 Jiayang Wu1†
Jiayang Wu1† Chengdi Wang
Chengdi Wang Weimin Li
Weimin Li
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet. , 18 April 2023
Sec. Stem Cell Research
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1120815
This article is part of the Research Topic Genetic and Epigenetic Regulation of Stem Cells by the Immune System in Homeostasis, Regeneration, and Oncogenesis View all 6 articles
Epigenetics plays an important role in regulating stem cell signaling, as well as in the oncogenesis of lung cancer and therapeutic resistance. Determining how to employ these regulatory mechanisms to treat cancer is an intriguing medical challenge. Lung cancer is caused by signals that cause aberrant differentiation of stem cells or progenitor cells. The different pathological subtypes of lung cancer are determined by the cells of origin. Additionally, emerging studies have demonstrated that the occurrence of cancer treatment resistance is connected to the hijacking of normal stem cell capability by lung cancer stem cells, especially in the processes of drug transport, DNA damage repair, and niche protection. In this review, we summarize the principles of the epigenetic regulation of stem cell signaling in relation to the emergence of lung cancer and resistance to therapy. Furthermore, several investigations have shown that the tumor immune microenvironment in lung cancer affects these regulatory pathways. And ongoing experiments on epigenetics-related therapeutic strategies provide new insight for the treatment of lung cancer in the future.
Lung cancer is the most prevalent and deadly cancer in the world. Surgery, chemotherapy, radiation therapy, targeted therapy, and immunotherapy, and targeted therapy are the primary treatments for lung cancer (Hirsch et al., 2017). Despite some success with these treatments in the clinic, a significant number of patients with lung cancer still can not benefit from these therapies.
Cancer is caused by the proliferation and differentiation of aberrant cells (Hanahan and Weinberg, 2000). Therefore, the signals that could drive cell proliferation and differentiation during cancer development and progression are of particular importance in the study of cancer.
The fundamental conditions for the formation of cancer are provided by stem cells, which are somatic cells with the capacity for self-renewal and differentiation (Soteriou and Fuchs, 2018). Cells that keep stemness characteristics even after lung carcinogenesis are known as cancer stem cells (CSCs) (Batlle and Clevers, 2017). These cells play a significant role in the spread of cancer and resistance to therapy (Yang et al., 2020). Stem cell differentiation is regulated by driving differentiation signals called stem cell signals (Huang et al., 2020). It follows that it is easy to understand the significant regulatory role of this driver signal in the onset and progression of cancer.
The entire tumor system is composed of the tumor microenvironment (TME) and cancer cells (Zhang et al., 2022). The tumor microenvironment contains a significant number of immune cells, cancer stem cells respond differently to signals as prompted by their unique microenvironment (Mao et al., 2021). Therefore, a new approach for treating lung cancer in the future may depend on the understanding of these regulatory processes. Here, we focused on how epigenetic inheritance modulates these stem cell signals in the immune microenvironment, leading to lung cancer development and treatment resistance.
Subpopulations of cells with undifferentiated or hypodifferentiated characteristics are likely to become cancerous. Both stem and progenitor cells could be employed as the original cells for lung cancer when risk factors for lung cancer are present (Sanchez-Danes and Blanpain, 2018). Cells that initiate cancer vary depending on the pathology. Lung cancer is usually divided into small-cell lung cancer (SCLC) and non-small cell lung cancer, based on the histology (Thai et al., 2021). Approximately 85% of cases are non-small cell carcinomas, the prevalent subtypes being adenocarcinomas (LUAD), and squamous cell carcinomas (LUSC). LUAD typically develops from peripherally arising bronchioles, bronchioles, and alveoli, whereas LUSC and SCLC frequently develop from the centrally originating proximal segment bronchus to the main bronchus (Herbst et al., 2008; Ernani et al., 2022) (Figure 1).
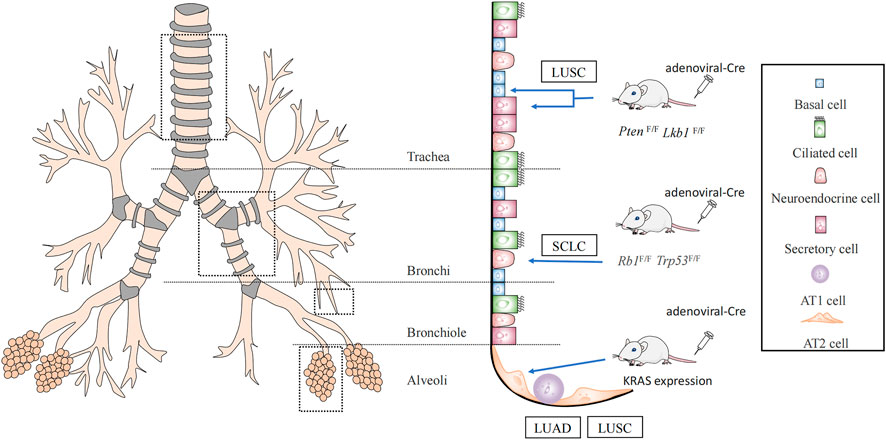
FIGURE 1. Lung cancer cells of origin. It was demonstrated by Cre mice that lung squamous cancer (LUSC) originates from basal cells with secretory cells, AT2 cells, while AT2 cells may also evolve into lung adenocarcinoma (LUAD). And small-cell lung cancer (SCLC) originates from neuroendocrine cells.
Using a Cre mouse model, it was discovered that non-squamous epithelial basal, secretory, and alveolar epithelial type Ⅱ cell (AT2) cells are the origin of LUSC (Ferone et al., 2016). One of the most frequent genetic changes in lung LUSC is SOX2. Deletion of SOX2 in human lung epithelial cells has been reported to reduce their ability to self-renew and repair tissue injury (Bass et al., 2009). Therefore, these key genes are crucial for preserving the phylogeny and stability of mature airways. SOX2 overexpression and deletion of the tumor suppressor genes, PTEN, CDKN2A, and/or CDKN2B resulted in the conversion of lung basal cells, secretory cells, and AT2 cells into lung squamous cells (Ferone et al., 2016; Sanchez-Danes and Blanpain, 2018). The dominant negative of the Maml1 mutation in secretory and AT2 cells, accompanied by KrasG12D expression and NOTCH inhibition, also causes SOX2 expression and alveolar proliferative lesions containing squamous markers (Xu et al., 2014). In addition, the activation of PIK3CA, P63, and deletion of LKB1 are mutations associated with squamous cell carcinoma (McCaughan et al., 2010; Mukhopadhyay et al., 2014) (Figure 1).
It has been demonstrated that AT2 cells serve as the main origin of LUAD, by using a KRAS-driven mouse model of lung cancer (Xu et al., 2012; Mainardi et al., 2014). The oncogenes, EGFR, KRAS, BRAF, and PIK3CA are frequently mutated in LUAD (Swanton and Govindan, 2016). LUAD can be triggered directly by independent KrasG12D expression. A limited number of LUAD could develop when SOX2 expression in combination with deletion of Trp53 or dual deletion of Trp53 and Rb1 in a mouse model that was constructed by the lentiviral approach. In mouse models constructed using a lentiviral approach, SOX2 expression and deletion of Trp53 or dual deletion of Trp53 and Rb1 could lead to a small number of mouses with LUAD (Sanchez-Danes and Blanpain, 2018).
