 Junhao Jiang1
Junhao Jiang1 Junxia Yan
Junxia Yan Junyu Liu
Junyu Liu- 1Hunan Normal University School of Medicine, Changsha, China
- 2Department of Epidemiology and Health Statistics, XiangYa School of Public Health, Central South University, Changsha, China
- 3Hunan Provincial Key Laboratory of Clinical Epidemiology, XiangYa School of Public Health, Central South University, Changsha, China
- 4Interventional Medical Center, Hunan Province People’s Hospital (The First Affiliated Hospital of Hunan Normal University), Changsha, China
- 5Department of Pharmacology, Kyoto University Graduate School of Medicine, Kyoto, Japan
Objectives: Rupture of a brain arteriovenous malformation (bAVM) can cause intracranial hemorrhage and severe clinical outcomes. At present, the mechanisms of bAVM-related hemorrhage are poorly understood. This study aimed to summarize the potential genetic risk factors for bAVM-related hemorrhage and appraise the methodological quality of existing genetic studies on bAVM-related hemorrhage using a cross-sectional design.
Methods: A systematic literature search was conducted on genetic studies associated with bAVM-related hemorrhage published in PubMed, Embase, Web of Science, China National Knowledge Internet, and Wangfang databases, up to November 2022. Subsequently, a cross-sectional study was performed to describe the potential candidate genetic variants of bAVM associated with risk of hemorrhage and to evaluate the methodological quality of the identified studies using the Newcastle–Ottawa quality assessment scale and Q-genie tool.
Results: Of the 1811 records identified in the initial search, nine studies met the filtering criteria and were included. Twelve single nucleotide polymorphisms (SNPs), including IL6 rs1800795, IL17A rs2275913, MMP9 rs9509, VEGFA rs1547651, and EPHB4 rs314353, rs314308, and rs314313, were associated with bAVM-related hemorrhage. However, only 12.5% of the evaluated SNPs showed statistical power> 0.80 (α = 0.05). Methodological quality assessment revealed significant flaws in the designs of the included studies, such as less reliable representativeness of recruited individuals, short follow-up periods in cohort studies, and less comparability between groups of hemorrhagic and non-hemorrhagic patients.
Conclusion: IL1B, IL6, IL17A, APOE, MMP9, VEGFA and EPHB4 were potentially associated with bAVM-related hemorrhage. The methodological designs of the analyzed studies required improvement in order to obtain more reliable results. Regional alliances and rare disease banks need to be established to recruit large numbers of bAVM patients (especially familial and extreme-trait cases) in a multicenter, prospective cohort study with an adequate follow-up period. Furthermore, it is important to use advanced sequencing techniques and efficient measures to filter candidate genetic variants.
1 Introduction
The most common and severe manifestation of a brain arteriovenous malformation (bAVM) is its rupture, which is also the leading cause of intracranial hemorrhage in children and young adults. High pressure blood flow from the feeding arteries of the bAVM floods directly to the draining veins through the malformed nidus, causing the development of abnormal shear stress due to lack of capillary structure within the anomalous nidus, ultimately resulting in its rupture (Rutledge et al., 2014). Past observational studies have reported a 1%–3% annual incidence of bAVM-related hemorrhage in unruptured and untreated patients, whereas the reported risk was much higher in individuals with ruptured bAVM(2). Current treatments, including microsurgery, endovascular embolization, and stereotactic radiosurgery aim to reduce the risk of hemorrhage and eradicate existing lesions. Although microsurgery offers the advantage of a higher rate of complete obliteration and elimination of bAVM-related hemorrhage compared to the other treatments, craniotomy is a highly traumatic procedure resulting in a longer hospitalization as well as substantial morbidity and mortality during the perioperative period (van Beijnum et al., 2011; Derdeyn et al., 2017). Thus, it is imperative to identify risk factors for bAVM rupture as early as possible.
Prior hemorrhage has been reported to be associated with a higher rate of subsequent hemorrhage as a strongly predictive factor (Chen et al., 2020). Existing evidence suggests that angioanatomic features of bAVM, including large size, deep venous drainage, few draining veins, and coexisting arterial aneurysm, contribute to its rupture (Krithika and Sumi, 2021). Several studies have investigated and discovered genetic variants of inflammation- or angiogenesis-related genes that could potentially influence bAVM rupture by accelerating growth and modifying lesion behavior to promote disease pathogenesis (Pawlikowska et al., 2004; Achrol et al., 2006; Pawlikowska et al., 2006; Kim et al., 2009; Weinsheimer et al., 2009; Gong et al., 2011; Li et al., 2012; Sun et al., 2012; Delev et al., 2017). However, due to the low prevalence and incidence of bAVM, most genetic studies on bAVM recruited small samples of patients and were prone to selection bias, resulting in inconsistent results. In addition, different research designs may yield conflicting results and may have varying methodological quality. Therefore, existing genetic studies on bAVM cannot always be considered as a reliable source of evidence. A well-performed research that provides reliable and high-quality information can help medical practitioners in improving their understanding of the nature of a particular disease as well as assist them in making appropriate treatment decisions. Before accepting and using any scientific evidence, healthcare professionals, medical managers, health policymakers, and even patients should evaluate the methodological quality of the referred studies.
The present study performed a cross-sectional survey to summarize the genetic factors influencing the risk of hemorrhage associated with bAVM and evaluate the methodological rigor of the included studies using the Newcastle–Ottawa quality assessment scale (NOS) and Q-genie tool. Our aim was to summarize the current information regarding genetic risk of bAVM-related hemorrhage, discuss potential research directions, and provide insights into how to improve the methodological quality in future studies investigating the risk of bAVM-related hemorrhage or hemorrhagic stroke caused by other diseases.
