 Yanjiao Cui1†
Yanjiao Cui1† Zhengli Liu
Zhengli Liu- 1Department of Life Sciences, Tangshan Normal University, Tangshan, China
- 2Tianjin Key Laboratory of Crop Genetics and Breeding, Institute of Crop Sciences, Tianjin Academy of Agricultural Sciences, Tianjin, China
Introduction: Spruce spider mite is a primary insect pest of Chinese chestnut in China and seriously influences its yield and quality. However, the current management against this mite is costly and poorly effective. In previous research, we bred several foxtail millet materials for interplanting with chestnut tree, and found that they had high levels of crude protein (CP) in leaves and attracted spruce spider mite to feed on the leaves, thereby reducing chestnut damage.
Methods: In this study, four foxtail millet varieties with significant differences in leaf crude protein content were used for high-throughput sequencing and identification of genes associated with leaf crude protein content. Gene enrichment analyses were carried out to comprehend the functions of these genes and the biological processes in which they are involved. In addition, transcription factors (TFs) were evaluated.
Results: 435 differentially expressed genes (DEGs) were identified, suggesting their potential role in crude protein accumulation. Some differentially expressed genes were found to be associated with nitrogen metabolism and ubiquitin-mediated proteolysis pathways. Moreover, we identified 40 TF genes categorized into 11 transcription factor families.
Discussion: Our findings represent an important resource that clarifies the mechanisms of accumulation and control of leaf crude protein in foxtail millet, and provide an opportunity for suppression of spruce spider mite attack on Chinese chestnut by interplanting with foxtail millet varieties with high concentrations of leaf crude protein.
Introduction
Chestnut has been cultivated in China for more than 2700 years (Adua, 1999). It is an economically valuable plant, its wood used for timber, its leaf and flower utilized as raw material in the pharmaceutical and cosmetic industries, and its fruit peel a source of extracted tannin. Most of all, its fruit is a popular food resource with high nutritional value. The genus Castanea comprises 12 world chestnut species, including four cultivated species: Chinese chestnut (Castanea mollissima Blume), Japanese chestnut (Castanea crenata Sieb. et Zucc.), American chestnut (Castanea dentata [Marsh.] Borkh), and European chestnut (Castanea sativa Miller) (Jiang et al., 2019). Among these, Chinese chestnut and Japanese chestnut are the most commercially important (Tanaka and Kotobuki, 1992).
Chinese chestnut is commonly grown in China for its fruits. However, several insect pests can seriously damage the tree’s trunk, buds, and fruit, and reduce nut production and quality (Gokce et al., 2020). One such pest is the spruce spider mite (Oligonychus ununguis Jacobi), a typical polyphagous mite that consumes the chestnut’s leaves (Wang et al., 1989; Wang et al., 2010). In the main chestnut-producing region of China, the occurrence of spruce spider mite generally leads to a 15%–50% decline in production (Wang et al., 1991).
To date, several biological and chemical control measures have been taken to prevent this mite from attacking trees in China, including the use of natural enemy species and pesticides (Wang et al., 2010; Zhao and Fan, 2019). Nevertheless, because Chinese chestnut trees are tall and mostly distributed in mountains, it is inconvenient to spray pesticides. Moreover, it often results in the development of resistance to pesticides and environmental pollution, thereby slowing the movement toward green, pollution-free production of crops and restricting the development of the Chinese chestnut industry. In sum, the control measures for spruce spider mite prevention are currently expensive and poorly effective.
In previous work, we developed a number of foxtail millet (Setaria italica (L.) P. Beauv.) materials for interplanting with chestnut. They were early-maturing, shade-tolerant, could thrive beneath Chinese chestnut trees, realizing the high-efficient utilization of land resources under the trees. Surprisingly, we discovered that the leaves of these materials were attacked by spruce spider mites. This decreased the rate of bad chestnut nuts resulting from mite attack by 5%–15%, while damage to the millet was minimal (Li et al., 2022). Furthermore, these materials had a high content of crude protein (CP) in their leaves.
Plant–insect interactions are intricate and dynamic. To overcome insect attack, plants develop different defense mechanisms, including physical and chemical defenses (War et al., 2012; Belete, 2018). The first mechanical barrier to feeding is formed by structural features on the leave surface, such as a waxy cuticle, spines and thorns, trichomes, and thick and lignified cell walls. Secondary metabolites, including phenolics, flavonoids, tannins, and lectins, provide the next barriers of protection. Among chemical constituents, nutrients such as amino acids, vitamins, proteins, and carbohydrates have been also reported to affect the relationship between plants and insects (Li and Jin, 2005; Yang et al., 2014; Li and Li, 2018). In one study, nymphal density and the leaf damage index of grape leafhopper (Erythroneura apicalis Nawa) were negatively correlated with CP levels in grape leaf (Fan et al., 2008). Another study reported that total soluble protein content in cotton leaves was positively associated with the population densities of thrip (Thrips tabaci), whitefly (Bemesia tabaci), and jassid (Amrasca devastans), and these pests were more prone to attack cotton genotypes with high concentrations of such proteins (Rizwan et al., 2021). Hence, the protein content of host plant leaves may be related to the degree of insect infestation, which could explain the effect of the foxtail millet materials we bred in previous work. Consequently, it is important to mine genes related to CP accumulation in foxtail millet leaves and to elucidate their regulation mechanisms. This would be helpful for breeding of foxtail millet varieties with high concentrations of leaf CP, and provide an effective, economically, and environmentally safe control measure for spruce spider mite through interplanting.
In this study, we performed transcriptome analyses on four foxtail millet varieties with significantly different leaf CP levels to identify genes associated with leaf CP content. We identified and characterized differentially expressed genes (DEGs) and conducted gene enrichment analyses to investigate their possible functions. We also predicted differentially expressed transcription factor (TF) genes. Our results lay a foundation for clarifying the biological processes and regulation mechanisms underlying leaf CP accumulation, and provide potentially precious gene resources for the future development of foxtail millet varieties with high concentrations of leaf CP.
Materials and methods
Below we provide a summary of our methods. More details can be found in the Supplementary Materials and Methods.
Plant materials
Four hybrid varieties of foxtail millet (51950, 12950, 1121, 57295) and a conventional variety (JG32) grown in an experimental field of Beiguan Village, Tangshan, China (40°10'N, 118°28'E) were utilized. The millet was sown in 10 m long rows with 0.5 m between rows. For each variety, three standard plants were chosen, and the middle parts of the first, second, and third leaves from the top of each were collected during the booting stage. Then we mixed and ground the sampled leaves for CP content analysis, RNA-seq analysis, and quantitative real-time PCR (qRT-PCR) validation.
