidea. PeerJ 8 (3), e8780. doi:10.7717/peerj.8780
Sun, Y., Wang, J., and Wang, X. (2022). Sequencing and analysis of the complete mitochondrial genome of Laothoe amurensis sinica (Lepidoptera: Sphingidae) from China and its phylogenetic analysis. Mitochondrial DNA Part B 7 (5), 750–752. doi:10.1080/23802359.2022.2070043
Talavera, G., Lukhtanov, V. A., Pierce, N. E., and Vila, R. (2013). Establishing criteria for higher-level classification using molecular data: The systematics of Polyommatus blue butterflies (Lepidoptera, Lycaenidae). Cladistics 29 (2), 166–192. doi:10.1111/j.1096-0031.2012.00421.x
Taylor, C. T. (2008). Mitochondria and cellular oxygen sensing in the HIF pathway. Biochem. J. 409 (1), 19–26. doi:10.1042/BJ20071249
Trifinopoulos, J., Lam-Tung, N., von Haeseler, A., and MinhW-Iq-Tree, B. Q. (2016). W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44 (W1), W232–W235. doi:10.1093/nar/gkw256
Tripodi, F., Castoldi, A., Nicastro, R., Reghellin, V., Lombardi, L., Airoldi, C., et al. (2018). Methionine supplementation stimulates mitochondrial respiration. Biochimica Biophysica Acta-Molecular Cell. Res. 1865 (12), 1901–1913. doi:10.1016/j.bbamcr.2018.09.007
Tyagi, K., Kumar, V., Poddar, N., Prasad, P., Tyagi, I., Kundu, S., et al. (2020). The gene arrangement and phylogeny using mitochondrial genomes in spiders (Arachnida: Araneae). Int. J. Biol. Macromol. 146, 488–496. doi:10.1016/j.ijbiomac.2020.01.014
Ugelvig, L. V., Vila, R., Pierce, N. E., and Nash, D. R. (2011). A phylogenetic revision of the Glaucopsyche section (Lepidoptera: Lycaenidae), with special focus on the Phengaris-Maculinea clade. Mol. Phylogenetics Evol. 61 (1), 237–243. doi:10.1016/j.ympev.2011.05.016
Wahlberg, N., Braby, M. F., Brower, A. V. Z., de Jong, R., Lee, M. M., Nylin, S., et al. (2005). Synergistic effects of combining morphological and molecular data in resolving the phylogeny of butterflies and skippers. Proc. R. Soc. B Biol. Sci. 272 (1572), 1577–1586. doi:10.1098/rspb.2005.3124
Wang, A. R., Jeong, H. C., Han, Y. S., and Kim, I. (2014a). The complete mitochondrial genome of the mountainous duskywing, Erynnis montanus (Lepidoptera: Hesperiidae): A new gene arrangement in Lepidoptera. Mitochondrial DNA 25 (2), 93–94. doi:10.3109/19401736.2013.784752
Wang, Y., Li, H., Wang, P., Song, F., and Cai, W. (2014b). Comparative mitogenomics of plant bugs (Hemiptera: Miridae): Identifying the AGG codon reassignments between serine and lysine. PLoS One 9 (7), e101375. doi:10.1371/journal.pone.0101375
Wang, Y., Shen, Y. J., Feng, C. G., Zhao, K., Song, Z. B., Zhang, Y. P., et al. (2016). Mitogenomic perspectives on the origin of Tibetan loaches and their adaptation to high altitude. Sci. Rep. 6 (1), 29690. doi:10.1038/srep29690
Wang, C., Lai, T. H., Ye, P. Y., Yan, Y. R., Feutry, P., He, B. Y., et al. (2022a). Novel duplication remnant in the first complete mitogenome of Hemitriakis japanica and the unique phylogenetic position of family Triakidae. Gene. 820 (4), 146232. doi:10.1016/j.gene.2022.146232
Wang, J., Xin, T. R., Li, Z. Z., Zhang, X. J., Zou, Z. W., and Xia, B. (2022b). Complete mitochondrial genome of Idea leuconoe (Lepidoptera: Danaidae) and related phylogenetic analyses. Archives Insect Biochem. Physiology 111 (1), e21868. doi:10.1002/arch.21868
