 Hong-Feng Liang1Wei-Min Liang2Wen-Guang Xie2Fen Lin3Li-Li Liu1Lie-Jun Li4Yi-Yuan Ge4Min Lu4Yu-Wei Liao1Guang-Kuan Zeng1Jin-Xiu Yao5Jing-Wei Situ5
Hong-Feng Liang1Wei-Min Liang2Wen-Guang Xie2Fen Lin3Li-Li Liu1Lie-Jun Li4Yi-Yuan Ge4Min Lu4Yu-Wei Liao1Guang-Kuan Zeng1Jin-Xiu Yao5Jing-Wei Situ5 Li-Ye Yang1*
Li-Ye Yang1*- 1Precision Medical Lab Center, People’s Hospital of Yangjiang, Yangjiang, Guangdong, China
- 2Medical Laboratory, Women and Children Hospital of Yangjiang, Yangjiang, Guangdong, China
- 3Precision Medical Lab Center, Chaozhou Central Hospital Affiliated to Southern Medical University, Chaozhou, Guangdong, China
- 4Guangdong Hybribio Limited Corporation, Chaozhou, Guangdong, China
- 5Laboratory Medical Center, People’s Hospital of Yangjiang, Yangjiang, Guangdong, China
Background: Thalassemia presents a higher incidence in southern China. The objective of this study is to analyze the genotype distribution of thalassemia in Yangjiang, a western city of Guangdong Province in China.
Methods: The genotypes of suspected cases with thalassemia were tested by PCR and reverse dot blot (RDB). Unidentified rare thalassemia genotypes of the samples were further ascertained by PCR and direct DNA sequencing.
Results: Among 22467 suspected cases with thalassemia, 7658 cases were found with thalassemia genotypes using our PCR-RDB kit. Among these 7658 cases, 5313 cases were found with α-thalassemia (α-thal) alone, --SEA/αα was the most common genotype, accounting for 61.75% of α-thal genotypes, and the following mutations were found: α3.7/αα, -α4.2/αα, αCSα/αα, αWSα/αα, and αQSα/αα. A total of 2032 cases were found with β-thalassemia (β-thal) alone. βCD41-42/βN, βIVS−II−654/βN, and β−28/βN accounted for 80.9% of all β-thal genotypes, and the following genotypes were found: βCD17/βN, βCD71-72/βN, and βE/βN. Compound heterozygotes of β-thal and β-thalassemia homozygotes were identified in 11 and five cases, respectively, in this study. α-thal combined with β-thal was identified in 313 cases, showing 57 genotype combinations of the coincidence of both Hb disorders; one extreme patient had a genotype of --SEA/αWSα and βCD41-42/β−28. In addition, four rare α-mutations (--THAI, HKαα, Hb Q-Thailand, and CD31 AGG>AAG) and six rare β-mutations (CD39 CAG>TAG, IVS-Ⅱ-2 (−T), −90(C>T), Chinese Gγ+(Aγδβ)0, CD104 (-G), and CD19 A>G) were also found in this study population.
Conclusion: This study provided detailed genotypes of thalassemia in Yangjiang of western Guangdong Province in China and reflected the complexity of genotypes in this high-prevalence region, and this would be valuable for diagnosis and counseling for thalassemia in this area.
1 Introduction
Thalassemia is one of the most common human genetic disorders that are characterized by decreased or absent synthesis of one or more hemoglobin polypeptide chains. Thalassemia carriers are found at variable frequencies in all subtropical and tropical populations. Southern China has a high incidence of thalassemia, with a carrier frequency of 3%–24% (Weatherall, 1997). Of all hemoglobin disorders, α-thalassemia (α-thal) and β-thalassemia (β-thal) are the most widely distributed, and therefore many individuals in these areas have complex combinations of these variants (Weatherall, 1997).
Clinical manifestations of α-thal range from asymptomatic cases to patients with lethal hydrops fetalis syndrome. Patients with β-thal homozygote present severe thalassemia, while the patients with compound heterozygosity, such as β-thal/Hb E, present variable severity of anemia (Weatherall, 2004). Severe thalassemia poses a major public health concern in areas where thalassemia is endemic.
Yangjiang, with an area of approximately 7,813.4 km2 and a population of 2.7 million, includes two counties and two prefectures, locates to the west of Guangdong Province in southern China, and borders Maoming to the southwest, Yunfu to the north, and Jiangmen to the east. All these places are known to be the regions of prevalence of thalassemia (Xu et al., 2004; Yin et al., 2014).
A previous study showed that the frequency of thalassemia in a county in Yangjiang was 20%, and about 5% of the population was β-thal carriers, which was the highest in Guangdong Province (Yin et al., 2014). Thalassemia has different carrier frequencies in different regions, with strong genetic heterogeneity and ethnic differences. To date, the gene distribution and complexity of thalassemia in whole Yangjiang have not been reported. Therefore, it is necessary to clarify the true distribution of various thalassemia genotypes and their co-inheritance patterns in this area of southern China.