Chemical analysis of crude protein concentration
Hebei Jintianfeng Grain Trading Co., Ltd. (China) conducted the CP concentration analysis. Total nitrogen content was obtained using the Kjeldahl method (Kjeldahl, 1883), and the value was multiplied by 6.25 to calculate CP content. Each sample had three biological replicates.
To screen varieties with different CP levels, JG32 was used as a control, which has been found to be suitable for use in interplanting with Chinese chestnut. Three varieties with high CP concentrations (51950, 12950, and 1121) and one with a low concentration (57295) were selected for high-throughput sequencing. The CP levels are listed in Supplementary Table S1.
Library construction, high-throughput sequencing, and transcriptome analysis
To mine genes associated with leaf CP content, the leaves of each variety were sampled as mentioned above. RNA extraction, library construction, high-throughput sequencing, and transcriptome assembly were carried out as described previously (Li et al., 2022).
Screening of differentially expressed genes
Fragments per kilobase of transcript per million fragments mapped (FPKM) values were used to determine the gene expression levels (Trapnell et al., 2010). To detect DEGs, the software edgeR version 3.3.3 (Robinson et al., 2009) was used, and genes with a |log2 (fold change)| value >1 and adjusted p-value (padj) < 0.05 were considered significant DEGs.
qRT-PCR validation
The qRT-PCR validation was conducted as described previously (Liu et al., 2021). Three independent replicates were analyzed, and the primers used are listed in Supplementary Table S2.
Functional enrichment analyses
GO functional enrichment analysis and KEGG pathway analysis of DEGs were conducted using topGO (Yu et al., 2010) and KOBAS 3.0 (Xie et al., 2011), respectively.
Identification of TFs
The online webserver PlantTFDB 5.0 (http://planttfdb.gao-lab.org/prediction.php) with default parameters was used to identify TF families (Jin et al., 2017). In this tool, ESTScan 3.0 was employed to analyze coding regions in the provided input sequences (Iseli et al., 1999). A heatmap of differentially expressed TF genes was generated using TBtools software (Chen et al., 2020).
Results
Transcriptome sequencing and assembly
To characterize genes associated with biosynthesis and accumulation of leaf CP, 12 cDNA libraries from leaf samples of the four varieties with different CP levels were used for RNA sequencing (three library repeats for each variety). Approximately 645.17 M raw reads were generated using an Illumina Novaseq PE150 platform. After quality control, we obtained 641.29 M clean reads and found that 94.76%–95.65% were mapped to the foxtail millet reference genome (Setaria_italica_v2.0, http://plants.ensembl.org/Setaria_italica/Info/Index), provided in a previous report (Liu et al., 2022). Furthermore, Pearson’s correlation coefficients between biological replicates showed that the biological replicates were highly correlated (Liu et al., 2022), indicating the reliability of the RNA-seq results for gene expression analysis. In total, 30,141 genes were discovered. The FPKM values for each gene were computed and are listed in Supplementary Table S3.
Identification of differentially expressed genes
As shown in Figure 1, compared to the low-CP variety 57295, 1726, 2235, and 961 DEGs were upregulated in the high-CP varieties 51950, 12950, and 1121, respectively. In all, 3467 upregulated DEGs were identified, 337 of which were shared among the three high-CP varieties. Likewise, 962, 808, and 718 DEGs were downregulated in 51950, 12950, and 1121, respectively. In all, 1904 downregulated genes were identified, among which 98 were shared. Differential expression analysis provided 435 common DEGs shared in all comparisons, representing possible candidate genes associated with CP accumulation in foxtail millet.
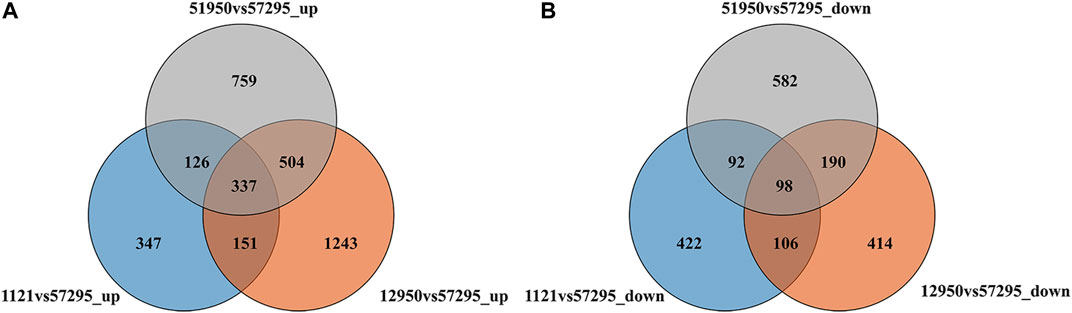
FIGURE 1. Venn map depicting the number of DEGs between foxtail millet varieties with high and low leaf crude protein content. (A) and (B) show the distribution of upregulated and downregulated DEGs in each comparison, respectively.
Validation of differentially expressed genes using qRT-PCR
To verify the accuracy of the RNA-seq data, the relative expression levels of eight randomly selected DEGs were examined via qRT-PCR. These eight DEGs included five genes that were predicted to be upregulated in varieties with high CP concentrations (SETIT_020998mg, SETIT_025437mg, SETIT_009199mg, SETIT_038934mg, SETIT_0403631 mg), and three genes predicted to be downregulated (SETIT_031738mg, SETIT_009896mg, SETIT_012108 mg). As shown in Figure 2, the expression profiles were in line with the RNA-seq data, further demonstrating the reliability of our RNA-seq data.