2 Materials and methods
2.1 Eligibility criteria
This study was registered with PROSPERO (CRD42021258353). All included articles were case-control or cohort studies recruiting individuals of any ethnic group and focused on the genetic risk factors associated with bAVM-related hemorrhage, using the methods of candidate gene association studies (CGAS), genome-wide association studies (GWAS) or whole exome sequencing (WES). Individuals diagnosed with bAVM based on a recognized criteria were recruited and divided into ruptured and unruptured groups (Atkinson et al., 2001). Only original papers with accurate and sufficient genotyping data that could allow the calculation of odds ratios (ORs) and 95% confidence intervals (95% CIs) were included in the analysis. Conference abstracts, reviews, meta-analyses, protocols, case reports, and animal studies were excluded. Whenever there were duplicate or overlapping papers published by the same researcher(s), the most up-to-date version was included for evaluation.
2.2 Literature search strategy
We identified potential eligible studies using the search terms (“Cerebral AVM” or “cerebral arteriovenous malformation” or “brain AVM” OR “brain arteriovenous malformation”) and (“gene” or “Variation” or “polymorphisms” or “SNPs”) across five electronic databases: PubMed, Embase, Web of Science, China National Knowledge Internet (CNKI) and Wanfang Data. The search was limited to published citations in English or Chinese. Thereafter, potentially relevant studies that were not obtained in the initial searches were manually retrieved from the references of the candidate papers.
2.3 Data extraction
The required information (e.g., author, publication year, study design, studied genes, and SNPs) was extracted from the selected studies. When genotype frequencies of variants could not be obtained from the published papers, the risk allele frequencies of SNPs were utilized to estimate the number of cases per genotype category and calculate the OR (95% CI) using STATA 14.0 (Stata Corporation, College Station, TX, USA).
2.4 Methodological quality assessment
Statistical power (1-β) was calculated according to the genetic models used in the original studies via a two-sided Z test, using an online software (http://powerandsamplesize.com/) to evaluate the quality of studies, with the type I error rate (α) set at 5%. The methodological quality assessment of all included studies was mainly based on the NOS (Stang, 2010) and Q-genie tool (version 1.1) (Sohani et al., 2015). NOS is a validated appraisal tool for non-randomized studies, with eight items categorized into three dimensions: selection, comparability, and outcome (in cohort studies) or exposure (in case-control studies). A score ≥6 is regarded as high quality. The Q-genie tool (version 1.1) was used to assess the quality of the genetic association studies. It is comprised of 11 items scored from 0–7, and a total score ≤35 indicates poor quality.
Study screening, data extraction, and methodological quality assessment were independently completed by two authors, and if there was a disagreement during the process, the third senior investigator resolved the issue through re-evaluation and discussion.
3 Results
3.1 Literature selection and characteristics of included studies
The initial search indicated 1881 records through a single database check until November 2022. Among these, nine studies met the eligibility criteria and were included in the final analysis. This was followed by a manual reference-list screening; however, no additional studies were found to satisfy the filtering criteria. The detailed procedure of literature selection followed in this study is displayed in Figure 1.
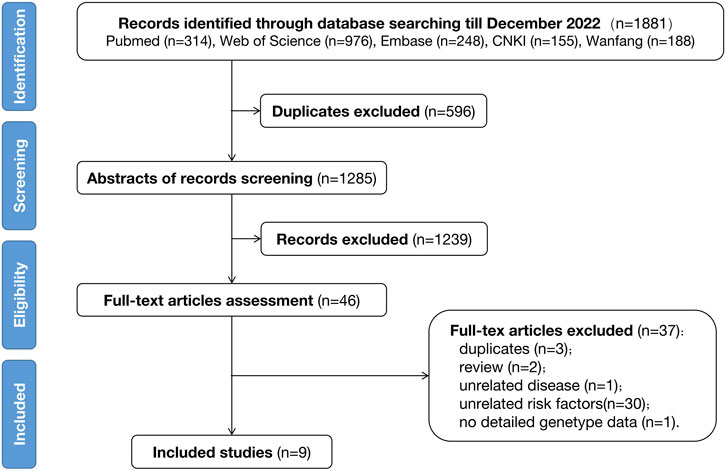
FIGURE 1. Flow diagram of the study selection process.
Six case-control studies and three cohort studies were identified, incorporating 1214 bAVM individuals from North America, Europe, and China (Table 1). The case-control studies divided patients into two groups based on the presence or absence of hemorrhage, and genotyping was performed to test the association between SNPs and bAVM rupture. In these studies (Pawlikowska et al., 2004; Weinsheimer et al., 2009; Gong et al., 2011; Li et al., 2012; Sun et al., 2012; Delev et al., 2017), a total of 984 cases had been recruited, including 529 with hemorrhage and 455 without hemorrhage. In contrast, the cohort studies relied on a prospective follow-up of the included bAVM patients until a new intracranial hemorrhage event occurred. These studies attempted to identify the association between genetic variants and the risk of new rupture during the natural process of bAVM. Furthermore, all three cohort studies (Achrol et al., 2006; Pawlikowska et al., 2006; Kim et al., 2009) belonged to the same research team, which initially recruited 237 non-hemorrhagic and 173 hemorrhagic cases. Less than 25% of the patients were followed-up for over 2 years, at the end of which twenty-seven patients had experienced bAVM rupture and new intracranial hemorrhage events. All included studies were CGAS, including four single-center studies and four multi-center studies. The last study did not mention the source of patient recruitment.