In this study, we performed genotyping of the six α-thal defects [--SEA, -α3.7, -α4.2, αCS (Constant Spring), αQS (Quong Sze), and αWS (Westmead)] and 19 common β-thal mutations in two hospital-based populations in Yangjiang City. An unidentified genotype of the samples was further ascertained by PCR and DNA sequencing. We aimed to get a better understanding of the gene distribution of thalassemia and its co-inheritance patterns in this area, which was a prerequisite to perform appropriate prevention and control programs.
2 Materials and methods
2.1 Study population
This study was performed in two local hospitals (People’s Hospital of Yangjiang and Women and Children Hospital of Yangjiang) between September 2014 and July 2021 in Yangjiang. The majority of the study subjects were couples who received thalassemia screening and those subjects either with suspicion of a thalassemia trait or with clinically recognized thalassemia syndrome of different degrees of severity. About one-third of the study subjects received thalassemia gene diagnosis directly for convenience; they did not receive blood routine test and Hb electrophoresis. Amniotic fluid, umbilical cord blood, and fetal villi were collected from 21 fetuses for prenatal diagnosis. All studies were approved by the Ethics Committee of People’s Hospital of Yangjiang and Women and Children Hospital of Yangjiang. All the patients or their guardians signed the informed consent forms.
2.2 Hematological analysis
A measure of 2–3 mL of peripheral blood samples of EDTA anticoagulants was taken from 22446 subjects for screening and genetic diagnosis of thalassemia. Hematological data of these subjects were obtained using an automated blood cell counter and by Hb electrophoresis. Subjects with reduced mean corpuscular volume (MCV<82 fl), reduced mean corpuscular hemoglobin (MCH<27 pg), and HbA2<2.5% were considered possible α-thal carriers. Subjects with MCV<82 fl, MCH<27 pg, and HbA2>3.5% were considered possible β-thal carriers. Some subjects who had undergone thalassemia screening in other hospitals did not receive the blood routine test and were only demanded for molecular diagnosis. Amniotic fluid, umbilical cord blood, and fetal villi were obtained from 21 fetuses for thalassemia gene diagnosis. Totally, 22467 samples were characterized by molecular diagnosis.
2.3 α/β-Thalassemia mutation analysis
Genomic DNA was extracted from all peripheral blood leukocytes, umbilical cord blood, fetal villi, and amniotic fluid. PCR and reverse dot blot (RDB) were used to detect the three commonly known α-thal deletions (--SEA, -α3.7, and -α4.2) and three known point mutations (Hb CS (αCSα) HBA2: c.427T>C; Hb QS (αQSα), HBA2: c.377T>C; Hb WS (αWSα), and HBA2: c.369C>G) responsible for α-thal, and 19 known β-thal mutations most commonly seen in the Chinese population [IVS-II-654 (C>T), HBB: c.316–197C>T; codons 41/42 (‒TCTT), HBB: c.126_129delCTTT; codon 17 (A>T), HBB: c.52A>T; −28 (A>G), HBB: c.‒78A>G; codon 26 (G>A), HBB: c.79G>A; codons 71/72 (+A), HBB: c.216_217insA; codons 27/28 (+C), HBB: c.84_85insC; −29 (A>G), HBB: c.‒79A>G; IVS-I-1 (G>A, G>T), HBB: c.92 + 1G>A, c.92 + 1G>T; codons 14/15 (+G), HBB: c.45_46insG; codon 43 (G>T), HBB: c.130G>T; Cap (-AAAC, A>C), HBB: c.-11_-8delAAAC; IVS-I-5 (G>C), HBB: c. 92 + 5G>C; codon 31 (‒C), HBB: c.94delC; initiation codon ATG>AGG, HBB: c.2T>G; −32 (C>A), HBB: c.‒82C>A; −30 (T>C), and HBB: c.‒80T>C] were also identified (Liang et al., 2022). All genotyping was analyzed using a thalassemia gene chip kit (Guangdong Hybribio Limited Corporation, Chaozhou, China), as described in our previous study (Liang et al., 2022).
Those cases with abnormal capillary electrophoresis behavior or thalassemia traits which could not be identified by this gene chip kit were further tested by gap-PCR and PCR sequencing based on their primary gene analysis result (Liang et al., 2022), and anti-3.7 and anti-4.2 α-globin gene triplications were identified by single-tube multiplex PCR (Wang et al., 2005). Only a small number of suspected cases with rare variants were further tested.
2.4 Statistical analysis
Statistical analysis was performed on the genotypic frequency of thalassemia by using the percentage method.
3 Results
First, genetic diagnosis of all 22467 possible thalassemia samples identified 19 known β-thal mutations and six kinds of α-thal mutations using a kit (Liang et al., 2022). We detected six common mutations that cause α-thal and 12 common mutations responsible for β-thal; five β-thal mutations of 19 known β-thal mutations were not detected by our kit (Tables 1, 2). Among 22467 possible thalassemia cases, 7658 cases were identified with thalassemia genotypes, including 5313 cases were identified with α-thal alone (Table 1), 2032 cases were identified with β-thal alone (Table 2), and 313 cases were identified with α-thal combined with β-thal (Table 3).