Inactivating mutations in the tumor suppressor genes TP53 and RB transcriptional corepressor 1 (RB1) are common in SCLC (Korsen et al., 2022). By conditional knockout of TP53 and RB1, it was confirmed that SCLC originates mainly from neuroendocrine cells (Sutherland et al., 2011) (Figure 1). These studies suggest that the cells of origin allow for specific types of lung carcinogenesis by selecting the expression of particular genes.
In addition to lung carcinogenesis being directly guided by genetic alterations, epigenetics could also control lung carcinogenesis by controlling the ability to initiate tumors. The patterns of epigenetic changes during cellular senescence and tumorigenesis are similar, with the major abnormalities being DNA and histone methylation (Xie et al., 2018; Yu et al., 2018). However, cellular senescence and tumorigenesis involve different pathways. When CPG islands are hypermethylated, they mostly affect genes that control cell synthesis and metabolism in senescent cells, and genes that control the growth and differentiation of malignant cells (Xie et al., 2018). According to this phenomenon, the epigenetic landscape is crucial because of its regulatory role in stem cell differentiation (Teschendorff and Feinberg, 2021).
Mutations in chromatin-modifying enzymes that lead to methylation modifications in DNA and histones are common in lung cancer. For example, lung cancer stem cells carry loss-of-function mutations in nuclear receptor-binding SET domain protein 1 (NSD1), which is capable of methylation-modifying histones (Garraway and Lander, 2013; Cancer Genome Atlas Research Network, 2014).
DNA methylation, histone modifications, and non-coding RNA alterations could activate multiple stem cell signals, including WNT, NOTCH, and Hedgehog, to regulate lung cancer oncogenesis and progression (Lytle et al., 2018).
This review describes how epigenetic mechanisms regulate the activation and expression of stem cell signaling pathways leading to lung carcinogenesis, mostly from two time periods: before and after gene transcription.
DNA methylation was one of the first epigenetic modifications to be identified even before the establishment of the double-helix structure of DNA. DNA methylation is the process by which DNA methyltransferase (DNMT) adds a methyl group to the cytosine 5th carbon atom in the DNA sequence to create 5-methylcytosine (5 mC) (Liu et al., 2022a). Most of these alterations occur in CpG islands (CGIs). The three methyltransferases linked to methylation in humans are DNMT1, DNMT3A, and DNMT3B. After DNA replication is complete, DNMT1 methylates hemimethylated DNA during the cell division cycle, whereas DNMT3A and DNMT3B are responsible for establishing fresh DNA methylation (He et al., 2011).
The downregulation of P53 and P21 expression through the IL-6/JAK2/STAT pathway could upregulate DNMT1, enhance tumor initiation, and promote lung cancer stem cell proliferation (Liu et al., 2015). This demonstrates that DNA hypermethylation is associated with the silencing of oncogenes and differentiation genes in lung cancer, and that the low expression of these genes may contribute to the formation of lung cancer stem cells.
Activation of the WNT signaling pathway has been shown to be associated with increased tumor initiation potential in mouse models (Nusse and Clevers, 2017). APC, LKB1, WNT inhibitor 1 (WNT-1), Disabled-2 (Dab2), secreted frizzled-related proteins (sFRPs), and members of the Dickkopf (Dkk) family are hypermethylated silencing factors that are involved in the abnormal activations of the Wnt/β-Catenin pathway, and these aberrant activations may lead to the development of LUAD (Duruisseaux and Esteller, 2018). Upregulated G9a may silence the expression of WNT-1 through DNA hypermethylation, leading to an abnormal WNT pathway, thus affecting the growth of lung cancer cells (Zhang et al., 2018). DKK1 is a secretory protein that negatively regulates WNT signaling. The DNA hypermethylation of the DKK1 promoter promotes lung cancer growth through the WNT signaling pathway (Park et al., 2012; Han et al., 2017).
NOTCH signaling is a highly conserved intercellular communication pathway that performs multiple functions during lung development (Siebel and Lendahl, 2017), including the regulation of cell differentiation, survival, and genealogical specification. Although normal NOTCH signaling is required to maintain homeostasis in vivo, its abnormal activity has been shown to be associated with the development and progression of lung cancer (Allen et al., 2011; Lim et al., 2017). ASCL1 hypomethylation is common in SCLC. As a direct target of ASCL1, DLL3 (a NOTCH inhibitor) was significantly correlated with its expression status (Sabari et al., 2017).
The Hedgehog (Hh) pathway is an evolutionarily conserved signaling axis that is essential for regulating a variety of fundamental biological processes. The Hh ligands, repair receptor (PTCH), smooth intermediator (SMO), and zinc finger-containing glioblastoma transcription factor (GLI) are the four main elements of the Hh pathway. In the absence of PTCH inhibition, SMO activates the Gli transcription factor, thereby activating cancer-related target genes (Kasiri et al., 2017). The human contains Gli1, Gli2, and Gli3 proteins. Gli2 and Gli3 primarily function as Shh-regulated transcriptional activators and repressors, respectively, whereas Gli1 primarily functions as a transcriptional activator and amplifies Shh signals in a positive feedback loop (Fu et al., 2016). A regulatory pathway consisting of the ERK/PIK3/Hedgehog signaling pathway is affected by aberrant DNA methylation of 256 negatively associated genes. This signaling pathway regulates cell death and survival, and is implicated in squamous cell tumorigenesis (Shi et al., 2017). Set7-mediated methylation of Gli3 at the K436 and K595 sites increases the stability of Gli3 and its ability to bind to DNA, thereby activating Shh signaling and contributing to the development of non-small cell lung cancer (Fu et al., 2016).
Histones could be modified in a diverse range of ways, including acetylation and methylation. The two major roles of covalent histone modifications are to silence the expression of specific genes and promote transcription. Specific histone lysine methyltransferases could methylate K4, K9, K27, K36, and K7 sites in histone H3 and K20 sites in histone H4 respectively (Wang et al., 2018b). While methylation of H3K9, H3K27, and H4K20 inhibits gene transcription, methylation of H3K4, H3K79, and H3K36 increases it (Guo et al., 2019). Histone acetylation leads to a decrease in the affinity between histones and DNA, thereby facilitating transcription. In contrast, histone deacetylation removes acetyl groups and inhibits transcription through HDACs (Montgomery and Srinivasan, 2019). The methylation of H3 histones, such as H3K79me2, was found to decrease the expression of several WNT repressors to increase WNT signaling in research on the epigenetic modification of H3 histones by triptolide in NSCLC (Liang et al., 2019). In addition to changing the level of H3K4 methylation and regulating the NOTCH pathway, KDM5A, a demethylase of H3K4, also exhibits reciprocal epistasis with NOTCH 2 in ASCL1 and neuroendocrine differentiation. This further demonstrates the significance of H3K4 methylation in SCLC formation (Oser et al., 2019). HDACs could affect lung carcinogenesis and progression in the Hh pathway by changing the acetylation status of histones in the promoter region. HDAC could interact with GLI1, causing SOX2 promoter activity to be expressed (Wei et al., 2021) (Table 1).