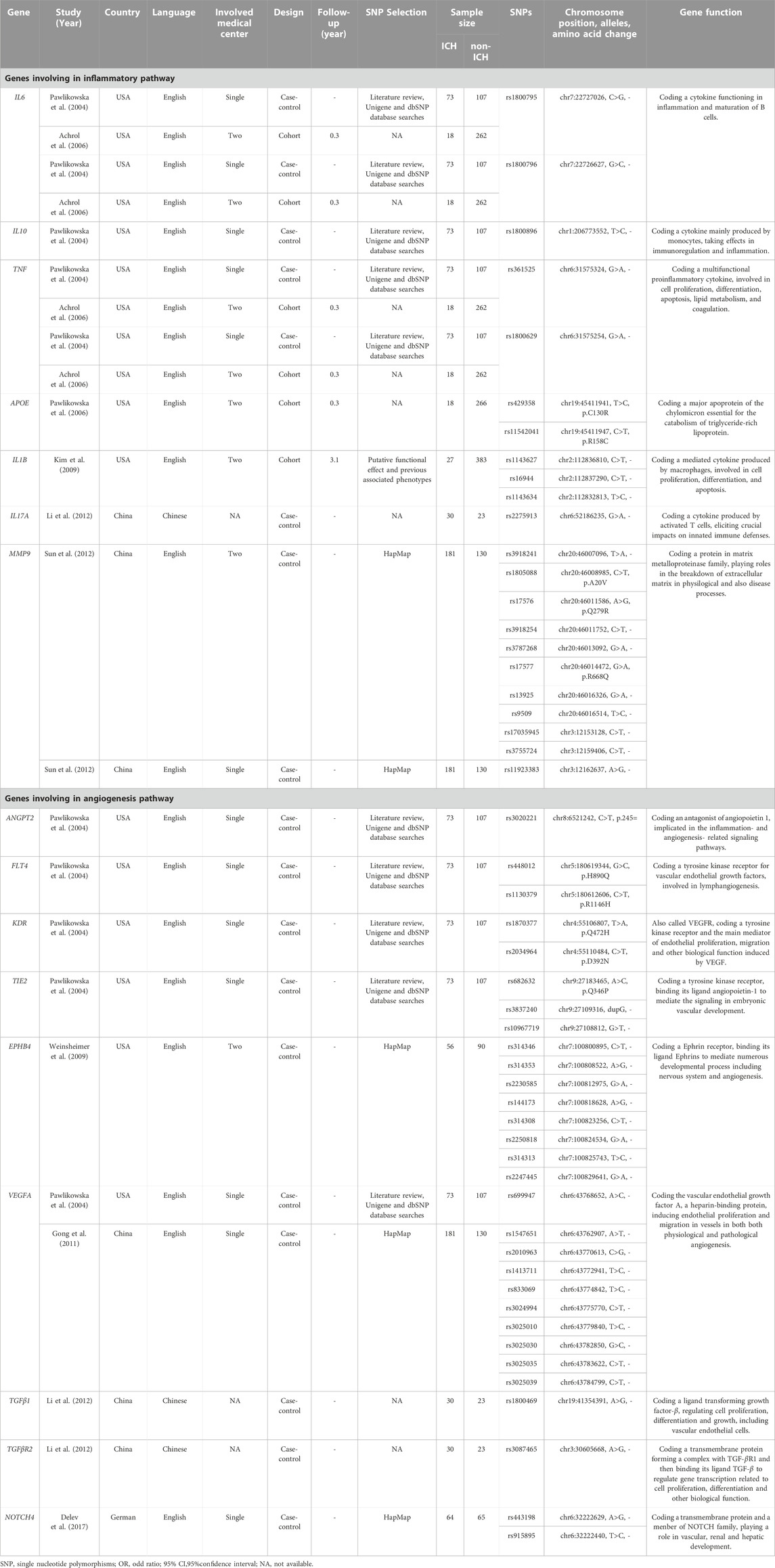
TABLE 1. Summary of the included genetic studies on brain arteriovenous malformation related hemorrhage.
3.2 Genetic characteristics
The nine CGASs evaluated SNPs in seven genes (IL6, IL10, TNF, APOE, IL1B, IL17A, MMP9) of the inflammatory pathway and nine genes (ANGPT2, FLT4, KDR, TIE2, EPHB4, VEGFA, TGFβ1, TGFβR2, NOTCH4) of the angiogenic pathway (Table 1; Figure 2). Seven SNPs in five inflammatory genes were reported to be significantly associated with bAVM rupture, including APOE rs429358 (OR, 5.09; 95% CI, 1.46–17.70), IL1B rs1143627 (OR, 4.01; 95% CI, 1.31–12.29), and IL17A rs2275913 (OR, 0.20; 95% CI, 0.05–0.66) in dominant models, and IL6 rs1800795 (OR, 2.43; 95% CI, 1.04–5.68), IL1B rs16944 (OR, 3.23; 95% CI, 1.70–6.14), IL1B rs1143634 (OR, 1.79; 95% CI, 1.21–2.66), and MMP9 rs9509 (OR, 0.19; 95% CI, 0.05–0.66) in recessive models. Five SNPs in two angiogenic genes were discovered to contribute to bAVM-related hemorrhage: VEGFA rs1547651 (OR, 2.11; 95% CI, 1.01–4.42) in dominant models, and rs314346 (OR, 1.67; 95% CI, 1.04–2.68), rs314353 (OR, 1.79; 95% CI, 1.11–2.89), rs314308 (OR, 0.36; 95% CI, 0.20–0.65), and rs314313 (OR, 0.45; 95% CI 0.25–0.79) in allelic models of EPHB4.