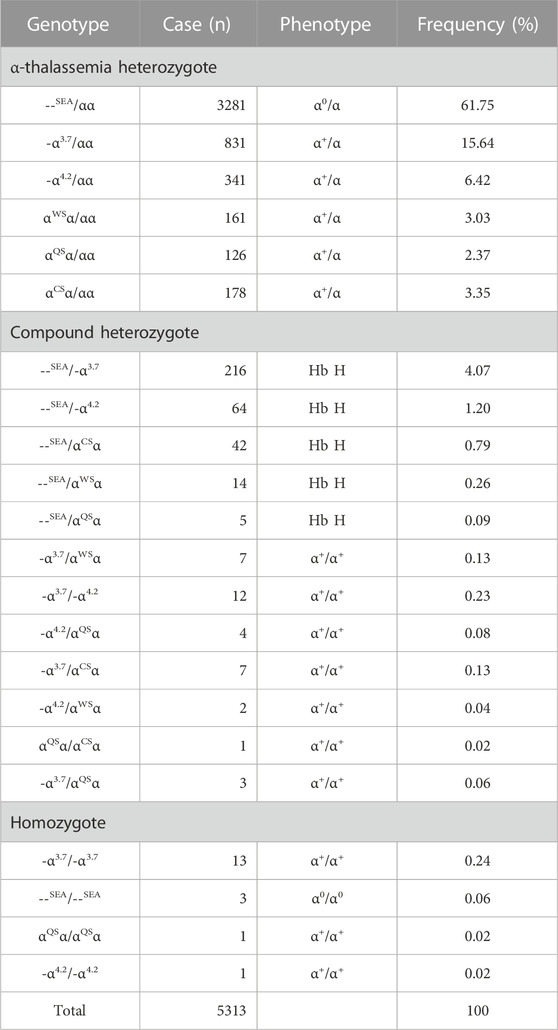
TABLE 1. Spectrum of α-thalassemia mutations alone.
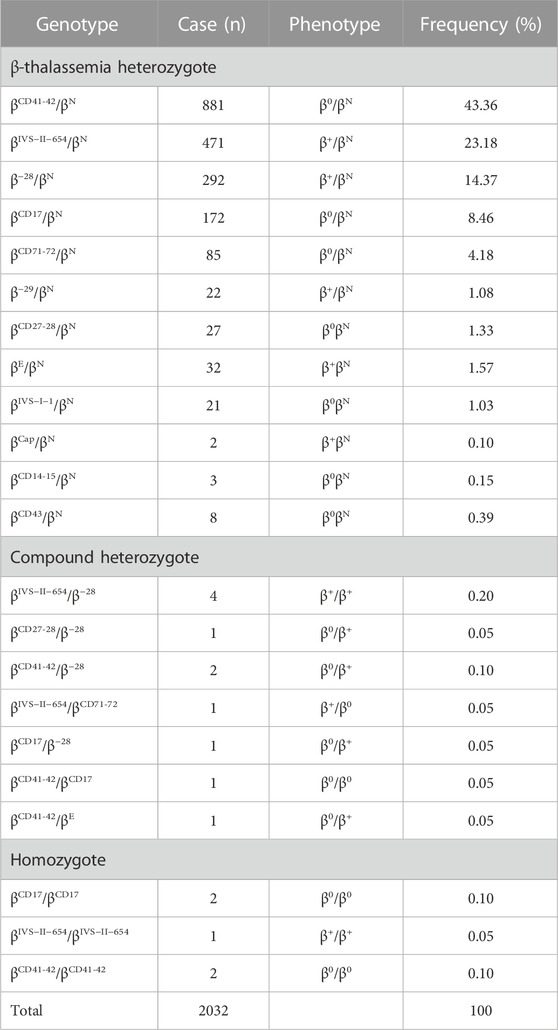
TABLE 2. Spectrum of β-thalassemia mutations alone.
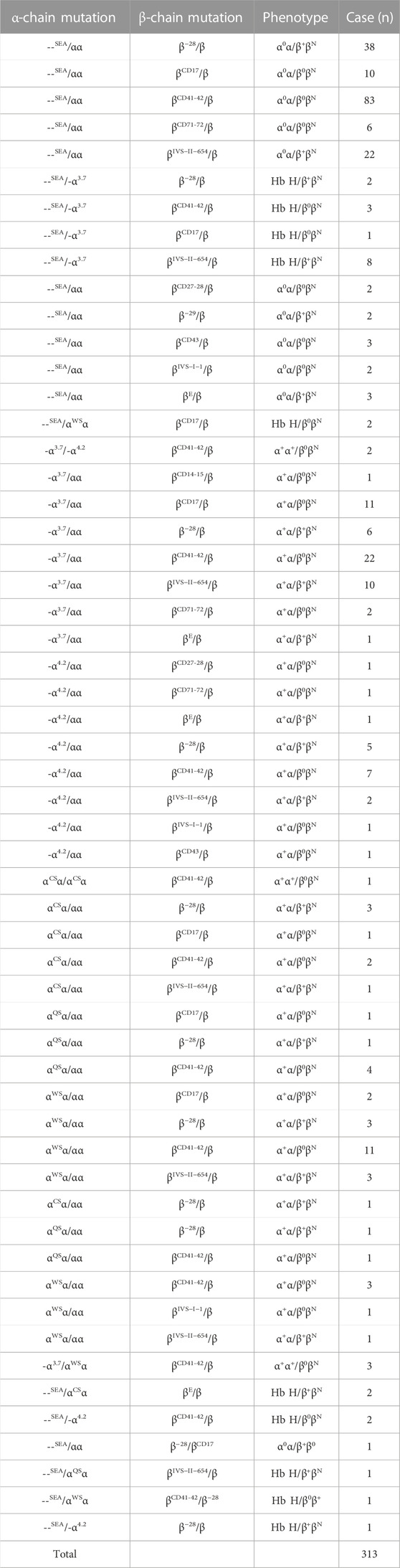
TABLE 3. Genotype distribution of β-thal combined with α-thal mutations.