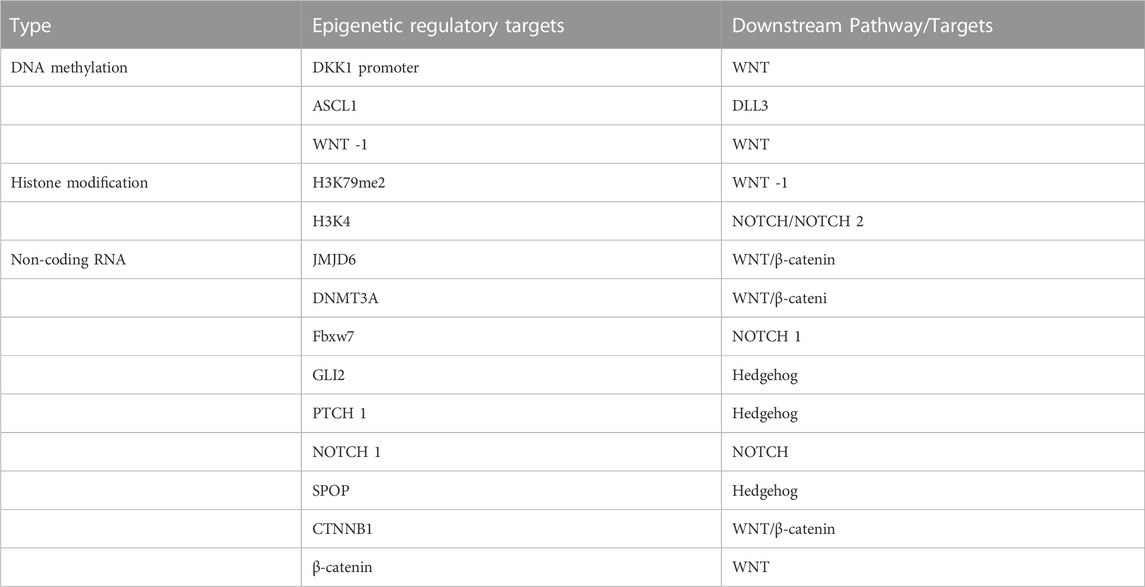
TABLE 1. Epigenetic targets acting on lung carcinogenesis and their downstream stem cell-related signaling pathways or targets.
In addition, pre-transcriptional DNA methylation with histone modifications may increase the sensitivity of cells to transformation. Therefore, understanding the mechanisms of the pre-transcriptional epigenetic regulation of stem cells is an important strategy for controlling the development of lung cancer.
Non-coding RNAs play an essential role in the pathology of cancer. The role of long non-coding RNAs (LncRNAs) is currently poorly understood, while microRNAs (miRNAs) are imbalance regulated non-coding RNA isoforms that have received the most research attention in lung cancer.
MiR-708-5p directly inhibits the translation of DNMT3A, leading to hypomethylation in A549 and Calu-3 cells and an increase in the expression of the tumor suppressor CDH1. This reduces the activity of the Wnt/β-catenin signaling pathway and affects the development of NSCLC by altering stem cell characteristics (Liu et al., 2018). miR-770 activates the WNT/β-catenin pathway by directly binding to the 3′-UTR of JMJD6 mRNA and downregulating JMJD6 expression which, leading to non-small cell carcinogenesis (Zhang et al., 2017). miR-27a plays an oncogenic role in human lung tumorigenesis. Fbxw7, which is inhibited by the overexpression of miR-27a, could regulate cell cycle progression, including c-Myc, c-Jun, cyclin E1, and NOTCH 1 (Wang et al., 2011a). Lung cancer considerably expresses the Hh pathway in comparison to nearby normal tissues. The miR-182-5p/GLI2 axis controls lung adenocarcinogenesis by influencing the Hh pathway (Seidl et al., 2020). miR-212 causes lung carcinogenesis by directly targeting the Hh pathway receptor, PTCH1, resulting in the inhibition of PTCH1 (Li et al., 2012). The expression of miR-520b was significantly upregulated in NSCLC samples compared with normal samples. Additionally, miR-520b was found to promote NSCLC tumorigenesis through the SPOP-GLI2/3 axis (Liu et al., 2019).
The Long non-coding RNA small nucleolar RNA host gene 11 (SNHG11) could promote lung carcinogenesis through two distinct WNT pathways. The first pathway activates the WNT/β-catenin pathway via the SNHG11/miR-4436a/CTNNB1 ceRNA axis. In the second pathway, SNHG11 directly binds to β-catenin and activates the WNT pathway (Liu et al., 2020). A novel long intergenic non-protein coding RNA (LINC01783) that suppresses miR-432-5p, a route that results in non-small cell carcinogenesis, activates the NOTCH pathway to increase DLL-1 expression and enhances the proliferation of NSCLC cells (Deng et al., 2021) (Table 1).
The gain and loss of epigenetic modifications at all stages of transcription may lead to the development of lung cancer. Lung cancer stem cells are known to be preferentially affected by this process. Therefore, the regulation of lung cancer stem cells using these modification pathways as targets would be very effective therapeutic strategy for lung cancer.
Therapy resistance is a challenge in the clinical treatment of lung cancer. Existing treatment regimens typically fail to eliminate all tumor cells, and residual cells are believed to be the key driver of cancer recurrence in patients (Lytle et al., 2018).
Lung cancer stem cells participate in drug resistance by hijacking the properties of normal stem cells. The three most common pathways are drug transport, DNA damage repair, and niche protection (Hovinga et al., 2010; Blanpain et al., 2011; Assaraf et al., 2019).
ATP-binding cassette (ABC) transport proteins are the main proteins that regulate the efflux of cytotoxic drugs (Greenwood et al., 2019), including ABCB1 and ABCG2. Most ABC transport proteins directly contribute to the development of drug resistance, and the enhanced efflux activity of these proteins positively regulates drug resistance (Wang et al., 2018a; Wu et al., 2019). Multidrug resistance (MDR) caused by lung cancer stem cells correlates with the expression of ABCB1 and ABCG2 (Bhukhai et al., 2018; Mohammad et al., 2018; Zhang et al., 2020; Cortes-Dericks and Galetta, 2022). Docetaxel is widely used as a third-generation chemotherapeutic agent for the treatment of patients with NSCLC. However, most patients exhibit drug resistance after a period of treatment. A study of the NSCLC docetaxel-resistant cell lines HCC827-DR found that this resistant cell lines exhibited CSC-like markers and high expression of ABCB1 in all cells (Chen et al., 2017). By pumping chemotherapeutic medications out of cells and lowering the concentration of intracellular pharmaceuticals, ABCG2 significantly increased the chemotherapy resistance of lung cancer stem cells (Huang and Fu, 2015) (Figure 2).