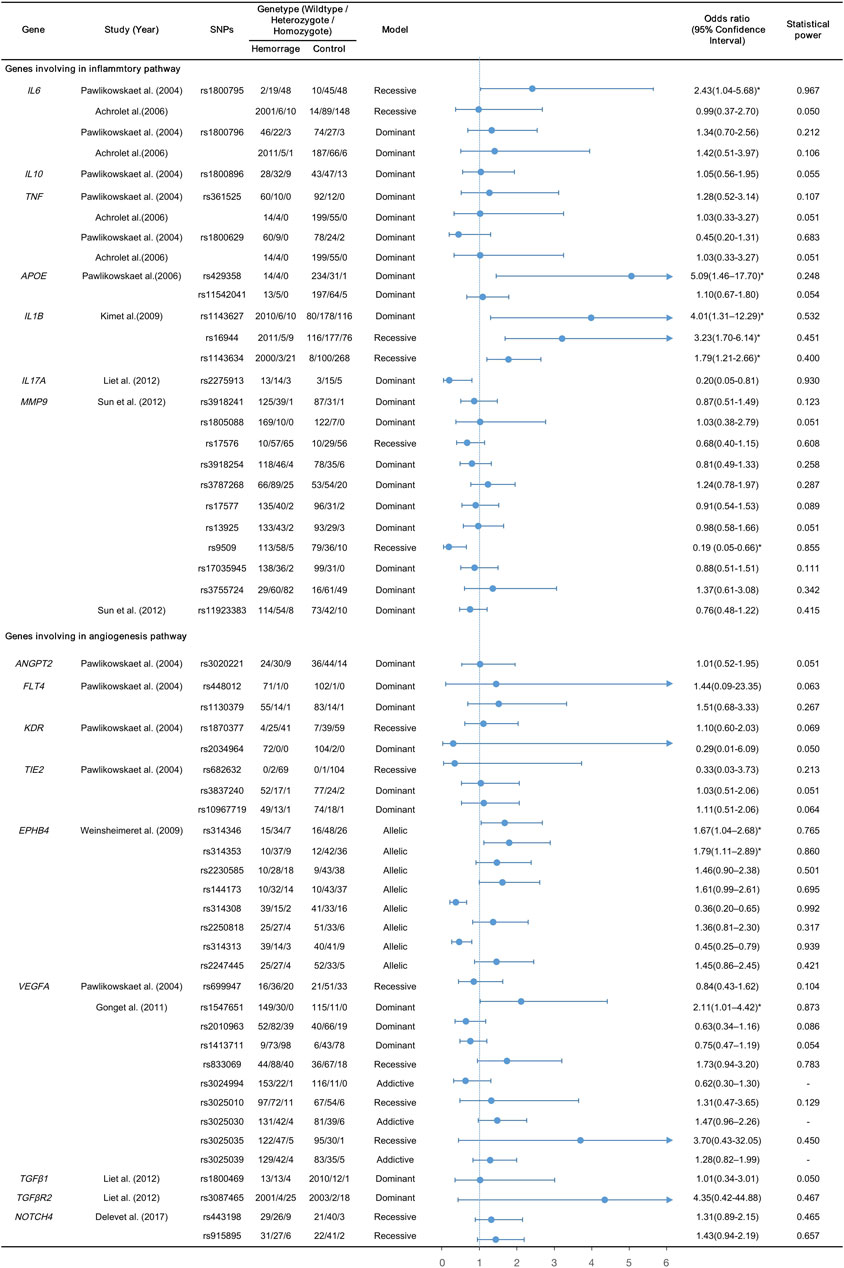
FIGURE 2. Summary and forest plots for the reported variants; *, calculated by multivariant analysis.
3.3 Methodological quality assessment
The statistical power of each polymorphism was calculated using the respective genotype model. The powers of the three SNPs in VGEFA could not be calculated as their ORs and 95% CIs were derived using additive models, with α set at 0.05. The 44 SNPs that were not associated with bAVM-related hemorrhage failed to reach sufficient statistical power (range, 0.050–0.783). Among the remaining 12 candidate variants, APOE rs429358 (power, 0.248), IL1B rs1143627 (power, 0.532), IL1B rs16944 (power, 0.451), IL1B rs1143634 (power, 0.400) and EPHB4 rs314346 (power, 0.765), there were risks of false negatives. Only seven SNPs (12.5%) demonstrated powers greater than 0.80, with IL6 rs1800795, IL17A rs2275913, EPHB4 rs314308, and rs314313 reaching powers >0.90.
Although two-thirds of the included studies were judged to be of high quality (average 6.1 stars) after assessment by NOS (Table 2), all of them were classified as high-bias studies using the Q-Genie tool (Figure 3). Regarding methodology, most studies performed well in identification of patients with bAVM, as well as the methods for identification of genetic variants. However, there was still much room for improvement in the following areas: representativeness for the gene and SNP selection procedure as well as patients with bAVM; comparability between hemorrhagic and non-hemorrhagic individuals as confounding factors, as these were hardly taken into sufficient consideration; and a longer follow-up period in cohort studies.