Amniotic fluid (19 cases), umbilical cord blood (one case), and fetal villi (one case) were obtained from 21 fetuses for thalassemia gene diagnosis. They were identified as --SEA/--SEA (three cases), --SEA/αα (five cases), --SEA/-α4.2 (one case), -α4.2/αα (one case), αα/αα (two cases), βCD41-42/βCD41-42 (one case), βCD41-42/βIVS−II−654 (one case), βIVS−II−654/β (two cases), βCD41-42/β (two cases), βCD17/β (one case), and β/β (two cases).
Among the 5313 α-thal cases, --SEA/αα was the most common genotype, accounting for 61.75% (3281/5313) of α-thal genotypes, and the following genotypes were identified: -α3.7/αα (15.64%, 831/5313), -α4.2/αα (6.42%, 341/5313), αCSα/αα (3.35%, 178/5313), αWSα/αα (3.03%, 161/5313), and αQSα/αα (2.37%, 126/5313). Compound heterozygotes of α-thal were found in 377 cases, and Hb H disease was found in 341 cases. As expected, the most common genotypes of patients with Hb H disease were --SEA/-α3.7 (63.34%, 216/341) and --SEA/-α4.2 (18.77%, 64/341). --SEA/αCSα, --SEA/αwSα, and --SEA/αQSα were also encountered in 42, 14, and 5 cases, respectively, in our study. In total, 18 cases were homozygotes, including 13 cases of -α3.7/-α3.7, one case of -α4.2/-α4.2, one case of αQSα/αQSα, and three cases of Hb Bart’s hydrops fetalis syndrome with the genotype of --SEA/--SEA (Table 1).
Of the study subjects, 2032 cases were β-globin mutation alone (Table 2). In total, 22 different genotypes were detected, including 2016 heterozygotes, 11 compound heterozygotes, and five homozygotes, accounting for 99.21%, 0.54%, and 0.25%, respectively (Table 2). βCD41-42/βN (43.36%, 881/2032), βIVS−II−654/βN (23.18%, 471/2032), and β−28/βN (14.37%, 292/2032) accounted for 80.9% (1644/2032) of all β-thal genotypes, and the following mutations were found: βCD17/βN, βCD71-72/βN, and β−29/βN. The initiation codon ATG>AGG, IVS-I-5 (G>C), codon 31 (‒C), −30 (T>C), and −32 (C>A) were not found in our study.
The very variable α-globin alterations in combination with β-globin mutations were found in 313 cases, and 57 genotype combinations of both Hb disorders were identified (Table 3). --SEA/αα combined with βCD41-42/βN was identified in 83 cases, which was the most common combination, and the other combinations found are as follows: --SEA/αα combined with β−28/βN, --SEA/αα combined with βIVS−II−654/βN, and -α3.7/αα combined with βCD41-42/βN (Table 3). Out of 313 cases of α-thal/β-thal, 30 cases were with at least three globin mutations, one case was with one α-globin mutation combined with two β-globin mutations, 28 cases were with two α-globin mutations combined with one β-globin mutation, and one extreme patient had four globin mutations with a genotype of --SEA/αWSα combined with βCD41-42/β−28 (Table 3).
As for the frequency of a specific type of mutation in all α (or β)-mutant chromosomes in this study population, 18 kinds of mutations were identified, including six α-gene mutations and 12 β-gene mutation (Tables 4, 5). The --SEA deletion was the most frequent, accounting for 63.63% of all α-mutant chromosomes; the following mutations were found: -α3.7, -α4.2, αWS, and αCS, with allele frequencies of 19.54%, 7.47%, 3.56%, and 3.30%, respectively (Table 4). Regarding β-globin mutant chromosomes, five mutations accounted for 94.16% of all β-thalassemia defects; these mutations, in the order of frequency, were CD41-42 (-CTTT) (43.76%), IVS-II-654(C>T) (22.26%), −28 (A>G) (15.36%), CD17 (A>T) (8.76%), and CD71-72 (+A) (4.25%). The other seven mutations with frequencies no more than 2% were −29 (A>G), CD27-28 (+C), CD26 (Hb E) (G>A), IVS-I-1 (G>T), CD43 (G>T), CD14-15 (+G), and Cap (Table 5).