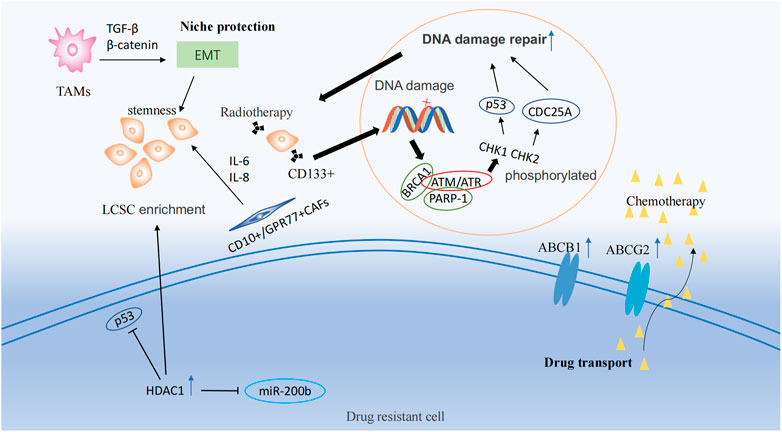
FIGURE 2. Drug resistance mechanisms and epigenetic regulation involving tumor stem cells. Drug resistance mechanisms include drug transport, DNA damage repair, and ecological niche protection processes. LSCS: lung cancer stem cell; TAMs: Tumor-associated macrophages; EMT: Epithelial-Mesenchymal Transition; CAFs: cancer associated fibroblasts.
CSCs have a higher capacity to repair DNA damage than other types of tumor cells (Masoumi et al., 2021). Ataxia telangiectasia mutated Protein (ATM) and ataxia telangiectasia and Rad3-related (ATR) are members of the phosphatidylinositol 3-kinase (PI3K)-related kinase (PIKK) protein family, and are frequently activated as checkpoint sensors during cancer treatment therapy. When DNA is damaged by chemoradiotherapy, ATM and ATR kinases form complexes with PARP-1 and BRCA1, phosphorylating CHK1 and CHK2 and then promoting the activation of target proteins, including p53 and CDC25A, temporarily halting the cell cycle, thus repairing most DNA damaged by chemoradiotherapy or directly inducing apoptosis (Zhou et al., 2021). In NSCLC, CD133+ lung cancer stem cells were observed to increase the expression of DNA damage response and repair in genes (Desai et al., 2014). Meanwhile, a study demonstrated that lung cancer stem cells could lead to therapy resistance through enhanced DNA repair and reduced uptake of cisplatin (Yu et al., 2017) (Figure 2).
The niche, also known as the tumor microenvironment (TME), is composed of multiple cell types, including tumor cells, endothelial cells, mesenchymal cells, immune cells, and fibroblasts, as well as non-cellular components such as the extracellular matrix (ECM). This niche helps to maintain the stem-like properties of lung cancer stem cells, leading to the development of therapeutic resistance (Plaks et al., 2015; Chiu et al., 2022). Tumor-associated macrophages (TAMs) could induce epithelial-mesenchymal transition (EMT) of cancer cells in NSCLC by activating TGF-β signaling and the β-catenin pathway (Zeng et al., 2020). EMT plays a significant role in cancer progression and is associated with the production of cancer stem-like cells (Shibue and Weinberg, 2017). Additionally, the use of EMT markers is a common approach for predicting drug resistance in cancer (Huang et al., 2020). TAMs and cancer-associated fibroblasts (CAFs) from primary NSCLC help generate lung cancer stem cells and maintain their stemness (Chen et al., 2014b). CD10+/GPR77+ CAFs could promote drug resistance in patients with lung cancer by secreting IL-6 and IL-8 to maintain the stemness of cancer stem cells (Su et al., 2018) (Figure 2).
Lung cancer stem cells lead to drug resistance by hijacking the properties of normal stem cells. Therefore, reducing the enrichment of lung cancer stem cells through epigenetic modulation is one way to improve therapeutic efficacy.
Histone deacetylase 1 (HDAC1) is highly expressed in cisplatin-resistant lung cancer cells (Wang et al., 2016). HDAC1, in combination with tribbles pseudokinase 1 (TRIB1), in cisplatin treatment, reduced the activity of oncogene p53 through its deacetylation and induced the enrichment of lung cancer stem cells. In contrast, the silencing of HDAC1 resulted in reduced expression of the transcription factors, Nanog and Oct4, in lung cancer stem cells and increased sensitivity to cisplatin treatment (Wang et al., 2017) (Figure 2). In addition, HDAC1 maintains lung cancer cell stemness and induces a drug-resistant phenotype in lung cancer cells by inhibiting miR-200b expression and reducing the targeting of miR-200b to Suz12 (Chen et al., 2014a). HDAC11 has an effect similar to that of HDAC1, which is highly expressed in the cancer stem cell population of LUAD, resulting in enhanced self-renewal of lung cancer stem cells and interaction with GLI1 to upregulate SOX2 expression (Zuo et al., 2020). MARCKSL1-2 is a long non-coding RNA that recruits Suz12 to the promoter of histone deacetylase 1 (HDAC1), increasing the level of H3K27me3 at the HDAC1 promoter, while decreasing HDAC1 expression. Thus, miR-200b expression is upregulated to reverse drug resistance (Jiang et al., 2022).
Although there are many epigenetic targets that have shown advantages in the treatment of lung cancer stem cells, the molecular mechanisms of the upstream and downstream regulation of most targets remain unclear. Additionally, the types of drug resistance that could be improved by these targets are limited.
The tumor microenvironment, especially the tumor immune microenvironment (TIME), is not only related to the resistance of lung cancer treatment, but also influences the whole process of lung cancer development (Ferguson et al., 2021).
The location, type, density, and functional status of immune cells (T cells, B cells, NK cells, DC cells, macrophages, neutrophils, monocytes, and mast cells) within the tumor immune microenvironment contribute to its diversity (Binnewies et al., 2018). Using single-cell technology, significant differences in the immune microenvironments of LUAD and LUSC have been confirmed (Wang et al., 2022). The occurrence, growth, and treatment of tumors are significantly affected by this diversity. Therefore, many studies have focused on the immune landscape of the tumor immune microenvironment. Patients who received neoadjuvant chemotherapy had higher levels of PD-L1 expression and T cell subsets regulation than those who did not receive neoadjuvant chemotherapy, according to a study based on the effects of multiple immunofluorescence and image analysis methods on the immune microenvironment of NSCLC (Parra et al., 2018; Chiu et al., 2022). An analysis of the number, density, and ratio of 26 kinds of immune cells in the tumor immunological microenvironment of 681 NSCLC cases revealed that patients with immunodeficient tumors had shorter disease-free survival and that their tumors had a high number of LCSC and macrophages (Peng et al., 2021). The overall proportion and characteristics of T cells within the TIME are major factors that determine the direction of tumor progression (Mohammad et al., 2018). T cell exhaustion occurs immediately after oncogenic initiation, and some irreversible T cell exhaustion is responsible for the insensitivity of patients to anti-PD-1/ PD-L1 therapy (Pauken et al., 2016; Guo et al., 2018). In the process of T cell exhaustion, inhibitory receptors such as CTLA-4, TIM-3, LAG-3, and PD-1 are usually over-expressed on T cells, and effector cytokines such as IFN-γ are down-regulated (Blank et al., 2019).