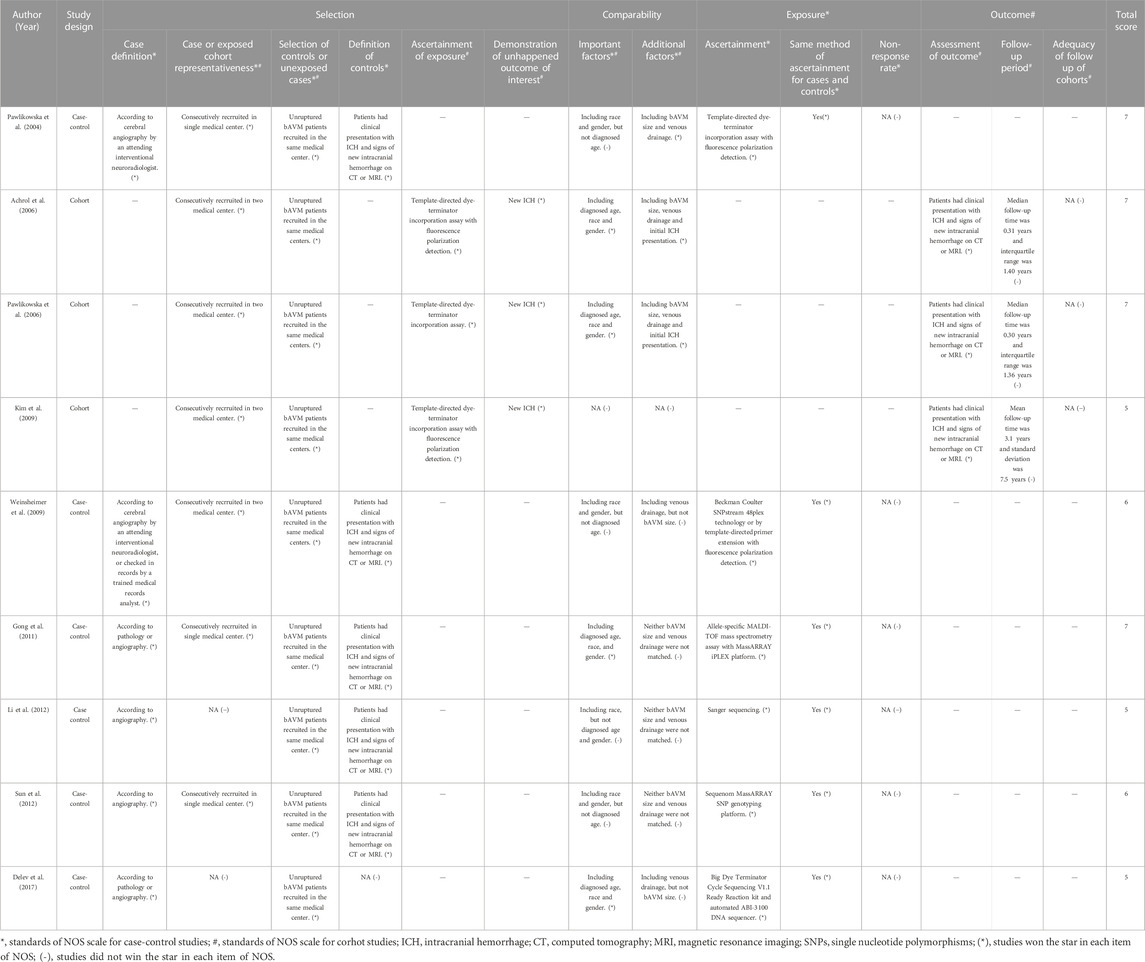
TABLE 2. Summary of the included genetic studies on brain arteriovenous malformation related hemorrhage.

FIGURE 3. Methodological quality assessment using Q-genie tool (version 1.1), including 11 items: Item 1, rationale for study; Item 2, selection and definition of outcome of interest; Item 3, selection and comparability of comparison groups; Item 4, technical classification of the exposure; Item 5, non-technical classification of the exposure; Item 6, other sources of bias; Item 7, sample size and power; Item 8, a priori planning of analyses; Item 9, statistical methods and control for confounding; Item 10, testing of assumptions and inferences for genetic analyses; and Item 11, appropriateness of inferences drawn from results.
4 Discussion
This cross-sectional study systematically reviewed the published studies on genetic factors associated with bAVM-related hemorrhage and identified nine correlative research works, which were examined for statistical power as well as methodological quality using the NOS and Q-Genie tool. This led to the identification of statistically significant association between bAVM-related hemorrhage and twelve heritable variants of seven genes (IL6, APOE, IL1B, IL17A, MMP9, EPHB4, and VEGFA) involved in the inflammatory and angiogenic signaling pathways. After methodological assessment, limitations were noted in the study designs of the included research works, indicating that the quality and reliability of these studies needed to be improved.
The biological functions of the identified candidate genes are known to be involved in inflammatory and angiogenic signaling pathways. We summarized that SNPs of five inflammatory genes (IL6, APOE, IL1B, IL17A, MMP9) were reported to be associated with bAVM-related hemorrhage. Additionally, it has been reported that these genes can increase the expression of inflammatory cytokines in bAVM tissues, leading to endothelial dysfunction and malformation of vasculature (Krithika and Sumi, 2021). It has been shown that inflammatory infiltration can be observed even in unruptured bAVM lesions, proving the role of inflammation in the development and rupture of the disease (Liu et al., 2022). As the disease progresses, endothelial lesions would weaken the vasculature, and once patients are exposed to a trigger, an acute hemorrhage event occurs. Many studies have investigated genes involved in angiogenesis signaling and their associations with bAVM(18). The reported genes (EPHB4, VEGFA, and also MMP9) are involved in the signaling of vascular endothelial growth factor (VEGF), a representative signaling molecule of angiogenesis. VEGF is highly expressed in endothelial cells of bAVM, especially in ruptured bAVM lesions (McDonald et al., 2015). Activation of this pathway could promote endothelial cell migration and recruitment of smooth muscle cells, resulting in pathological angiogenesis (Liu et al., 2022). A recent study using mouse bAVM models demonstrated that an elevated VEGF level could contribute to bAVM hemorrhage by exposure to variable degrees of higher intraluminal flow and hypertension in the venous system (Cheng et al., 2019).