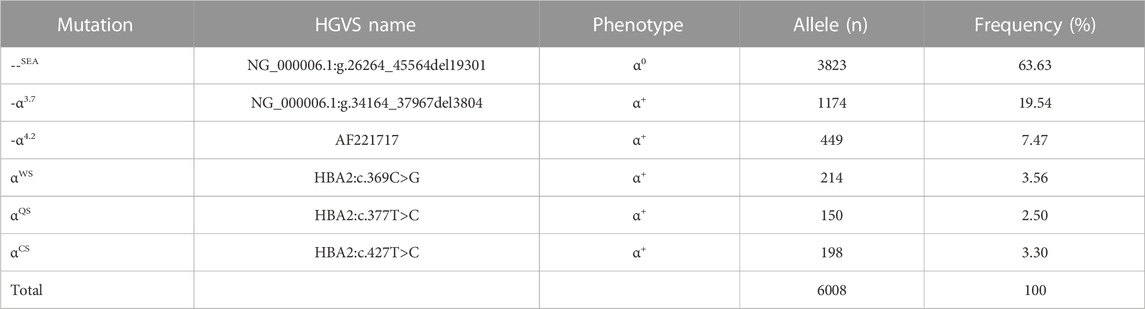
TABLE 4. Allele frequency of α-thal in individuals of Yangjiang region.
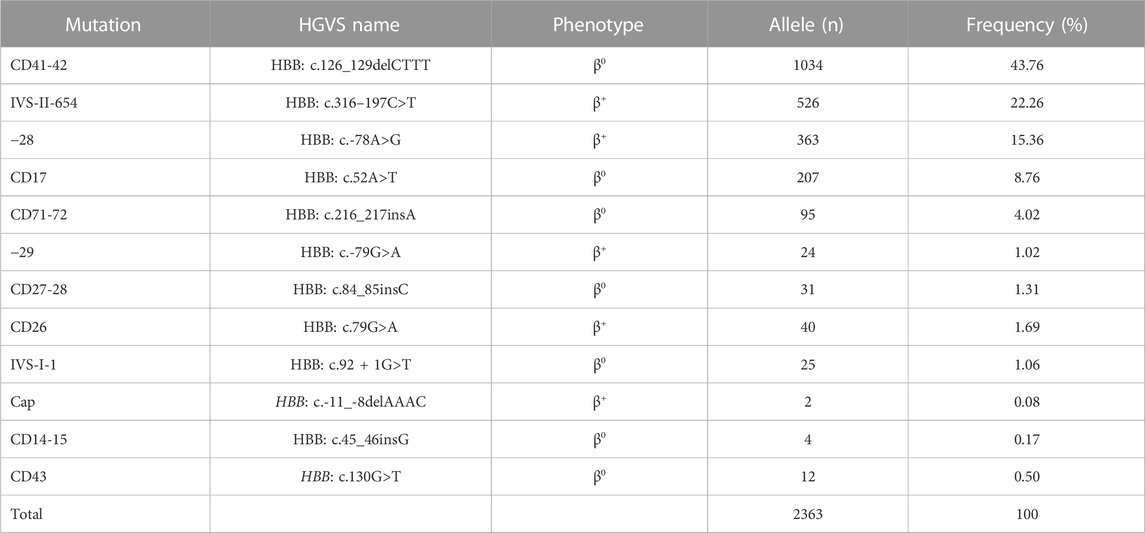
TABLE 5. Allele frequency of β-thal in individuals of Yangjiang region.
Furthermore, a small number of cases suspected with rare genotypes were referred to undergo gene sequencing or gap-PCR (Liang et al., 2022). Rare thalassemia genotypes were identified in 19 cases which were not included in our detection kit; they included four types of rare α-globin genotypes of αα/--THAI (two cases), HKαα/--SEA (four cases), αWSαCD74 GAC>CAC/-α4.2 (two cases), and αCD31 AGG>AAGα/--SEA (one case). Six rare β-globin genotypes were identified: CD39 CAG>TAG (one case), IVS-Ⅱ-2 (−T) (two cases), −90(C>T) (three cases), Chinese Gγ+(Aγδβ)0 (two cases), CD104 (-G) (one case), and CD19 (A>G) (one case) (Table 6).