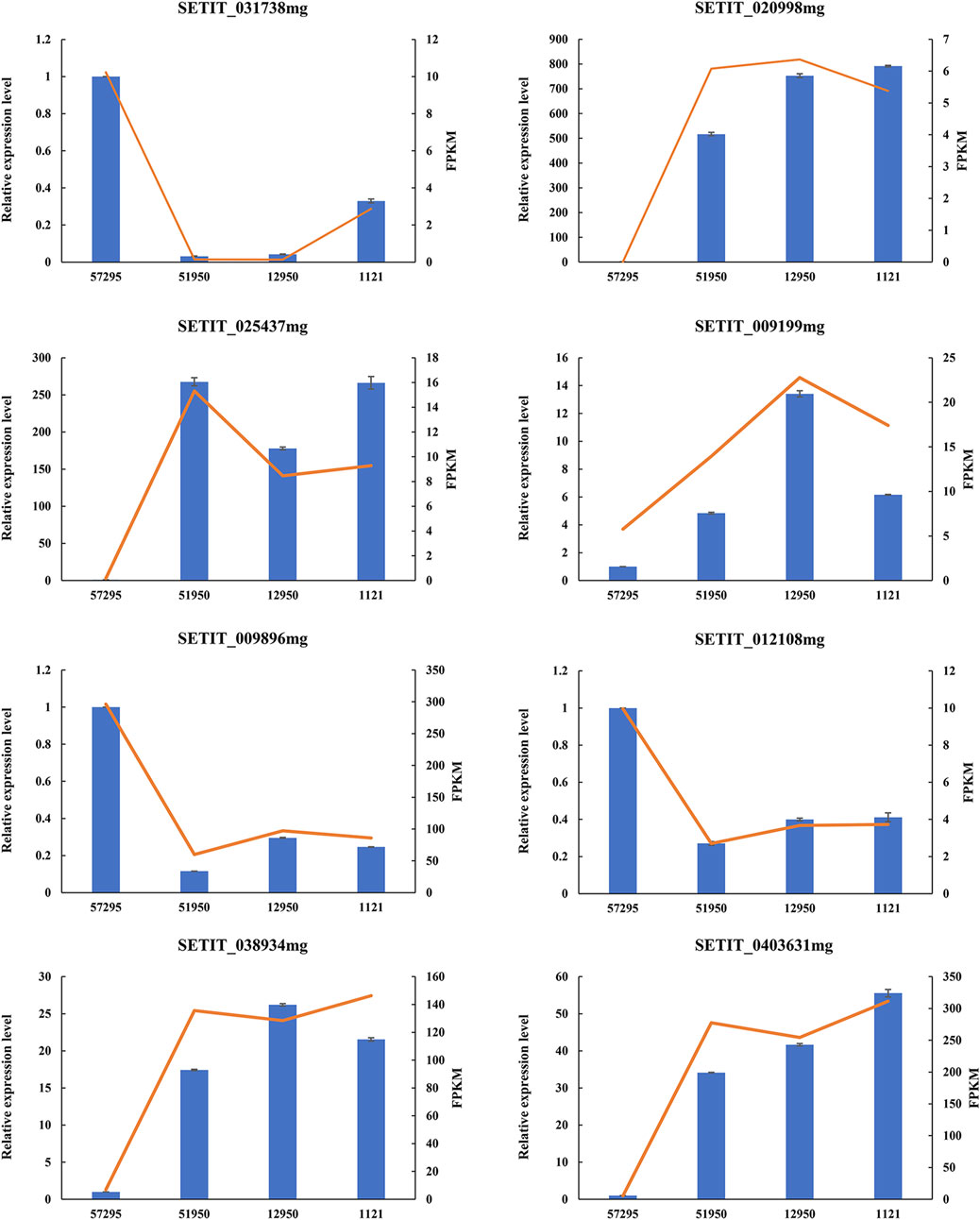
FIGURE 2. Validation of eight DEGs by qRT-PCR. The X-axis represents foxtail millet varieties with different levels of leaf crude protein content, and the Y-axis (left side) indicates the relative expression level of selected genes determined by qRT-PCR (blue columns). The SiACTIN gene was used as an internal control, and the transcript level of genes in variety 57295 was set as 1.0. Error bars represent standard error (n = 3). The Y-axis (right side) depicts the expression level of genes in RNA-seq data (evaluated by FPKM, red lines).
Gene enrichment analysis of differentially expressed genes
For 435 common DEGs in all comparisons, GO category enrichment analysis was conducted using topGO (Yu et al., 2010) to determine their potential biological processes and functions (Supplementary Table S4). These DEGs were annotated to three categories and 40 GO terms: 42.5% of the terms were classified as biological processes, 27.5% were molecular functions, and 30% were cellular components (Figure 3). In the biological process category, genes were most enriched in metabolic processes (GO:0008152) and cellular processes (GO:0009987). In the molecular function category, most genes fell into the binding (GO:0005488) and catalytic activity (GO:0003824) subgroups. In the cellular component category, the genes mainly belonged to cell (GO:0005623) and cell part (GO:0044464) subgroups.
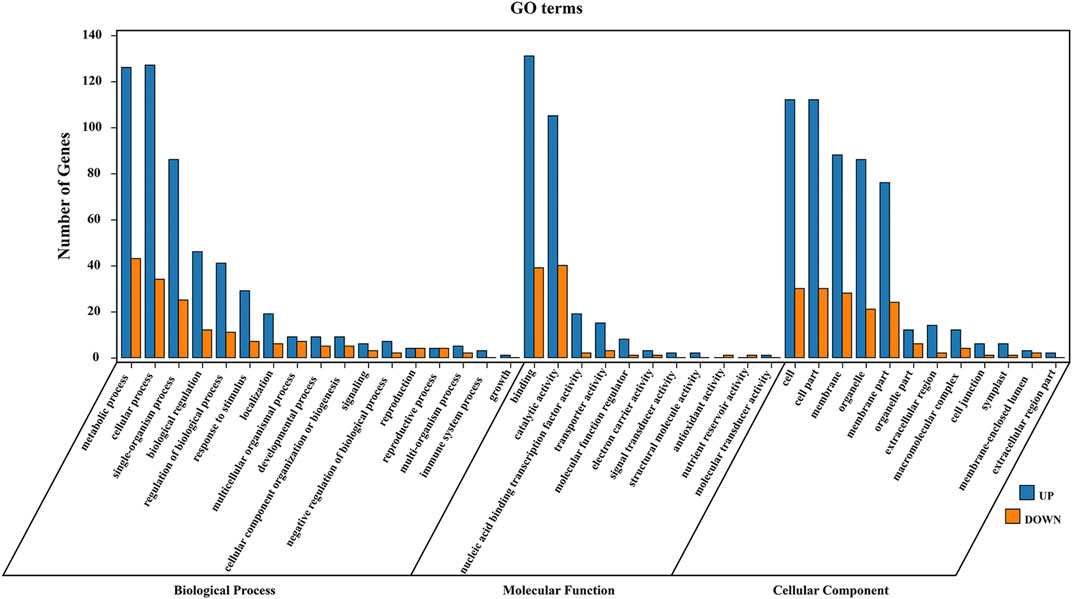
FIGURE 3. GO enrichment analysis of DEGs. Enriched GO terms of biological processes, molecular function, and cellular components are shown. Blue represents upregulated DEGs in varieties with high leaf crude protein content, and red represents downregulated DEGs.
To further understand the functional categorizations, the 435 DEGs were also subjected to KEGG enrichment analysis (Table S5); 174 DEGs were assigned to 60 enrichment pathways categories, of which the top five were metabolic pathways (35 genes, 20.11%), biosynthesis of secondary metabolites (28, 16.09%), plant–pathogen interactions (8, 4.60%), cyanoamino acid metabolism (7, 4.02%), and phenylpropanoid biosynthesis (7, 4.02%). These pathways were classified into five main categories and 16 subcategories, with the top five subcategories being global and overview maps, biosynthesis of other secondary metabolites, carbohydrate metabolism, metabolism of other amino acids, and environmental adaptation (Figure 4). The enrichment analysis illustrated that changes in leaf CP content significantly impacted the life processes of foxtail millet.