Almost all of the included studies discussed the limitation of their small sample size (mean, 223.78; standard deviation, 106.40). Only seven (12.50%) of the 52 calculated statistical powers for each SNP were more than 0.80, indicating a lower risk of Type II error. Therefore, a large study cohort would be preferable in order to achieve a statistical power 0.80 or more and to detect relatively reliable association of bAVM-related hemorrhage with the genetic variants of bAVM. All nine included studies used a CGAS design for the genotyping of individuals. The selection of genes and SNPs was based on their known biological functions. Four studies selected tagging SNPs from the HapMap project data (http://www.hapmap.org), which takes the initiative of genotyping human populations around the world and narrows down the significant loci associated with reviewed diseases (Patnala et al., 2013). The other five studies chose SNPs located in the promoters or exomes of inflammatory/angiogenesis genes to explore their associations with bAVM-related hemorrhage. However, the researchers ignored the possibility that several genes with unknown functions may also be involved in the pathogenic process of bAVM. Thus, with the development of sequencing platforms and techniques, advanced methods should be used to detect the increasing number of genetic associations of bAVMs involving GWAS and WES (McCarthy et al., 2008; Wijmenga and Zhernakova, 2018; Tam et al., 2019). After obtaining sufficiently large whole-genome sequencing datasets, machine learning can be a practical tool to extract key information efficiently (Powell et al., 2021). Based on a variatiy of statistical approaches and biological processes of genes involved in development, signaling, disease, and homeostasis, unsupervised machine learning approaches are not biased by allele frequencies, even without reliance on prior knowledge, to identify heretofore unrecognized genetic risk factors (Powell et al., 2021; Mizikovsky et al., 2022). In addition, the representativeness of the included patients with bAVM should be mentioned. Although most of the studies recruited their cohorts consecutively, the limited number of patients who only came from one or two medical centers failed to represent the populations of their regions or countries, contributing to selection bias. Therefore, we advocate for multi-center cohorts of bAVM patients with large sample sizes to improve the representativeness of the studies.
Another issue that demands greater attention is how to avoid confounding risk factors to ensure comparability. During the study design process, four studies used the strategy of matching and taking baseline characteristics (sex, age, and race) into consideration to reduce confounding bias and improve reliability (Zeldow and Hatfield, 2021). Seven studies were able to achieve statistically significant results by performing multivariate analysis to adjust for not only for the above mentioned baseline characteristics, but also for the identified morphological risk factors, such as the size and draining veins of bAVM. Three included cohort studies were conducted by the same research team; two of which considered prior hemorrhage an independent risk and confounding factor to assess the associations between genetic factors and bAVM-related hemorrhage, therefore, only new hemorrhagic events were evaluated their association with genetic risk factors, but not the overall risk of hemorrhage. Hence, it would be preferable to recruit patients with unruptured bAVM in future cohort studies. Additionally, it should be noted that the follow-up period was too short (median follow-up period was 4 months in two studies, and average follow-up was 3.1 years in one) to obtain reliable results, since some patients may have experienced hemorrhagic events after the last follow-up, resulting in a misclassification. Furthermore, the researchers used the time point and clinical information of the last follow-up, instead of regarding them as lost subjects. Calculating the rate of loss to follow-up and evaluating the adequacy of the follow-up stage are challenging tasks. Thus, in order to improve the research quality and accuracy of results, it is necessary to set an appropriate follow-up duration so that outcome events can be observed as much as possible, while preventing excessive environmental confounding factors.
To promote the genetic study on bAVM-related hemorrhage, the methodological issues in other diseases are also worth referring to. Two strategies are usually performed to identify the disease-causing variants: sequencing affected individuals with a family history and extreme-trait sequencing (Cirulli and Goldstein, 2010). The first strategy is widely adopted for rare neurological and cerebrovascular diseases, such as moyamoya disease and hereditary hemorrhagic telangiectasia (Liu et al., 2011; Benzinou et al., 2012). It is easier to detect co-effected members within families to identify overlapping variants. Considering a relatively low incidence of bAVM and its related hemorrhage, as well as its rare familial cases, it is of more value for clinicians to consciously collect these individuals for genetic study in their clinical practice (Chen et al., 2020). Extreme-trait sequencing is based on a hypothesis that the enrichment of rare variants would result in an extreme phenotype (Cirulli and Goldstein, 2010). Thus, it could be efficient to filter damaging variants by recruiting individuals with extreme-traits, including: large lesions, epileptic symptoms or an early onset of hemorrhage.
This study had several limitations. First, owing to the low incidence of bAVM, only a small number of studies with small sample sizes and insufficient statistical powers were included in the assessment. Second, the included studies published only in English or Chinese, resulting in a reduction of representativeness. Third, we calculated the statistical powers assuming α = 0.05, while the results would be more reliable by using the Bonferroni correction and multiple comparisons similar to those used in GWAS. However, the sample sizes of the existing studies did not reach a statistical significance. Thus, multi-center studies are warranted to increase the sample size and improve comparability.