Recent studies have shown that tumor immune microenvironment could be reshaped by epigenetic immune editing (Gangoso et al., 2021). Epigenetic changes could be triggered by inflammation (Karin and Shalapour, 2022). The hypoxia-adapted cellular phenotype is sustained in the tumor microenvironment by the synergistic effect of epigenetic factors and hypoxia-inducible transcription factors (HIF). Extensive DNA methylation and histone modifications occur in the hypoxic TME, promoting tumor growth, increasing invasiveness, and maintaining cancer cell stemness (Wang et al., 2011b; Hu et al., 2021) (Figure 3).
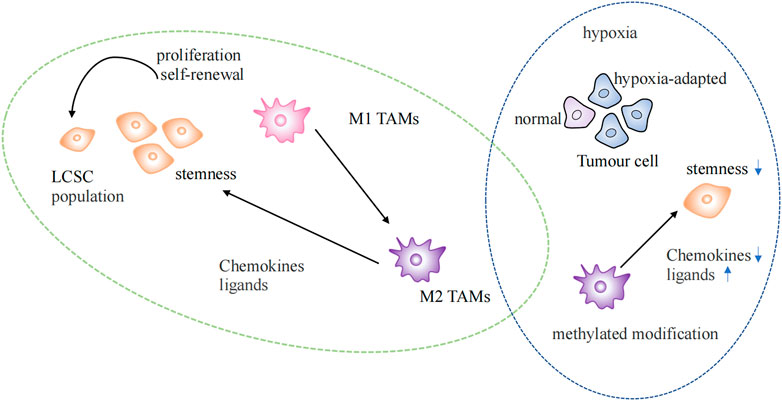
FIGURE 3. Epigenetic regulation in the tumor microenvironment. Regulation of lung cancer stem cells by TAMs of the M2 phenotype in normal and hypoxic environments. LSCS: lung cancer stem cell; TAMs: Tumor-associated macrophages.
TAMs are currently the most widely studied immunosuppressive cell at the TIME (Mohammad et al., 2018). TAMs gather at the injury site after identifying signals, such as chemokines, cytokines, inflammatory mediators, pathogens, or damage-associated molecular patterns (DAMP), which trigger the inflammatory response. M1 and M2 phenotypes of TAMs exist. The M1 phenotype is characterized by its anticancer activity and typically activated macrophages. After epigenetic reprogramming, M2 phenotype macrophages are formed by differentiation and polarization, which have the potential to promote tumors (Netea et al., 2016). M2 phenotypic TAMs maintain tumor stem cell populations by secreting chemokines and ligands that activate stem cell pathways (Huang et al., 2020) (Figure 3). Enhanced methylation modifications and diminished chemokine expression within TAMs in hypoxic environments alter the immune landscape within the TME (Tausendschon et al., 2011). LARRPM (lncRNA) reduces TET1 binding to the CSF1 promoter in LUAD, resulting in increased DNA methylation of the CSF1 promoter and the inhibition of CSF1 transcription, thereby reducing macrophage M2 polarization and infiltration. At the same time, discovered that negative regulation of TAM contributes to carcinogenesis (Li et al., 2022). The differentiation of T helper (TH) cells are more stable under epigenetic regulation, and the direction of T helper cell differentiation is determined by different histone modification levels at IFNG sites (Karin and Shalapour, 2022). NEAT1 was found highly expressed in lung cancer and interacts with DNA methyltransferase DNMT1 to regulate cytotoxic T cell infiltration in lung cancer by inhibiting the cGAS/STING pathway (Ma et al., 2020). The proliferation, differentiation, and survival of T cells depend on the activity of EZH2 enhancers, which are important epigenetic regulators of gene expression. Notably, GSK126, an EZH2 inhibitor, could alter the TIME, encourage the synthesis of the Th1 chemokines CXCL9 and CXCL10 in tumors, and boost CD8+ T cell infiltration (Huang et al., 2019). The presence of tumor-infiltrating B lymphocytes could be observed at all stages of lung cancer development, and it has been found that histone modification could also increase the infiltration of B cells (Wang et al., 2019; Karin and Shalapour, 2022). The epigenetic silencing of NKG2DL in SCLC results in a lack of stimulatory signals to activate NK cells, thereby increasing the aggressiveness and metastasis of SCLC (Zhu et al., 2021).
These studies illustrate that the tumor microenvironment plays an essential role in the progression of lung cancer. In particular, the status of lung cancer stem cells, which is influenced by epigenetic alterations in the tumor microenvironment, is an important cause of treatment resistance and cancer recurrence.
Epigenetic-based therapeutic strategies aim to regulate the transcriptional programming of various signaling pathways in immune cells, and cancer cells, thereby affecting the fate of each of these cell populations (Dai et al., 2021) (Table 2). The major epigenetic targets associated with lung cancer treatment are DNA methyltransferase (DNMT), histone lysine methyltransferase (KMT), and histone lysine acetyltransferase. Epigenetic-based drugs are often used in combination with targeted therapies and chemotherapy to enhance their efficacy and reduce drug resistance.
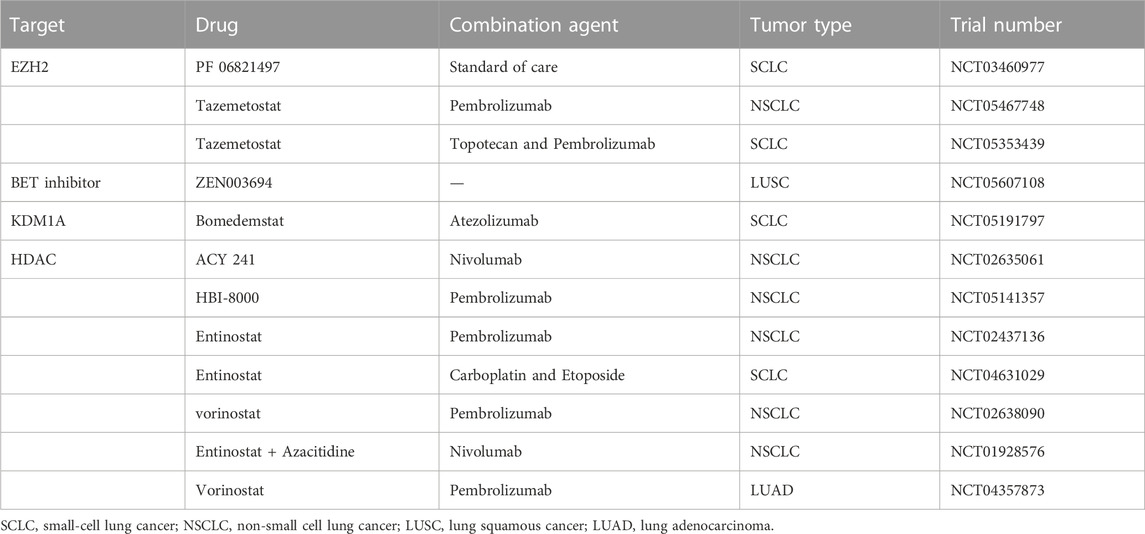
TABLE 2. Ongoing clinical trials of epigenetic drugs in lung cancer.