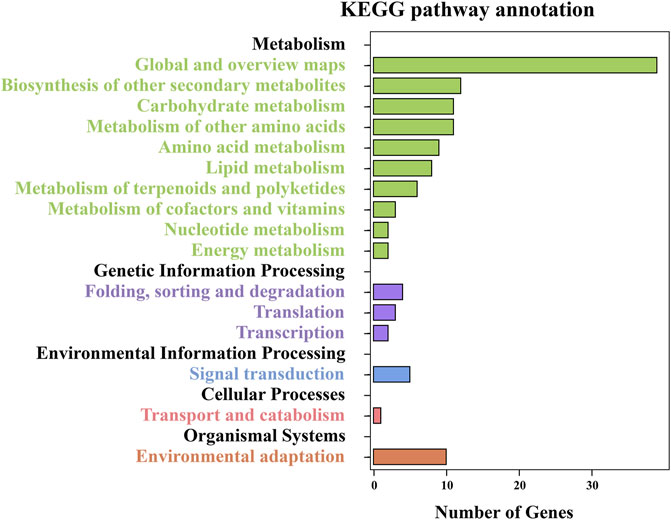
FIGURE 4. KEGG pathway analysis of enriched DEGs. The numbers of DEGs are shown on the X-axis, and the KEGG pathway terms are shown on the Y-axis.
Amino acids are necessary for the formation of proteins and other related compounds, with nitrogen being the essential constituent (Mokhele et al., 2012). Due to the close relationship between nitrogen and protein and other nitrogenous compounds, nitrogen metabolism in plants directly affects CP content. According to the KEGG pathway analysis, the genes SETIT_014128 mg and SETIT_014124 mg present in carbonic anhydrase (EC:4.2.1.1) were enriched in the nitrogen metabolism pathway and both were upregulated in leaf tissues of foxtail millet varieties with high CP concentrations.
As one of the most prevalent protein posttranslational modifiers, ubiquitin (Ub) is a small peptide of 76 amino acids that can be attached to target proteins via ubiquitination (Hershko et al., 1998). Protein ubiquitination is involved in the regulation of proteolysis, subcellular localization, and the stability and activity of substrate proteins (Zhou et al., 2017). As per the KEGG analysis, two genes, SETIT_004756 mg present in E3 ubiquitin-protein ligase SIAH1 (EC:2.3.2.27) and SETIT_025290 mg present in ubiquitin-conjugating enzyme E2 J2 (EC:2.3.2.23), were annotated to ubiquitin-mediated proteolysis, a major pathway of degradation of cellular proteins. Based on differential expression analysis, SETIT_004756 mg and SETIT_025290 mg were downregulated and upregulated in varieties with high CP concentrations, respectively.
TF analysis
The GO enrichment analysis revealed that several DEGs may function in regulation of transcription and have TF activity (Supplementary Table S4). TFs are DNA-binding proteins that mediate many processes by playing a crucial role in gene transcription and expression. As a consequence, to more fully understand the functions of DEGs and analyze their regulatory mechanisms, we predicted possible TF genes from among the 435 common DEGs shared in all comparisons using the plant TF database PlantTFDB (Jin et al., 2017). In total, 40 genes were identified, including 33 upregulated genes and seven downregulated genes, and they were grouped into 11 different TF families (Figure 5; Figure 6, Table S6). Among them, the ERF family was the most abundant family (13 genes). The remaining top 4 TF families were bHLH (seven genes), NAC (five genes), C2H2 (four genes), and B3 (three genes).
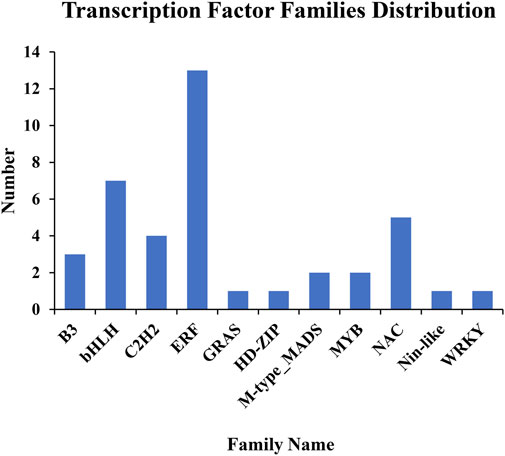
FIGURE 5. Graphical distribution of transcription factor families enriched in DEGs.
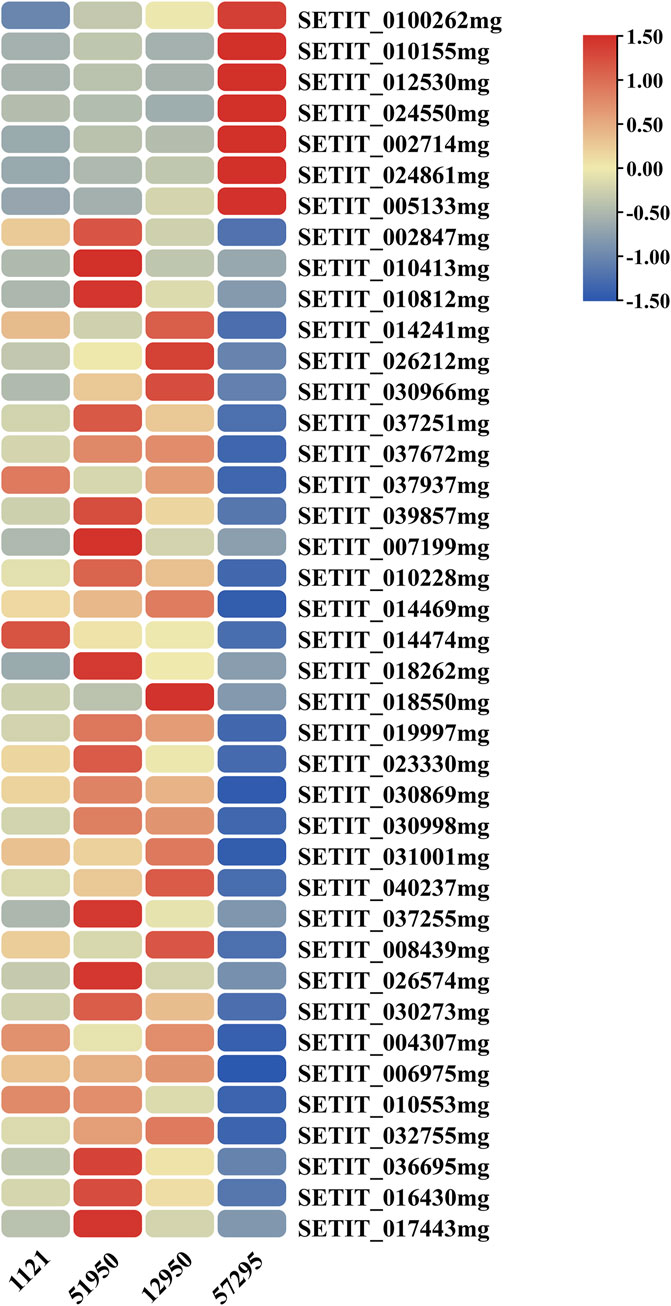
FIGURE 6. Heatmap of the differentially expressed transcription factor genes. The color gradient indicates the FPKM value of genes; red and blue represent high and low expression, respectively.