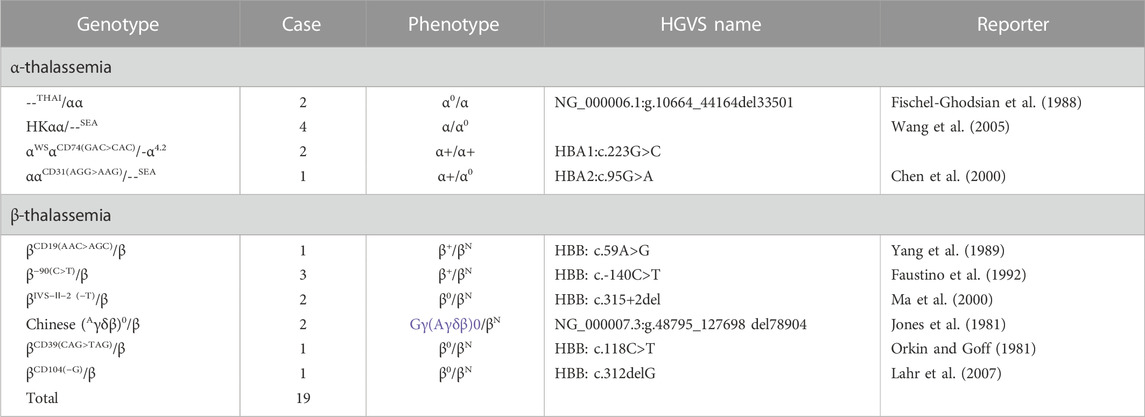
TABLE 6. Summary of rare thalassemia genotypes.
4 Discussion
Thalassemia is an autosomal recessive hematologic disease, in which one of the genes that encode for the hemoglobin components, the α- and β-globin chains, is reduced or deleted; its clinical phenotype ranges from asymptomatic forms to fatal hemolytic anemia. The globin genes can be affected by a variety of alterations, and two main types were α- and β-thalassemia (α-thal and β-thal). Thalassemia is prevalent in Middle East, Mediterranean countries, Central Asia, India, and southern China, as well as countries along the north coast of Africa and in South America; about 5% of the world’s population is a carrier of thalassemia (Weatherall, 1981; Weatherall, 1997; Lai et al., 2017). Previous studies indicated that there was a high frequency of thalassemia cases in southern China, especially in Guangxi, Guangdong, Jiangxi, and Hainan provinces (Xu et al., 2004; Xiong et al., 2010; Lin et al., 2014; Yao et al., 2014; Yin et al., 2014; Lai et al., 2017; Liang et al., 2022). Patients of each ethnic population carry their own specific types of mutations, including a few very common ones and a variable number of rare ones.
A county in Yangjiang has a high prevalence of thalassemia, which has become a major social and medical concern (Yin et al., 2014). In this study, we performed genetic diagnosis to determine the spectrum of α-thal and β-thal among patients in Yangjiang and set up the foundation for genetic counseling and performing comprehensive care programs. Our study showed that the most common mutation causes of α-thal in Yangjiang were --SEA, -α3.7, -α4.2, and αCS. A similar distribution was found in Guangdong, Jiangxi, whole Guangxi Province, and Meizhou and Chaoshan of eastern Guangdong Province (Xu et al., 2004; Xiong et al., 2010; Lin et al., 2013; Lin et al., 2014; Zheng et al., 2016). This pattern was different from that of Hainan Li with fewer --SEA and that of Yunnan with fewer -α4.2 (Zhang et al., 2012; Yao et al., 2014).
β-thalassemia is very heterogeneous, both in the molecular defects and the clinical manifestations. More than 200 different mutations in the β-globin gene have been characterized worldwide (Lai et al., 2017). In this study, of the 17 common mutations in Chinese people, we detected 12 mutations in the Yangjiang population. The three common mutations 41/42 (-TCTT), IVS-II-654 (C>T), and −28 (A>G) accounted for 81.11% of total β-globin gene mutations. We compared the genotype distribution of β-thal mutations in this study with data reported in the population of other areas in China. IVS-II-654(C>T) and 41/42 (-TCTT) were also the most common mutations in Guangdong, mainland China, Jiangxi, and Han nationality in Hainan (Lin et al., 2014; Yao et al., 2014; Yin et al., 2014; Lai et al., 2017). Different from other areas, 41/42 (-TCTT) and CD17 (A>T) were the most common β-thal mutations in Guangxi Province, whereas Hb E and codon 17 (A>T) were the dominant genotypes in Yunnan (Zhang et al., 2012) and codon 17 (A>T) and 41/42 (-TCTT) were the dominant genotypes in Guizhou (Huang et al., 2013). 41/42 (-TCTT) was the only β-thal mutation in Li people of Hainan Province (Yao et al., 2014). β-thal genotypes are relatively population-specific, and each ethnic group has its own set of common mutants (Lai et al., 2017).
Hemoglobin (Hb) Malay was first described in the Malaysian population in 1989 (Yang et al., 1989). Hb Malay results from an AAC→AGC (HBB: c.59A>G, p.Asn19Ser) mutation in codon 19 of the β-globin gene. This mutation creates an alternate splicing site between codons 17 and 18, reducing the efficiency of the β-chain globin to about 60% of the normal level (Yang et al., 1989). Hb Malay carriers do not present any clinical symptoms, only present increased HbA2 and decreased MCV, and are often diagnosed during thalassemia screening. However, compound heterozygosity for Hb Malay and β0-thal mutation shows β-thal intermedia clinically (Yang et al., 1989). CD104 (-G) (HBB: c.312delG), a heterozygous frameshift mutation in exon 2 of the β-globin gene, results in a dominantly inherited β0-phenotype with mild anemia (Lin et al., 2017). To date, there has been no report of CD104 (-G) and Hb Malay in mainland China in the literature.