Some HDAC and DNMT inhibitors are currently clinically approved, such as the histone deacetylase inhibitors Vorinostat, Romidepsin, Belinostat, and DNA methylation inhibitor diecitabine. Additionally, multiple clinical trials are underway for EZH2 inhibitors, KDM1A inhibitors, and BET proteins (Table 2). Although these drugs currently show a partial advantage, in a study of the efficacy of histone methyltransferase G9a in lung cancer, Rowbotham found that G9a may increase the number of lung cancer stem cells and thus promote lung cancer progression (Rowbotham et al., 2018). In addition, Hypomethylation therapy may lead to demethylation and upregulation of oncogene expression (Liu et al., 2022b). Current studies cannot explain these phenomena; therefore, more in-depth studies on these regulatory mechanisms are warranted in the future.
As mentioned previously, the stem cell programs control the growth and therapy resistance of lung cancer through epigenetic inheritance. Numerous studies have also shown the significance of WNT, NOTCH, and other traditional stem cell pathways in the onset and progression of lung cancer. Epigenetic modification enzymes, such as DNA methylase, histone deacetylase, and their inhibitory enzymes, play a role in different stem cell pathways. The three main causes of resistance to lung cancer treatment are drug transport, DNA damage repair, and niche protection. ATP-binding cassette transporters greatly increase the chemotherapeutic resistance of lung cancer stem cells by pumping chemotherapeutic medications out of cells and lowering intracellular drug concentrations. ATM and ATR kinases associate with PARP-1 and BRCA1 to form complexes that phosphorylate CHK1 and CHK2, boosting the activation of target proteins and preserving the stemness of LCSC. Through the spatial distribution and composition of different cells, the niche maintains the stemness of lung cancer stem cells. Additionally, epigenetic regulation of the immune microenvironment could also affect the outcome of lung cancer. Although the epigenetic regulation in lung cancer stem cell-related drug resistance regulation is still insufficient compared to genetic regulation, existing research shows great potential.
As expected, the modulation of various cell fates through epigenetic modulators is an effective strategy for lung cancer treatment. However, this type of drug is a double-edged sword. It may also increase the number of lung cancer stem cells, making it difficult for the cancer to be completely eliminated. Therefore, the combination of epigenetic drugs with other drugs or other treatments may be the future direction in lung cancer treatment.
WL and CW conceived the project. JW and JF wrote the manuscript. QZ designed the chart. YH and CX reviewed and edited the manuscript. And all the authors have read and edited the manuscript.
This study was supported by the Science and Technology Project of Sichuan (grant 2022ZDZX0018, 2020YFG0473); Chinese Postdoctoral Science Foundation (2022T150451).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Allen, T. D., Rodriguez, E. M., Jones, K. D., and Bishop, J. M. (2011). Activated Notch1 induces lung adenomas in mice and cooperates with Myc in the generation of lung adenocarcinoma. Cancer Res. 71 (18), 6010–6018. doi:10.1158/0008-5472.CAN-11-0595
Assaraf, Y. G., Brozovic, A., Goncalves, A. C., Jurkovicova, D., Line, A., Machuqueiro, M., et al. (2019). The multi-factorial nature of clinical multidrug resistance in cancer. Drug Resist Updat 46, 100645. doi:10.1016/j.drup.2019.100645
Bass, A. J., Watanabe, H., Mermel, C. H., Yu, S., Perner, S., Verhaak, R. G., et al. (2009). SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat. Genet. 41 (11), 1238–1242. doi:10.1038/ng.465
Batlle, E., and Clevers, H. (2017). Cancer stem cells revisited. Nat. Med. 23 (10), 1124–1134. doi:10.1038/nm.4409
Bhukhai, K., de Dreuzy, E., Giorgi, M., Colomb, C., Negre, O., Denaro, M., et al. (2018). Ex vivo selection of transduced hematopoietic stem cells for gene therapy of β-Hemoglobinopathies. Mol. Ther. 26 (2), 480–495. doi:10.1016/j.ymthe.2017.10.015
Binnewies, M., Roberts, E. W., Kersten, K., Chan, V., Fearon, D. F., Merad, M., et al. (2018). Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 24 (5), 541–550. doi:10.1038/s41591-018-0014-x
Blank, C. U., Haining, W. N., Held, W., Hogan, P. G., Kallies, A., Lugli, E., et al. (2019). Defining 'T cell exhaustion'. Nat. Rev. Immunol. 19 (11), 665–674. doi:10.1038/s41577-019-0221-9
Blanpain, C., Mohrin, M., Sotiropoulou, P. A., and Passegue, E. (2011). DNA-damage response in tissue-specific and cancer stem cells. Cell. Stem Cell. 8 (1), 16–29. doi:10.1016/j.stem.2010.12.012
Cancer Genome Atlas Research Network (2014). Comprehensive molecular profiling of lung adenocarcinoma. Nature 511 (7511), 543–550. doi:10.1038/nature13385
Chen, D. Q., Huang, J. Y., Feng, B., Pan, B. Z., De, W., Wang, R., et al. (2014a). Histone deacetylase 1/Sp1/microRNA-200b signaling accounts for maintenance of cancer stem-like cells in human lung adenocarcinoma. PLoS One 9 (10), e109578. doi:10.1371/journal.pone.0109578
Chen, H., Shien, K., Suzawa, K., Tsukuda, K., Tomida, S., Sato, H., et al. (2017). Elacridar, a third-generation ABCB1 inhibitor, overcomes resistance to docetaxel in non-small cell lung cancer. Oncol. Lett. 14 (4), 4349–4354. doi:10.3892/ol.2017.6678
Chen, W. J., Ho, C. C., Chang, Y. L., Chen, H. Y., Lin, C. A., Ling, T. Y., et al. (2014b). Cancer-associated fibroblasts regulate the plasticity of lung cancer stemness via paracrine signalling. Nat. Commun. 5, 3472. doi:10.1038/ncomms4472