Discussion
In the natural environment, plants are constantly threatened by herbivorous insects. For Chinese chestnut, damage from spruce spider mite is a primary challenge restricting its output and quality, and generally leads to a serious commercial loss. In previous work, we developed several shade-tolerant foxtail millet varieties for interplanting with chestnut trees, and found that it improved the quality of chestnut fruits by attracting spruce spider mite to feed on their leaves. This strategy could become a new, effective, and environmentally friendly, pest-management option for chestnut tree.
Nutrients of host plants, such as proteins and carbohydrates, are essential sources of energy for the growth and development of herbivorous insects. CP content in leaves affects the lifespan, development duration, and egg laying outcomes of mites, and in certain plant species the degree of leaf damage by insects is higher for leaves with higher CP levels (Wu et al., 1995; Fan et al., 2008; Wei et al., 2009; Yang et al., 2014; Sonalkar, 2020). The foxtail millet varieties we previously bred for interplanting with chestnut tree all had high CP levels in leaves, which may be one major reason why they attracted the spider mites. In present study, four foxtail millet varieties with significant differences in leaf CP content were used to identify genes related to CP levels via transcriptome sequencing technology. We successfully identified 435 DEGs, including 337 upregulated DEGs and 98 downregulated DEGs in varieties with high CP concentrations. Our findings provide a precious resource for research on the mechanisms that control the accumulation of CP, and offer a chance to effectively control spruce spider mite outbreaks among chestnut trees.
Protein is an important product of nitrogen metabolism, and therefore CP levels are closely associated with nitrogen metabolism. For example, an increase in CP levels of vegetable leaves is correlated with an increase in N uptake (Mhlontlo et al., 2007). Most plants assimilate inorganic and organic forms of nitrogen, including nitrate and ammonium (Temple et al., 1998). Metabolism normally begins with nitrate being converted into nitrite in the presence of nitrate reductase, and then the nitrite is reduced to form ammonia by nitrite reductase. Following its formation, ammonia is assimilated by a number of pathways, including reductive amination and transpiration, to yield a variety of amino acids (Mokhele et al., 2012; Kishorekumar et al., 2020). In the current study, two DEGs, SETIT_014128 mg and SETIT_014124mg, were enriched in the nitrogen metabolism pathway and may code for carbonic anhydrase (CA). The reversible conversion of carbon dioxide to bicarbonate (CO2+H2O
In eukaryotes, the ubiquitin-proteasome system (UPS) is responsible for the majority of cytosolic protein breakdown, accomplished through the degradation of the substrate protein by ATP-dependent 26S proteasome after covalent attachment of many Ub molecules to the substrate protein (Ciechanover et al., 2000; Nandi et al., 2006). Binding of Ub to target proteins includes the following steps. First, the C-terminal Gly of Ub is activated and transferred to a carrier E2 protein (Ub-conjugating enzyme) by the Ub-activating enzyme E1. Then with the aid of E3 (a Ub-protein ligase enzyme), the E2 protein tags Ub to the target protein. Various E3 ligases within the UPS recognize substrates carrying diverse degradation signals, giving the system high specificity and selectivity. In this study, we identified two DEGs that encode Ub-conjugating enzyme E2 J2 (UBE2J2) and E3 Ub-protein ligase SIAH1, namely, SETIT_025290 mg and SETIT_004756mg, respectively. The UBE2J2 protein is a Ub-conjugating enzyme which, in association with Ub ligases including TEB4, PARKIN, CHIP, and cIAP1, promotes proteasomal degradation in mammals and responds to proteotoxic stress (Imai et al., 2002; Lenk et al., 2002; Kim et al., 2003; Wu et al., 2005; Choi et al., 2010; Claessen et al., 2010; Burr et al., 2011). Nevertheless, no UBE2J2 proteins have been characterized in plants to date. The seven in absentia (SINA)/seven in absentia homolog (SIAH) family proteins are E3 Ub-protein ligases with a RING domain and are highly conserved from plants to mammals (Polekhina et al., 2002). This superfamily was first characterized in Drosophila melanogaster and plays a crucial role in the regulation of development and proteasome-mediated protein degradation (Hu and Fearon Eric, 1999). To date, 18 SINA proteins homologous to the Drosophila SINA protein have been defined in Arabidopsis, among which SINAT5 targets the noapical meristem/cup-shaped cotyledon 1 (NAC1) TF and late elongated hypocotyl (LHY) for degradation and plays a role in various developmental processes in plants (Xie et al., 2002; Park et al., 2010). Therefore, SETIT_025290 mg and SETIT_004756 mg may be involved in specific ubiquitination and subsequent proteolysis of proteins, and have an impact on leaf CP content in foxtail millet by regulating protein levels and the stability of related proteins. However, their biological functions and regulation mechanisms remain to be elucidated in future work.
TFs regulate a variety of biological processes, such as plant metabolism, growth, and development, by binding to the promoters (or intragenic regions) of target genes (Kaufmann et al., 2018; Burgess et al., 2019). Many TFs have been found to control nitrogen assimilation. Recently, Wei et al. reported that overexpression of the dehydration-responsive element-binding protein 1C (OsDREB1C) gene in rice (Oryza sativa), a member of the AP2/ERF TF family, improved nitrogen content, nitrogen use efficiency (NUE), and protein abundance in leaves by elevating nitrogen uptake and transport activity (Wei et al., 2022). Furthermore, a few TFs affect protein synthesis in plants. For example, overexpressing the MYB family TF TaODORANT1 in common wheat (Triticum aestivum) and its homolog in Triticum urartu reduces the transcription levels of seed storage protein (SSP) genes and total SSP levels of mature grains, revealing an inhibition effect on SSP synthesis (Luo et al., 2021). OPAQUE 11 (O11) encodes a seed-specific bHLH TF in maize, and the protein levels of loss-of-function mutant o11 endosperm per kernel are significantly decreased compared to wild type (Feng et al., 2018), revealing regulatory role in protein accumulation. In addition, several NAC TFs have also been reported to regulate protein accumulation and content in grains of wheat, rice, and maize (Zea mays) (Uauy et al., 2006; Liang et al., 2014; Zhang et al., 2019; Wang et al., 2020). An endosperm-specific NAC TF, TaNAC019, modulates SSP accumulation in wheat seeds (Gao et al., 2021). It directly binds to the promoters of high-molecular-weight glutenin (HMW-GS) genes and activates their expression, and triple knock-out mutants of TaNAC019 homologs have lower gluten levels. In the current research, 40 DEGs were identified as TF genes, containing several members of the bHLH, MYB, NAC, and especially ERF gene families (Figure 5). It can be speculated that these genes may function as TFs and improve CP accumulation in leaves of foxtail millet by modulating nitrogen assimilation and protein synthesis. However, the detailed underlying molecular mechanisms involved require further research, and will be a focus of ours in the future.