Hb H disease was diagnosed in 341 patients, and β-thal major or intermedia were found in 16 patients in this study. Our results revealed that the public health burden imposed by the high prevalence of thalassemia was still severe in Yangjiang. As we all know, the only curative therapy for β-thalassemia major is allogeneic hematopoietic stem cell transplantation; however, the difficulty to find matched donors and economic burden have limited this therapeutic option (Zhou et al., 2008). Due to the large number of thalassemia patients and limited medical resources, it is difficult to give regular blood transfusions to the majority of patients. Therefore, the most effective strategy for this area is to develop an effective prevention program, which includes carrier screening, prenatal diagnosis, and genetic counseling for the couples at risk of giving birth to new cases of thalassemia.
5 Conclusion
In conclusion, --SEA was the most common α-allele in Yangjiang, and the following alleles were found: -α3.7, -α4.2, αCS, αWS, and αQS. CD41/42 (−TCTT), IVS-II-654(C>T), and −28 (A>G) were the main alleles of all β-thal alleles. This study provided detailed genotypes of thalassemia in Yangjiang of western Guangdong Province and reflected the complexity of genotypes in this high-prevalence region, and this would be valuable for diagnosis and counseling for thalassemia in this area.
Data availability statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee of People’s Hospital of Yangjiang and Women and Children Hospital of Yangjiang. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
L-YY designed the study. H-FL and FL analyzed the data and wrote the first draft of the manuscript. W-ML, W-GX, L-LL, Y-YG, ML, Y-WL, G-KZ, J-XY, and J-WS collected the samples and clinical data. FL, ML, and L-JL performed gene assay. All authors discussed the results and commented on the manuscript. All authors read and approved the final manuscript.
Funding
This study was supported by the Natural Science Foundation of Guangdong Province (No. 2016A030307035) and High Level Development Plan of People’s Hospital of Yangjiang (Nos G2020007 and 2021007).
Conflict of interest
Authors L-JL, Y-YG, and ML were employed by Guangdong Hybribio Limited Corporation.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Chen, F. E., Ooi, C., Ha, S. Y., Cheung, B. M., Todd, D., Liang, R., et al. (2000). Genetic and clinical features of hemoglobin H disease in Chinese patients. N. Engl. J. Med. 343 (8), 544–550. doi:10.1056/NEJM200008243430804
Faustino, P., Osório-Almeida, L., Barbot, J., Espírito-Santo, D., Gonçalves, J., Romão, L., et al. (1992). Novel promoter and splice junction defects add to the genetic, clinical or geographic heterogeneity of beta-thalassaemia in the Portuguese population. Hum. Genet. 89 (5), 573–576. doi:10.1007/BF00219188
Fischel-Ghodsian, N., Vickers, M. A., Seip, M., Winichagoon, P., and Higgs, D. R. (1988). Characterization of two deletions that remove the entire human zeta-alpha globin gene complex (- -Thai and - -fil). Br. J. Haematol. 70 (2), 233–238. doi:10.1111/j.1365-2141.1988.tb02469.x
Huang, S. W., Liu, X. M., Li, G. F., Su, L., Wu, X., and Wang, R. L. (2013). Spectrum of β-thalassemia mutations in Guizhou Province, PR China, including first observation of codon 121 (GAA>TAA) in Chinese population. Clin. Biochem. 46 (18), 1865–1868. doi:10.1016/j.clinbiochem.2013.09.014
Jones, R. W., Old, J. M., Trent, R. J., Clegg, J. B., and Weatherall, D. J. (1981). Restriction mapping of a new deletion responsible for G gamma (delta beta)o thalassemia. Nucleic Acids Res. 9 (24), 6813–6825. doi:10.1093/nar/9.24.6813
Lahr, G., Brintrup, J., Over, S., Feurle, G. E., Debatin, K. M., and Kohne, E. (2007). Codon 104(-G), a dominant beta0-thalassemia-like phenotype in a German Caucasian family is associated with mild chronic hemolytic anemia but influenced in severity by co-inherited genetic factors. Haematologica 92 (9), 1264–1265. doi:10.3324/haematol.11383
Lai, K., Huang, G., Su, L., and He, Y. (2017). The prevalence of thalassemia in mainland China: Evidence from epidemiological surveys. Sci. Rep. 7 (1), 920. doi:10.1038/s41598-017-00967-2
Liang, H. F., Li, L. J., Yang, H., Zheng, X. B., Lu, M., Ge, Y. Y., et al. (2022). Clinical validation of a single-tube PCR and reverse dot blot assay for detection of common α-thalassaemia and β-thalassaemia in Chinese. J. Int. Med. Res. 50 (2), 3000605221078785. doi:10.1177/03000605221078785