Chiu, K. J., Chiou, H. C., Huang, C. H., Lu, P. C., Kuo, H. R., Wang, J. W., et al. (2022). Natural compounds targeting cancer-associated fibroblasts against digestive system tumor progression: Therapeutic insights. Biomedicines 10 (3), 713. doi:10.3390/biomedicines10030713
Cortes-Dericks, L., and Galetta, D. (2022). Impact of cancer stem cells and cancer stem cell-driven drug resiliency in lung tumor: Options in sight. Cancers (Basel) 14 (2), 267. doi:10.3390/cancers14020267
Dai, E., Zhu, Z., Wahed, S., Qu, Z., Storkus, W. J., and Guo, Z. S. (2021). Epigenetic modulation of antitumor immunity for improved cancer immunotherapy. Mol. Cancer 20 (1), 171. doi:10.1186/s12943-021-01464-x
Deng, Y., Zhang, L., and Luo, R. (2021). LINC01783 facilitates cell proliferation, migration and invasion in non-small cell lung cancer by targeting miR-432-5p to activate the notch pathway. Cancer Cell. Int. 21 (1), 234. doi:10.1186/s12935-021-01912-0
Desai, A., Webb, B., and Gerson, S. L. (2014). CD133+ cells contribute to radioresistance via altered regulation of DNA repair genes in human lung cancer cells. Radiother. Oncol. 110 (3), 538–545. doi:10.1016/j.radonc.2013.10.040
Duruisseaux, M., and Esteller, M. (2018). Lung cancer epigenetics: From knowledge to applications. Seminars Cancer Biol. 51, 116–128. doi:10.1016/j.semcancer.2017.09.005
Ernani, V., Du, L., Ross, H. J., Yi, J. E., Wampfler, J. A., Schild, S. E., et al. (2022). Gastroesophageal reflux disease and paraneoplastic neurological syndrome associated with long-term survival in limited stage small-cell lung cancer. Thorac. Cancer 13 (7), 925–933. doi:10.1111/1759-7714.14318
Ferguson, L. P., Diaz, E., and Reya, T. (2021). The role of the microenvironment and immune system in regulating stem cell fate in cancer. Trends Cancer 7 (7), 624–634. doi:10.1016/j.trecan.2020.12.014
Ferone, G., Song, J. Y., Sutherland, K. D., Bhaskaran, R., Monkhorst, K., Lambooij, J. P., et al. (2016). SOX2 is the determining oncogenic switch in promoting lung squamous cell carcinoma from different cells of origin. Cancer Cell. 30 (4), 519–532. doi:10.1016/j.ccell.2016.09.001
Fu, L., Wu, H., Cheng, S. Y., Gao, D., Zhang, L., and Zhao, Y. (2016). Set7 mediated Gli3 methylation plays a positive role in the activation of sonic Hedgehog pathway in mammals. Elife 5, e15690. doi:10.7554/eLife.15690
Gangoso, E., Southgate, B., Bradley, L., Rus, S., Galvez-Cancino, F., McGivern, N., et al. (2021). Glioblastomas acquire myeloid-affiliated transcriptional programs via epigenetic immunoediting to elicit immune evasion. Cell. 184 (9), 2454–2470. e26. doi:10.1016/j.cell.2021.03.023
Garraway, L. A., and Lander, E. S. (2013). Lessons from the cancer genome. Cell. 153 (1), 17–37. doi:10.1016/j.cell.2013.03.002
Greenwood, H. E., McCormick, P. N., Gendron, T., Glaser, M., Pereira, R., Maddocks, O. D. K., et al. (2019). Measurement of tumor antioxidant capacity and prediction of chemotherapy resistance in preclinical models of ovarian cancer by positron emission tomography. Clin. Cancer Res. 25 (8), 2471–2482. doi:10.1158/1078-0432.CCR-18-3423
Guo, C., Dong, G., Liang, X., and Dong, Z. (2019). Epigenetic regulation in AKI and kidney repair: Mechanisms and therapeutic implications. Nat. Rev. Nephrol. 15 (4), 220–239. doi:10.1038/s41581-018-0103-6
Guo, X., Zhang, Y., Zheng, L., Zheng, C., Song, J., Zhang, Q., et al. (2018). Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 24 (7), 978–985. doi:10.1038/s41591-018-0045-3
Han, S., Yang, X., Pan, Y., Qi, Q., Shen, J., Fang, H., et al. (2017). L-securinine inhibits the proliferation of A549 lung cancer cells and promotes DKK1 promoter methylation. Oncol. Lett. 14 (4), 4243–4248. doi:10.3892/ol.2017.6693
Hanahan, D., and Weinberg, R. A. (2000). The hallmarks of Cancer. Cell. 100 (1), 57–70. doi:10.1016/s0092-8674(00)81683-9
He, X. J., Chen, T., and Zhu, J. K. (2011). Regulation and function of DNA methylation in plants and animals. Cell. Res. 21 (3), 442–465. doi:10.1038/cr.2011.23
Herbst, R. S., Heymach, J. V., and Lippman, S. M. (2008). Lung cancer. N. Engl. J. Med. 359 (13), 1367–1380. doi:10.1056/NEJMra0802714
Hirsch, F. R., Scagliotti, G. V., Mulshine, J. L., Kwon, R., Curran, W. J., Wu, Y. L., et al. (2017). Lung cancer: Current therapies and new targeted treatments. Lancet 389 (10066), 299–311. doi:10.1016/S0140-6736(16)30958-8
Hovinga, K. E., Shimizu, F., Wang, R., Panagiotakos, G., Van Der Heijden, M., Moayedpardazi, H., et al. (2010). Inhibition of notch signaling in glioblastoma targets cancer stem cells via an endothelial cell intermediate. Stem Cells 28 (6), 1019–1029. doi:10.1002/stem.429
Hu, S., Cao, P., Kong, K., Han, P., Deng, Y., Li, F., et al. (2021). MicroRNA-449a delays lung cancer development through inhibiting KDM3A/HIF-1α axis. J. Transl. Med. 19 (1), 224. doi:10.1186/s12967-021-02881-8
Huang, L., and Fu, L. (2015). Mechanisms of resistance to EGFR tyrosine kinase inhibitors. Acta Pharm. Sin. B 5 (5), 390–401. doi:10.1016/j.apsb.2015.07.001
Huang, S., Wang, Z., Zhou, J., Huang, J., Zhou, L., Luo, J., et al. (2019). EZH2 inhibitor GSK126 suppresses antitumor immunity by driving production of myeloid-derived suppressor cells. Cancer Res. 79 (8), 2009–2020. doi:10.1158/0008-5472.CAN-18-2395
Huang, T., Song, X., Xu, D., Tiek, D., Goenka, A., Wu, B., et al. (2020). Stem cell programs in cancer initiation, progression, and therapy resistance. Theranostics 10 (19), 8721–8743. doi:10.7150/thno.41648