Conclusion
Transcriptome analysis of genes in foxtail millet associated with CP accumulation in leaves was performed and 435 DEGs were identified. Several DEGs related to nitrogen metabolism and ubiquitin-mediated proteolysis pathways were characterized. We also predicted the TFs that participate in the control of CP levels. These findings provide a resource that clarifies the accumulation and control mechanisms of CP levels in leaves. This information can be used to develop foxtail millet varieties with high CP concentrations for interplanting with chestnut, to improve production of this important crop.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA772942.
Author contributions
The research was conceived and designed by ZL and SL. DL, ZZ, SL, YC, and JZ performed the experiments. Data was analyzed by DL, ZZ, and YC. YC wrote the original draft. ZL and DL made revisions to the final manuscript. The final manuscript was read and approved by all authors.
Funding
This work was supported by S&T Program of Hebei (21326337D, 21326302D), Tangshan Municipal Science and Technology Project (20130218b) and Scientific Research Fund Project of Tangshan Normal University (2020A06).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1122212/full#supplementary-material
References
Adua, M. (1999). The sweet chestnut throughout history from the Miocene to the third millennium. Acta Hortic. 494, 29–36. doi:10.17660/actahortic.1999.494.2
Belete, T. (2018). Defense mechanisms of plants to insect pests: From morphological to biochemical approach. Trends Tech. Sci. Res. 2 (2), 30–38. doi:10.19080/ttsr.2018.02.555584
Burgess, S. J., Reyna-Llorens, I., Stevenson, S. R., Singh, P., Jaeger, K., and Hibberd, J. M. (2019). Genome-wide transcription factor binding in leaves from C3 and C4 grasses. Plant Cell. 31 (10), 2297–2314. doi:10.1105/tpc.19.00078
Burr, M. L., Cano, F., Svobodova, S., Boyle, L. H., Boname, J. M., and Lehner, P. J. (2011). HRD1 and UBE2J1 target misfolded MHC class I heavy chains for endoplasmic reticulum-associated degradation. Proc. Natl. Acad. Sci. U. S. A. 108 (5), 2034–2039. doi:10.1073/pnas.1016229108
Chatterjee, J., Coe, R. A., Acebron, K., Thakur, V., Yennamalli, R. M., Danila, F., et al. (2021). A low CO2-responsive mutant of Setaria viridis reveals that reduced carbonic anhydrase limits C4 photosynthesis. J. Exp. Bot. 72 (8), 3122–3136. doi:10.1093/jxb/erab039
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13 (8), 1194–1202. doi:10.1016/j.molp.2020.06.009
Cheng, Y., Wang, Y., Han, Y., Li, D., Zhang, Z., Zhu, X., et al. (2019). The stimulatory effects of nanochitin whisker on carbon and nitrogen metabolism and on the enhancement of grain yield and crude protein of winter wheat. Molecules 24 (9), 1752. doi:10.3390/molecules24091752
Choi, Y. L., Soda, M., Yamashita, Y., Ueno, T., Takashima, J., Nakajima, T., et al. (2010). EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. New Engl. J. Med. 363 (18), 1734–1739. doi:10.1056/NEJMoa1007478
Ciechanover, A., Orian, A., and Schwartz, A. L. (2000). Ubiquitin-mediated proteolysis: Biological regulation via destruction. Bioessays 22 (5), 442–451. doi:10.1002/(SICI)1521-1878(200005)22:5<442:AID-BIES6>3.0.CO;2-Q
Claessen, J. H., Mueller, B., Spooner, E., Pivorunas, V. L., and Ploegh, H. L. (2010). The transmembrane segment of a tail-anchored protein determines its degradative fate through dislocation from the endoplasmic reticulum. J. Biol. Chem. 285 (27), 20732–20739. doi:10.1074/jbc.M110.120766
Ebbs, S. (2004). Biological degradation of cyanide compounds. Curr. Opin. Biotechnol. 15 (3), 231–236. doi:10.1016/j.copbio.2004.03.006
Fan, Y., Jiang, X., Hao, J., Liu, Z., Wang, H., Wu, Y., et al. (2008). Correlation analysis between damage of the grape leafhopper and the chemicals of grape varieties in Xinjiang. Acta Agric. Boreali-occidentalis Sin. 17 (1), 70–73. doi:10.3969/j.issn.1004-1389.2008.01.016
Feng, F., Qi, W., Lv, Y., Yan, S., Xu, L., Yang, W., et al. (2018). OPAQUE11 is a central hub of the regulatory network for maize endosperm development and nutrient metabolism. Plant Cell. 30 (2), 375–396. doi:10.1105/tpc.17.00616
Gao, Y., An, K., Guo, W., Chen, Y., Zhang, R., Zhang, X., et al. (2021). The endosperm-specific transcription factor TaNAC019 regulates glutenin and starch accumulation and its elite allele improves wheat grain quality. Plant Cell. 33 (3), 603–622. doi:10.1093/plcell/koaa040
Gokce, M. P., Karagoz, M., Faraji, F., and Cakmak, I. (2020). Mite species composition and their population densities on chestnut trees in Turkey. Int. J. Acarol. 46 (4), 247–253. doi:10.1080/01647954.2020.1752796
Hershko, A. (1998). “The ubiquitin system,” in Ubiquitin and the biology of the cell. Editors J-M. Peters, J. R. Harris, and D. Finley (Boston, MA: Springer US), 1–17.
Hu, G., and Fearon Eric, R. (1999). Siah-1 N-terminal RING domain is required for proteolysis function, and C-terminal sequences regulate oligomerization and binding to target proteins. Mol. Cell. Biol. 19 (1), 724–732. doi:10.1128/mcb.19.1.724
Imai, Y., Soda, M., Hatakeyama, S., Akagi, T., Hashikawa, T., Nakayama, K-I., et al. (2002). CHIP is associated with Parkin, a gene responsible for familial Parkinson's disease, and enhances its ubiquitin ligase activity. Mol. Cell. 10 (1), 55–67. doi:10.1016/s1097-2765(02)00583-x
Iseli, C., Jngeneel, C. V., and Bucher, P. (1999). ESTScan: A program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 99, 138–148.