5 Conclusion
Several genes were identified to be associated with bAVM-related hemorrhage, including: IL6, IL17A, MMP9, VEGFA, EPHB4. Efforts are needed to investigate the mechanism of bAVM-related hemorrhage in the future. We call for the establishment of regional alliances and rare disease banks in order to perform a multicenter prospective cohort study with a large sample size of patients with bAVM and adequate follow-up. Familial and extreme-trait cases are precious genetic resources for high-throughput sequencing. Furthermore, it may be more efficient to investigate genetic risks capitalizing on machine learning, Multi-omic systems-based approaches should be utilized to uncover the roles of candidate genes in bAVM development. Designers and researchers should strive to improve the methodological quality of their studies according to the demands of assessment tools, especially avoiding confounding risk factors, to ensure comparability.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Author contributions
JJ: Data curation, statistical analysis, resources and software, writing for original draft; ZQ: Data curation, resources and software; JY: Administration and supervision, methodology, original draft review and editing; JL: Administration and supervision, methodology, statistical analysis, resources and software, writing for original draft and review and editing.
Funding
This study was supported by Central South University Case Database Construction Project for Graduate Students (2020ALK24).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
bAVM, brain arteriovenous malformation; CGAS, candidate gene association studies; CNKI, China National Knowledge Infrastructure; GWAS, genome-wide association studies; NOS, Newcastle–Ottawa scale; OR, odds ratio; SNPs, single nucleotide polymorphisms; VEGF, vascular endothelial growth factor; WES, whole exome sequencing; 95% CI, 95% confidence interval.
References
Achrol, A. S., Pawlikowska, L., McCulloch, C. E., Poon, K. Y., Ha, C., Zaroff, J. G., et al. (2006). Tumor necrosis factor-alpha-238G>A promoter polymorphism is associated with increased risk of new hemorrhage in the natural course of patients with brain arteriovenous malformations. UCSF BAVM Study Project. Stroke 37 (1), 231–234. doi:10.1161/01.STR.0000195133.98378.4b
Atkinson, R. P., Awad, I. A., Batjer, H. H., Dowd, C. F., Furlan, A., Giannotta, S. L., et al. (2001). Reporting terminology for brain arteriovenous malformation clinical and radiographic features for use in clinical trials. Stroke 32 (6), 1430–1442. doi:10.1161/01.str.32.6.1430
Benzinou, M., Clermont, F. F., Letteboer, T. G., Kim, J. H., Espejel, S., Harradine, K. A., et al. (2012). Mouse and human strategies identify PTPN14 as a modifier of angiogenesis and hereditary haemorrhagic telangiectasia. Nat. Commun 3, 616. doi:10.1038/ncomms1633
Chen, C. J., Ding, D., Derdeyn, C. P., Lanzino, G., Friedlander, R. M., Southerland, A. M., et al. (2020). Brain arteriovenous malformations: A review of natural history, pathobiology, and interventions. Neurology 95 (20), 917–927. doi:10.1212/WNL.0000000000010968
Cheng, P., Ma, L., Shaligram, S., Walker, E. J., Yang, S. T., Tang, C., et al. (2019). Effect of elevation of vascular endothelial growth factor level on exacerbation of hemorrhage in mouse brain arteriovenous malformation. J. Neurosurg. 132 (5), 1566–1573. doi:10.3171/2019.1.JNS183112
Cirulli, E. T., and Goldstein, D. B. (2010). Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat. Rev. Genet. 11 (6), 415–425. doi:10.1038/nrg2779
Delev, D., Pavlova, A., Grote, A., Bostrom, A., Hollig, A., Schramm, J., et al. (2017). NOTCH4 gene polymorphisms as potential risk factors for brain arteriovenous malformation development and hemorrhagic presentation. J. Neurosurg. 126 (5), 1552–1559. doi:10.3171/2016.3.JNS151731
Derdeyn, C. P., Zipfel, G. J., Albuquerque, F. C., Cooke, D. L., Feldmann, E., Sheehan, J. P., et al. (2017). Management of brain arteriovenous malformations: A scientific statement for healthcare professionals from the American heart association/American stroke association. Stroke 48 (8), e200–e224. doi:10.1161/STR.0000000000000134
Gong, Z. P., Qiao, N. D., Gu, Y. X., Song, J. p., Li, P. l., Qiu, H. j., et al. (2011). Polymorphisms of VEGFA gene and susceptibility to hemorrhage risk of brain arteriovenous malformations in a Chinese population. Acta Pharmacol. Sin. 32 (8), 1071–1077. doi:10.1038/aps.2011.76
Kim, H., Hysi, P. G., Pawlikowska, L., Poon, A., Burchard, E. G., Zaroff, J. G., et al. (2009). Common variants in interleukin-1-Beta gene are associated with intracranial hemorrhage and susceptibility to brain arteriovenous malformation. Cerebrovasc. Dis. 27 (2), 176–182. doi:10.1159/000185609
Krithika, S., and Sumi, S. (2021). Neurovascular inflammation in the pathogenesis of brain arteriovenous malformations. J. Cell Physiol. 236 (7), 4841–4856. doi:10.1002/jcp.30226
Li, X. S., Jiang, N., Guo, S. L., Liang, F., and Qi, T. W. (2012). Correlation of single nucleotide polymorphisms of three genes and hemorrhage risk in patients with the BAVM. J. Trop. Med. 12 (8), 977–979.