Wei, Z. X., Sun, G., Shiu, J. Y., Fang, Y., and Shi, Q. H. (2021). The complete mitochondrial genome sequence of Dodona eugenes (Lepidoptera: Riodinidae). Mitochondrial DNA Part B 6 (3), 816–818. doi:10.1080/23802359.2021.1884014
Wu, Y. P., Li, J., Zhao, J. L., Su, T. J., Luo, A. R., Fan, R. J., et al. (2012). The complete mitochondrial genome of the rice moth, Corcyra cephalonica. J. Insect Sci. 12 (1), 72. doi:10.1673/031.012.7201
Wu, Y. P., Liu, X. R., Zhang, Y. L., Fang, H., Lu, J. J., and Wang, J. P. (2022). Characterization of four mitochondrial genomes of Crambidae (Lepidoptera, Pyraloidea) and phylogenetic implications. Archives Insect Biochem. Physiology 112 (1), e21914. doi:10.1002/arch.21914
Xia, X. H. (2013). DAMBE5: A comprehensive software package for data analysis in molecular biology and evolution. Mol. Biol. Evol. 30 (7), 1720–1728. doi:10.1093/molbev/mst064
Yang, M., Gong, L., Sui, J. X., and Li, X. Z. (2019a). The complete mitochondrial genome of Calyptogena marissinica (Heterodonta: Veneroida: Vesicomyidae): Insight into the deep-sea adaptive evolution of vesicomyids. PLoS One 14 (9), e0217952. doi:10.1371/journal.pone.0217952
Yang, M. S., Song, L., Shi, Y. X., Yin, Y. J., Wang, Y. Y., Zhang, P. P., et al. (2019b). The complete mitochondrial genome of a medicinal insect, Hydrillodes repugnalis (Lepidoptera: Noctuoidea: Erebidae), and related phylogenetic analysis. Int. J. Biol. Macromol. 123, 485–493. doi:10.1016/j.ijbiomac.2018.10.149
Yang, M. S., Zhang, H. F., Song, L., Shi, Y. X., and Liu, X. M. (2019c). The complete mitochondrial genome of Mahanta tanyae compared with other zygaenoid moths (Lepidoptera: Zygaenoidea). J. Asia-Pacific Entomology 22 (2), 513–521. doi:10.1016/j.aspen.2019.03.010
Yang, Z. H. (2007). PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24 (8), 1586–1591. doi:10.1093/molbev/msm088
Yu, L., Wang, X. P., Ting, N., and Zhang, Y. P. (2011). Mitogenomic analysis of Chinese snub-nosed monkeys: Evidence of positive selection in NADH dehydrogenase genes in high-altitude adaptation. Mitochondrion 11 (3), 497–503. doi:10.1016/j.mito.2011.01.004
Yu, J. N., Chung, C. U., and Kwak, M. (2015). The complete mitochondrial genome sequence of the Korean hare (Lepus coreanus). Mitochondrial DNA 26 (1), 129–130. doi:10.3109/19401736.2013.815170
Yuan, M. L., Zhang, Q. L., Guo, Z. L., Wang, J., and Shen, Y. Y. (2015). Comparative mitogenomic analysis of the superfamily Pentatomoidea (Insecta: Hemiptera: Heteroptera) and phylogenetic implications. BMC Genomics 16 (1), 460. doi:10.1186/s12864-015-1679-x
Yuan, M. L., Zhang, Q. L., Zhang, L., Guo, Z. L., Liu, Y. J., Shen, Y. Y., et al. (2016). High-level phylogeny of the Coleoptera inferred with mitochondrial genome sequences. Mol. Phylogenetics Evol. 104, 99–111. doi:10.1016/j.ympev.2016.08.002
Yuan, M. L., Zhang, Q. L., Zhang, L., Jia, C. L., Li, X. P., Yang, X. Z., et al. (2018). Mitochondrial phylogeny, divergence history and high-altitude adaptation of grassland caterpillars (Lepidoptera: Lymantriinae: Gynaephora) inhabiting the Tibetan Plateau. Mol. Phylogenetics Evol. 122, 116–124. doi:10.1016/j.ympev.2018.01.016