Jiang, N., Fan, X. L., and Tian, C. M. (2019). Identification and pathogenicity of Cryphonectriaceae species associated with chestnut canker in China. Plant Pathol. 68 (6), 1132–1145. doi:10.1111/ppa.13033
Jin, J., Tian, F., Yang, D. C., Meng, Y. Q., Kong, L., Luo, J., et al. (2017). PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 45 (D1), D1040–D1045. doi:10.1093/nar/gkw982
Johnson, W. V., and Anderson, P. M. (1987). Bicarbonate is a recycling substrate for cyanase. J. Biol. Chem. 262 (19), 9021–9025. doi:10.1016/s0021-9258(18)48040-4
Kaufmann, K., and Airoldi, C. A. (2018). “Master regulatory transcription factors in plant development: A blooming perspective,” in Plant transcription factors: Methods and protocols. Editor N. Yamaguchi (New York, NY: Springer New York), 3–22.
Kim, B. W., Zavacki, A. M., Curcio-Morelli, C., Dentice, M., Harney, J. W., Larsen, P. R., et al. (2003). Endoplasmic reticulum-associated degradation of the human type 2 iodothyronine deiodinase (D2) is mediated via an association between mammalian UBC7 and the carboxyl region of D2. Mol. Endocrinol. 17 (12), 2603–2612. doi:10.1210/me.2003-0082
Kishorekumar, R., Bulle, M., Wany, A., and Gupta, K. J. (2020). An overview of important enzymes involved in nitrogen assimilation of plants. Methods Mol. Biol. 2057, 1–13. doi:10.1007/978-1-4939-9790-9_1
Kjeldahl, J. (1883). Neue Methode zur Bestimmung des Stickstoffs in organischen Körpern. Z Anal. Chem. 22 (1), 366–382. doi:10.1007/bf01338151
Kozliak, E. I., Fuchs, J. A., Guilloton, M. B., and Anderson, P. M. (1995). Role of bicarbonate/CO2 in the inhibition of Escherichia coli growth by cyanate. J. Bacteriol. 177 (11), 3213–3219. doi:10.1128/jb.177.11.3213-3219.1995
Lenk, U., Yu, H., Walter, J., Gelman, M. S., Hartmann, E., Kopito, R. R., et al. (2002). A role for mammalian Ubc6 homologues in ER-associated protein degradation. J. Cell. Sci. 115 (14), 3007–3014. doi:10.1242/jcs.115.14.3007
Li, S., Cui, Y., Liu, D., Zhao, Z., Zhang, J., and Liu, Z. (2022). Transcriptome analysis and characterization of genes associated to leaf tannin content in foxtail millet [Setaria italica (L.) P. Beauv.]. BMC Genom 23 (1), 512. doi:10.1186/s12864-022-08746-8
Li, L., and Jin, D. (2005). Main factors on interrelationship between phytophagous mites and parasite plants. Guizhou Agr. Sci. 33 (4), 95–97. doi:10.3969/j.issn.1001-3601.2005.04.045
Li, W., and Li, S. (2018). Advances in insect resistance of forest trees. J. Southwest For. Univ. 38 (5), 180–190.
Liang, C., Wang, Y., Zhu, Y., Tang, J., Hu, B., Liu, L., et al. (2014). OsNAP connects abscisic acid and leaf senescence by fine-tuning abscisic acid biosynthesis and directly targeting senescence-associated genes in rice. Proc. Natl. Acad. Sci. U. S. A. 111 (27), 10013–10018. doi:10.1073/pnas.1321568111
Liu, D., Cui, Y., Zhao, Z., Li, S., Liang, D., Wang, C., et al. (2021). Genome-wide identification and characterization of the BES/BZR gene family in wheat and foxtail millet. BMC Genom 22 (1), 682. doi:10.1186/s12864-021-08002-5
Liu, D., Cui, Y., Zhao, Z., Zhang, J., Li, S., and Liu, Z. (2022). Transcriptome analysis and mining of genes related to shade tolerance in foxtail millet (Setaria italica (L.) P. Beauv.). Roy. Soc. Open Sci. 9 (10), 220953. doi:10.1098/rsos.220953
Ludwig, M. (2016). Evolution of carbonic anhydrase in C4 plants. Curr. Opin. Plant Biol. 31, 16–22. doi:10.1016/j.pbi.2016.03.003
Luo, G., Shen, L., Song, Y., Yu, K., Ji, J., Zhang, C., et al. (2021). The MYB family transcription factor TuODORANT1 from Triticum urartu and the homolog TaODORANT1 from Triticum aestivum inhibit seed storage protein synthesis in wheat. Plant Biotechnol. J. 19 (9), 1863–1877. doi:10.1111/pbi.13604
Mhlontlo, S., Muchaonyerwa, P., and Mnkeni, P. (2007). Effects of sheep kraal manure on growth, dry matter yield and leaf nutrient composition of a local <i>Amaranthus</i> accession in the central region of the Eastern Cape Province, South Africa. Water sa. 33 (3), 363–368. doi:10.4314/wsa.v33i3.180597
Mokhele, B., Zhan, X., Yang, G., and Zhang, X. (2012). Review: Nitrogen assimilation in crop plants and its affecting factors. Can. J. Plant Sci. 92 (3), 399–405. doi:10.4141/cjps2011-135
Nandi, D., Tahiliani, P., Kumar, A., and Chandu, D. (2006). The ubiquitin-proteasome system. J. Biosci. 31 (1), 137–155. doi:10.1007/BF02705243
Park, B. S., Eo, H. J., Jang, I. C., Kang, H. G., Song, J. T., and Seo, H. S. (2010). Ubiquitination of LHY by SINAT5 regulates flowering time and is inhibited by DET1. Biochem. Biophys. Res. Commun. 398 (2), 242–246. doi:10.1016/j.bbrc.2010.06.067
Polekhina, G., House, C. M., Traficante, N., Mackay, J. P., Relaix, F., Sassoon, D. A., et al. (2002). Siah ubiquitin ligase is structurally related to TRAF and modulates TNF-alpha signaling. Nat. Struct. Biol. 9 (1), 68–75. doi:10.1038/nsb743
Rizwan, M., Abro, S., Asif, M. U., Hameed, A., Mahboob, W., Deho, Z. A., et al. (2021). Evaluation of cotton germplasm for morphological and biochemical host plant resistance traits against sucking insect pests complex. J. Cotton Res. 4 (1), 18–8. doi:10.1186/s42397-021-00093-5
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2009). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26 (1), 139–140. doi:10.1093/bioinformatics/btp616
Shinano, T., Nakajima, K., Wasaki, J., Mori, H., Zheng, T., and Osaki, M. (2006). Developmental regulation of photosynthate distribution in leaves of rice. Photosynthetica 44 (1), 1–10. doi:10.1007/s11099-005-0151-6
Sonalkar, V. U. (2020). Biochemicals in cotton hybrids and varieties and their correlation with sucking insect pests. Inte J. Curr. Microbiol. Appl. Sci. 9 (1), 1172–1183. doi:10.20546/ijcmas.2020.901.132
Tanaka, K., and Kotobuki, K. (1992). Studies on peeling characteristics of Japanese chestnut (Castanea crenata Sieb. et Zucc), Chinese chestnut (C. mollissima BL.) and their hybrids. Acta Hortic. 317, 175–180. doi:10.17660/actahortic.1992.317.20
Temple, S. J., Vance, C. P., and Stephen Gantt, J. (1998). Glutamate synthase and nitrogen assimilation. Trends Plant Sci. 3 (2), 51–56. doi:10.1016/s1360-1385(97)01159-x
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., et al. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28 (5), 511–515. doi:10.1038/nbt.1621
Uauy, C., Distelfeld, A., Fahima, T., Blechl, A., and Dubcovsky, J. (2006). A NAC gene regulating senescence improves grain protein, zinc, and iron content in wheat. Science 314, 1298–1301. doi:10.1126/science.1133649
Wang, Y., Wang, Y., and Xu, J. (1989). Seasonal occurrence and control of Spruce spider mite, Oligonychus ununguis (Jacobi) on chestnut trees. Acta Agric. boreali-sinica. 4 (4), 98–104. doi:10.3321/j.issn:1000-7091.1989.04.019
Wang, B., Jiang, Y., Zhang, F., and Ren, W. (1991). Two pestcides for prevention of spruce spider mite on Chinese chestnut. For. Pest Dis. (4), 52.