Liu, J., Li, Y., Zhang, H., Luo, C., Yuan, D., Jiang, W., et al. (2022). Associated genetic variants and potential pathogenic mechanisms of brain arteriovenous malformation. J. Neurointerv Surg., neurintsurg-2022-018776. doi:10.1136/neurintsurg-2022-018776
Liu, W., Morito, D., Takashima, S., Mineharu, Y., Kobayashi, H., Hitomi, T., et al. (2011). Identification of RNF213 as a susceptibility gene for moyamoya disease and its possible role in vascular development. PLoS One 6 (7), e22542. doi:10.1371/journal.pone.0022542
McCarthy, M. I., Abecasis, G. R., Cardon, L. R., Goldstein, D. B., Little, J., Ioannidis, J. P. A., et al. (2008). Genome-wide association studies for complex traits: Consensus, uncertainty and challenges. Nat. Rev. Genet. 9 (5), 356–369. doi:10.1038/nrg2344
McDonald, J., Wooderchak-Donahue, W., VanSant Webb, C., Whitehead, K., Stevenson, D. A., and Bayrak-Toydemir, P. (2015). Hereditary hemorrhagic telangiectasia: Genetics and molecular diagnostics in a new era. Front. Genet. 6, 1. doi:10.3389/fgene.2015.00001
Mizikovsky, D., Naval Sanchez, M., Nefzger, C. M., Cuellar Partida, G., and Palpant, N. J. (2022). Organization of gene programs revealed by unsupervised analysis of diverse gene-trait associations. Nucleic Acids Res. 50 (15), e87. doi:10.1093/nar/gkac413
Patnala, R., Clements, J., and Batra, J. (2013). Candidate gene association studies: A comprehensive guide to useful in silico tools. BMC Genet. 14, 39. doi:10.1186/1471-2156-14-39
Pawlikowska, L., Poon, K. Y., Achrol, A. S., McCulloch, C. E., Ha, C., Lum, K., et al. (2006). Apolipoprotein E epsilon 2 is associated with new hemorrhage risk in brain arteriovenous malformations. Neurosurgery 58 (5), 838–843. ; discussion 838-43. doi:10.1227/01.NEU.0000209605.18358.E5
Pawlikowska, L., Tran, M. N., Achrol, A. S., McCulloch, C. E., Ha, C., Lind, D. L., et al. (2004). Polymorphisms in genes involved in inflammatory and angiogenic pathways and the risk of hemorrhagic presentation of brain arteriovenous malformations. Stroke 35 (10), 2294–2300. doi:10.1161/01.STR.0000141932.44613.b1
Powell, S. K., O'Shea, C., Brennand, K. J., and Akbarian, S. (2021). Parsing the functional impact of noncoding genetic variants in the brain epigenome. Biol. Psychiatry 89 (1), 65–75. doi:10.1016/j.biopsych.2020.06.033
Rutledge, W. C., Ko, N. U., Lawton, M. T., and Kim, H. (2014). Hemorrhage rates and risk factors in the natural history course of brain arteriovenous malformations. Transl. Stroke Res. 5 (5), 538–542. doi:10.1007/s12975-014-0351-0
Sohani, Z. N., Meyre, D., de Souza, R. J., Joseph, P. G., Gandhi, M., Dennis, B. B., et al. (2015). Assessing the quality of published genetic association studies in meta-analyses: The quality of genetic studies (Q-Genie) tool. BMC Genet. 16, 50. doi:10.1186/s12863-015-0211-2
Stang, A. (2010). Critical evaluation of the Newcastle-Ottawa scale for the assessment of the quality of nonrandomized studies in meta-analyses. Eur. J. Epidemiol. 25, 603–605. doi:10.1007/s10654-010-9491-z
Sun, B., Qiu, H., Zhao, F., Qiao, N., Fan, W., Lu, D., et al. (2012). The rs9509 polymorphism of MMP-9 is associated with risk of hemorrhage in brain arteriovenous malformations. J. Clin. Neurosci. 19 (9), 1287–1290. doi:10.1016/j.jocn.2011.09.036
Tam, V., Patel, N., Turcotte, M., Bossé, Y., Paré, G., and Meyre, D. (2019). Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 20 (8), 467–484. doi:10.1038/s41576-019-0127-1
van Beijnum, J., van der Worp, H. B., Buis, D. R., Al-Shahi Salman, R., Kappelle, L. J., Rinkel, G. J. E., et al. (2011). Treatment of brain arteriovenous malformations: A systematic review and meta-analysis. JAMA 306 (18), 2011–2019. doi:10.1001/jama.2011.1632
Weinsheimer, S., Kim, H., Pawlikowska, L., Chen, Y., Lawton, M. T., Sidney, S., et al. (2009). EPHB4 gene polymorphisms and risk of intracranial hemorrhage in patients with brain arteriovenous malformations. Circ. Cardiovasc Genet. 2 (5), 476–482. doi:10.1161/CIRCGENETICS.109.883595
Wijmenga, C., and Zhernakova, A. (2018). The importance of cohort studies in the post-GWAS era. Nat. Genet. 50 (3), 322–328. doi:10.1038/s41588-018-0066-3
Keywords: brain arteriovenous malformation, intracranial hemorrhage, genetics, methodological quality, rupture
Citation: Jiang J, Qin Z, Yan J and Liu J (2023) Methodological quality assessment of genetic studies on brain arteriovenous malformation related hemorrhage: A cross-sectional study. Front. Genet. 14:1123898. doi: 10.3389/fgene.2023.1123898
Received: 14 December 2022; Accepted: 07 March 2023;
Published: 23 March 2023.
Edited by:
Farren Briggs, Case Western Reserve University, United StatesReviewed by:
Jun Wang, Huazhong University of Science and Technology, ChinaDaniel Cooke, University of California, San Francisco, United States
Copyright © 2023 Jiang, Qin, Yan and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junyu Liu, eXVtZTA1MDdAZ21haWwuY29t; Junxia Yan, MjA0NTc0NTZAcXEuY29t