Yuan, M. L., Zhang, L. J., Zhang, Q. L., Zhang, L., Li, M., Wang, X. T., et al. (2020). Mitogenome evolution in ladybirds: Potential association with dietary adaptation. Ecol. Evol. 10 (2), 1042–1053. doi:10.1002/ece3.5971
Zardoya, R., and Suadrez, M. (2008). Sequencing and phylogenomic analysis of whole mitochondrial genomes of animals. Methods Mol. Biol. 422, 185–200. doi:10.1007/978-1-59745-581-7_12
Zhang, J. Z., Nielsen, R., and Yang, Z. H. (2005). Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 22 (12), 2472–2479. doi:10.1093/molbev/msi237
Zhang, L. L., Hao, J. S., Huang, D. Y., Sun, X. Y., Hao, J. J., Peng, C. M., et al. (2013). Complete mitochondrial genomes of the bright sunbeam Curetis bulis and the small copper Lycaena phlaeas (Lepidoptera: Lycaenidae) and their phylogenetic implications. Genet. Mol. Res. 12 (4), 4434–4445. doi:10.4238/2013.October.10.9
Zhang, B., Ma, C., Edwards, O., Fuller, S., and Kang, L. (2014). The mitochondrial genome of the Russian wheat aphid Diuraphis noxia: Large repetitive sequences between trnE and trnF in aphids. Gene 533 (1), 253–260. doi:10.1016/j.gene.2013.09.064
Zhang, Q. L., Zhang, L., Yang, X. Z., Wang, X. T., Li, X. P., Wang, J., et al. (2017). Comparative transcriptomic analysis of Tibetan Gynaephora to explore the genetic basis of insect adaptation to divergent altitude environments. Sci. Rep. 7 (1), 16972. doi:10.1038/s41598-017-17051-4
Zhang, L. J., Li, Y. J., Ge, X. Y., Li, X. Y., Yang, Y. X., Bai, M., et al. (2022). Mitochondrial genomes of sternochetus species (Coleoptera: Curculionidae) and the phylogenetic implications. Archives Insect Biochem. Physiology 111 (1), e21898. doi:10.1002/arch.21898
Zhou, T. C., Shen, X. J., Irwin, D. M., Shen, Y. Y., and Zhang, Y. P. (2014). Mitogenomic analyses propose positive selection in mitochondrial genes for high-altitude adaptation in galliform birds. Mitochondrion 18, 70–75. doi:10.1016/j.mito.2014.07.012
Zhou, Y., Wang, S. Q., Wang, N., Liang, Z. Y., Zhong, H. H., Liu, Y. L., et al. (2020). Phylogenetic inference of Plebejus argus (Lepidoptera: Lycaenidae) using its complete mitochondrial genome with an extra copy of tRNASer. Mitochondrial DNA Part B 5 (2), 1584–1585. doi:10.1080/23802359.2020.1742615
Zhou, J. H., Li, H., Li, X. R., Gao, J., Long, X., Han, S. Y., et al. (2021). Tracing Brucella evolutionary dynamics in expanding host ranges through nucleotide, codon and amino acid usages in genomes. J. Biomol. Struct. Dyn. 39 (11), 3986–3995. doi:10.1080/07391102.2020.1773313
Keywords: insects, Lycaenidae, comparative mitogenomics, phylogeny, high-altitude adaptation, non-coding regions
Citation: Chen W-T, Li M, Hu S-Y, Wang S-H and Yuan M-L (2023) Comparative mitogenomic and evolutionary analysis of Lycaenidae (Insecta: Lepidoptera): Potential association with high-altitude adaptation. Front. Genet. 14:1137588. doi: 10.3389/fgene.2023.1137588
Received: 04 January 2023; Accepted: 03 April 2023;
Published: 18 April 2023.
Edited by:
Liandong Yang, Chinese Academy of Sciences (CAS), ChinaReviewed by:
Zhiqiang Ye, Arizona State University, United StatesKai Jun Zhang, Southwest University, China
Copyright © 2023 Chen, Li, Hu, Wang and Yuan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ming-Long Yuan, eXVhbm1sQGx6dS5lZHUuY24=
†These authors have contributed equally to this work