Wang, X., Wen, X., and Lu, C. (2010). Research advance in controlling insect pests on Castanea mollissima in China. J. Hebei Normal Univ. Science&Technology 24 (1), 39–43. doi:10.3969/j.issn.1672-7983.2010.01.011
Wang, J., Chen, Z., Zhang, Q., Meng, S., and Wei, C. (2020). The NAC transcription factors OsNAC20 and OsNAC26 regulate starch and storage protein synthesis. Plant Physiol. 184 (4), 1775–1791. doi:10.1104/pp.20.00984
War, A. R., Paulraj, M. G., Ahmad, T., Buhroo, A. A., Hussain, B., Ignacimuthu, S., et al. (2012). Mechanisms of plant defense against insect herbivores. Plant Signal Behav. 7 (10), 1306–1320. doi:10.4161/psb.21663
Wei, W., Qin, W., Wei, M., Lu, L., Tang, X., He, H., et al. (2009). Resistances of different cassava varieties to Tetranychus urticae. J. South. Agric. 40 (5), 504–506. doi:10.3969/j.issn.2095-1191.2009.05.013
Wei, S., Li, X., Lu, Z., Zhang, H., Ye, X., Zhou, Y., et al. (2022). A transcriptional regulator that boosts grain yields and shortens the growth duration of rice. Science 377 (6604), eabi8455. doi:10.1126/science.abi8455
Wu, Y., Liu, Q., Liu, H., and Wu, W. (1995). Induction of nitric oxide synthase and motoneuron death in newborn and early postnatal rats following spinal root avulsion. Acta Gossypii Sin. 7 (2), 109–112. doi:10.1016/0304-3940(95)11741-e
Wu, C. J., Conze, D. B., Li, X., Ying, S. X., Hanover, J. A., and Ashwell, J. D. (2005). TNF-alpha induced c-IAP1/TRAF2 complex translocation to a Ubc6-containing compartment and TRAF2 ubiquitination. EMBO J. 24 (10), 1886–1898. doi:10.1038/sj.emboj.7600649
Xie, Q., Guo, H-S., Dallman, G., Fang, S., Weissman, A. M., and Chua, N-H. (2002). SINAT5 promotes ubiquitin-related degradation of NAC1 to attenuate auxin signals. Nature 419 (6903), 167–170. doi:10.1038/nature00998
Xie, C., Mao, X., Huang, J., Ding, Y., Wu, J., Dong, S., et al. (2011). Kobas 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 39, W316–W322. doi:10.1093/nar/gkr483
Yang, N., Wu, S., Shen, L., Zhang, S., and Yang, B. (2014). A review on plant resistance to insect pests. Chin. J. Trop. Agric. 34 (9), 61–68+89. doi:10.3969/j.issn.1009-2196.2014.09.014
Yu, G., Li, F., Qin, Y., Bo, X., Wu, Y., and Wang, S. (2010). GOSemSim: an R package for measuring semantic similarity among GO terms and gene products. Bioinformatics 26 (7), 976–978. doi:10.1093/bioinformatics/btq064
Yu, X. Z., Lei, S. Y., Lin, Y. J., and Zhang, Q. (2019). Interaction of cyanate uptake by rice seedlings with nitrate assimilation: Gene expression analysis. Environ. Sci. Pollut. Res. Int. 26 (20), 20208–20218. doi:10.1007/s11356-019-05407-4
Zhang, Z., Dong, J., Ji, C., Wu, Y., and Messing, J. (2019). NAC-type transcription factors regulate accumulation of starch and protein in maize seeds. Proc. Natl. Acad. Sci. U. S. A. 116 (23), 11223–11228. doi:10.1073/pnas.1904995116
Zhao, H., and Fan, R. (2019). Occurrence and control methods of red spiders of chestnut. Mod. Rural Sci. Technol. 9, 34. doi:10.3969/j.issn.1674-5329.2019.09.028
Keywords: foxtail millet, Chinese chestnut, spruce spider mite, crude protein content, transcriptome analysis
Citation: Cui Y, Liu D, Zhao Z, Zhang J, Li S and Liu Z (2023) Transcriptome analysis and identification of genes associated with leaf crude protein content in foxtail millet [Setaria italica (L.) P. Beauv.]. Front. Genet. 14:1122212. doi: 10.3389/fgene.2023.1122212
Received: 12 December 2022; Accepted: 06 January 2023;
Published: 20 January 2023.
Edited by:
Jinbo Li, Luoyang Normal University, ChinaReviewed by:
Pooja Choudhary, Jaypee Institute of Information Technology, IndiaXianghui Zhang, Jilin University, China
Copyright © 2023 Cui, Liu, Zhao, Zhang, Li and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Suying Li, bGlzdXlpbmc2NUAxMjYuY29t; Zhengli Liu, bGl1emhlbmdsaTY1QDEyNi5jb20=
†These authors have contributed equally to this work