Lin, F., Yang, L., Lin, M., Zheng, X., Lu, M., Qiu, M., et al. (2017). Rare thalassemia mutations among southern Chinese population. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 34 (6), 792–796. doi:10.3760/cma.j.issn.1003-9406.2017.06.002
Lin, M., Wen, Y. F., Wu, J. R., Wang, Q., Zheng, L., Liu, G. R., et al. (2013). Hemoglobinopathy: Molecular epidemiological characteristics and health effects on Hakka people in the Meizhou region, southern China. PLoS One 8, e55024. doi:10.1371/journal.pone.0055024
Lin, M., Zhong, T. Y., Chen, Y. G., Wang, J. Z., Wu, J. R., Lin, F., et al. (2014). Molecular epidemiological characterization and health burden of thalassemia in Jiangxi Province, P. R. China. P. R. China. PLoS One 9, e101505. doi:10.1371/journal.pone.0101505
Ma, S. K., Ha, S. Y., Chan, A. Y., Chan, G. C., Lau, Y. L., and Chan, L. C. (2000). Two novel beta-thalassemia alleles in the Chinese: The IVS-II-2 (-T) and nucleotide +8 (C-->T) beta-globin gene mutations. Hemoglobin 24 (4), 327–332. doi:10.3109/03630260008993141
Orkin, S. H., and Goff, S. C. (1981). Nonsense and frameshift mutations in beta 0-thalassemia detected in cloned beta-globin genes. J. Biol. Chem. 256 (19), 9782–9784. doi:10.1016/s0021-9258(19)68689-8
Wang, W., Chan, A. Y., Chan, L. C., Ma, E. S., and Chong, S. S. (2005). Unusual rearrangement of the alpha-globin gene cluster containing both the -alpha 3.7 and alphaalphaalpha -4.2 crossover junctions: Clinical diagnostic implications and possible mechanisms. Clin. Chem. 51, 216–270.
Weatherall, D. J. (2004). Thalassaemia: The long road from bedside to genome. Nat. Rev. Genet. 5, 625–631. doi:10.1038/nrg1406
Xiong, F., Sun, M., Zhang, X., Cai, R., Zhou, Y., Lou, J., et al. (2010). Molecular epidemiological survey of haemoglobinopathies in the Guangxi Zhuang Autonomous Region of southern China. Clin. Genet. 78, 139–148. doi:10.1111/j.1399-0004.2010.01430.x
Xu, X. M., Zhou, Y. Q., Luo, G. X., Liao, C., Zhou, M., Chen, P. Y., et al. (2004). The prevalence and spectrum of alpha and beta thalassaemia in Guangdong province: Implications for the future health burden and population screening. J. Clin. Pathol. 57, 517–522. doi:10.1136/jcp.2003.014456
Yang, K. G., Kutlar, F., George, E., Wilson, J. B., Kutlar, A., Stoming, T. A., et al. (1989). Molecular characterization of beta-globin gene mutations in Malay patients with Hb E-beta-thalassaemia and thalassaemia major. Br. J. Haematol. 72 (1), 73–80. doi:10.1111/j.1365-2141.1989.tb07655.x
Yao, H., Chen, X., Lin, L., Wu, C., Fu, X., Wang, H., et al. (2014). The spectrum of α- and β-thalassemia mutations of the Li people in Hainan Province of China. Blood Cells Mol. Dis. 53 (1-2), 16–20. doi:10.1016/j.bcmd.2014.01.003
Yin, A., Li, B., Luo, M., Xu, L., Wu, L., Zhang, L., et al. (2014). The prevalence and molecular spectrum of α- and β-globin gene mutations in 14,332 families of Guangdong Province, China. China. PLoS One 9 (2), e89855. doi:10.1371/journal.pone.0089855
Zhang, J., Zhu, B. S., He, J., Zeng, X. H., Su, J., Xu, X. H., et al. (2012). The spectrum of α- and β-thalassemia mutations in yunnan province of southwestern China. Hemoglobin 36 (5), 464–473. doi:10.3109/03630269.2012.717327
Zheng, X., Lin, M., Yang, H., Pan, M. C., Cai, Y. M., Wu, J. R., et al. (2016). Molecular epidemiological characterization and health burden of thalassemias in the chaoshan region, people's republic of China. Hemoglobin 40, 138–142. doi:10.3109/03630269.2015.1137933
Keywords: thalassemia, gene testing, genotype, China, Guangdong Province
Citation: Liang H-F, Liang W-M, Xie W-G, Lin F, Liu L-L, Li L-J, Ge Y-Y, Lu M, Liao Y-W, Zeng G-K, Yao J-X, Situ J-W and Yang L-Y (2023) The gene spectrum of thalassemia in Yangjiang of western Guangdong Province. Front. Genet. 14:1126099. doi: 10.3389/fgene.2023.1126099
Received: 17 December 2022; Accepted: 18 January 2023;
Published: 13 February 2023.
Edited by:
Corrado Romano, University of Catania, ItalyReviewed by:
Ammar Husami, Cincinnati Children’s Hospital Medical Center, United StatesAnil Pathare, Sultan Qaboos University, Oman
Copyright © 2023 Liang, Liang, Xie, Lin, Liu, Li, Ge, Lu, Liao, Zeng, Yao, Situ and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li-Ye Yang, eWFuZ2xlZXllZUBzaW5hLmNvbQ==