Jiang, M., Qi, F., Zhang, K., Zhang, X., Ma, J., Xia, S., et al. (2022). MARCKSL1-2 reverses docetaxel-resistance of lung adenocarcinoma cells by recruiting SUZ12 to suppress HDAC1 and elevate miR-200b. Mol. Cancer 21 (1), 150. doi:10.1186/s12943-022-01605-w
Karin, M., and Shalapour, S. (2022). Regulation of antitumor immunity by inflammation-induced epigenetic alterations. Cell. Mol. Immunol. 19 (1), 59–66. doi:10.1038/s41423-021-00756-y
Kasiri, S., Shao, C., Chen, B., Wilson, A. N., Yenerall, P., Timmons, B. C., et al. (2017). GLI1 blockade potentiates the antitumor activity of PI3K antagonists in lung squamous cell carcinoma. Cancer Res. 77 (16), 4448–4459. doi:10.1158/0008-5472.CAN-16-3315
Korsen, J. A., Gutierrez, J. A., Tully, K. M., Carter, L. M., Samuels, Z. V., Khitrov, S., et al. (2022). Delta-like ligand 3-targeted radioimmunotherapy for neuroendocrine prostate cancer. Proc. Natl. Acad. Sci. U. S. A. 119 (27), e2203820119. doi:10.1073/pnas.2203820119
Li, Y., Chen, C., Liu, H. L., Zhang, Z. F., and Wang, C. L. (2022). LARRPM restricts lung adenocarcinoma progression and M2 macrophage polarization through epigenetically regulating LINC00240 and CSF1. Cell. Mol. Biol. Lett. 27 (1), 91. doi:10.1186/s11658-022-00376-y
Li, Y., Zhang, D., Chen, C., Ruan, Z., Li, Y., and Huang, Y. (2012). MicroRNA-212 displays tumor-promoting properties in non-small cell lung cancer cells and targets the hedgehog pathway receptor PTCH1. Mol. Biol. Cell. 23 (8), 1423–1434. doi:10.1091/mbc.E11-09-0777
Liang, X., Xie, R., Su, J., Ye, B., Wei, S., Liang, Z., et al. (2019). Inhibition of RNA polymerase III transcription by triptolide attenuates colorectal tumorigenesis. J. Exp. Clin. Cancer Res. 38 (1), 217. doi:10.1186/s13046-019-1232-x
Lim, J. S., Ibaseta, A., Fischer, M. M., Cancilla, B., O'Young, G., Cristea, S., et al. (2017). Intratumoural heterogeneity generated by Notch signalling promotes small-cell lung cancer. Nature 545 (7654), 360–364. doi:10.1038/nature22323
Liu, C. C., Lin, J. H., Hsu, T. W., Su, K., Li, A. F., Hsu, H. S., et al. (2015). IL-6 enriched lung cancer stem-like cell population by inhibition of cell cycle regulators via DNMT1 upregulation. Int. J. Cancer 136 (3), 547–559. doi:10.1002/ijc.29033
Liu, S., Yang, N., Wang, L., Wei, B., Chen, J., and Gao, Y. (2020). lncRNA SNHG11 promotes lung cancer cell proliferation and migration via activation of Wnt/β-catenin signaling pathway. J. Cell. Physiol. 235 (10), 7541–7553. doi:10.1002/jcp.29656
Liu, T., Guo, W., Luo, K., Li, L., Dong, J., Liu, M., et al. (2022a). Smoke-induced SAV1 gene promoter hypermethylation disrupts YAP negative feedback and promotes malignant progression of non-small Cell Lung Cancer. Int. J. Biol. Sci. 18 (11), 4497–4512. doi:10.7150/ijbs.73428
Liu, T., Wu, X., Chen, T., Luo, Z., and Hu, X. (2018). Downregulation of DNMT3A by miR-708-5p inhibits lung cancer stem cell-like phenotypes through repressing wnt/β-catenin signaling. Clin. Cancer Res. 24 (7), 1748–1760. doi:10.1158/1078-0432.CCR-17-1169
Liu, X., Liu, J., Zhang, X., Tong, Y., and Gan, X. (2019). MiR-520b promotes the progression of non-small cell lung cancer through activating Hedgehog pathway. J. Cell. Mol. Med. 23 (1), 205–215. doi:10.1111/jcmm.13909
Lytle, N. K., Barber, A. G., and Reya, T. (2018). Stem cell fate in cancer growth, progression and therapy resistance. Nat. Rev. Cancer 18 (11), 669–680. doi:10.1038/s41568-018-0056-x
Ma, F., Lei, Y. Y., Ding, M. G., Luo, L. H., Xie, Y. C., and Liu, X. L. (2020). LncRNA NEAT1 interacted with DNMT1 to regulate malignant phenotype of cancer cell and cytotoxic T cell infiltration via epigenetic inhibition of p53, cGAS, and STING in lung cancer. Front. Genet. 11, 250. doi:10.3389/fgene.2020.00250
Mainardi, S., Mijimolle, N., Francoz, S., Vicente-Duenas, C., Sanchez-Garcia, I., and Barbacid, M. (2014). Identification of cancer initiating cells in K-Ras driven lung adenocarcinoma. Proc. Natl. Acad. Sci. U. S. A. 111 (1), 255–260. doi:10.1073/pnas.1320383110
Mao, X., Xu, J., Wang, W., Liang, C., Hua, J., Liu, J., et al. (2021). Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: New findings and future perspectives. Mol. Cancer 20 (1), 131. doi:10.1186/s12943-021-01428-1
Masoumi, J., Jafarzadeh, A., Abdolalizadeh, J., Khan, H., Philippe, J., Mirzaei, H., et al. (2021). Cancer stem cell-targeted chimeric antigen receptor (CAR)-T cell therapy: Challenges and prospects. Acta Pharm. Sin. B 11 (7), 1721–1739. doi:10.1016/j.apsb.2020.12.015
McCaughan, F., Pole, J. C., Bankier, A. T., Konfortov, B. A., Carroll, B., Falzon, M., et al. (2010). Progressive 3q amplification consistently targets SOX2 in preinvasive squamous lung cancer. Am. J. Respir. Crit. Care Med. 182 (1), 83–91. doi:10.1164/rccm.201001-0005OC
Mohammad, I. S., He, W., and Yin, L. (2018). Understanding of human ATP binding cassette superfamily and novel multidrug resistance modulators to overcome MDR. Biomed. Pharmacother. 100, 335–348. doi:10.1016/j.biopha.2018.02.038
Montgomery, M., and Srinivasan, A. (2019). Epigenetic Gene Regulation by dietary compounds in cancer prevention. Adv. Nutr. 10 (6), 1012–1028. doi:10.1093/advances/nmz046
Mukhopadhyay, A., Berrett, K. C., Kc, U., Clair, P. M., Pop, S. M., Carr, S. R., et al. (2014). Sox2 cooperates with Lkb1 loss in a mouse model of squamous cell lung cancer. Cell. Rep. 8 (1), 40–49. doi:10.1016/j.celrep.2014.05.036
Netea, M. G., Joosten, L. A., Latz, E., Mills, K. H., Natoli, G., Stunnenberg, H. G., et al. (2016). Trained immunity: A program of innate immune memory in health and disease. Science 352, aaf1098. doi:10.1126/science.aaf1098
Nusse, R., and Clevers, H. (2017). Wnt/β-Catenin signaling, disease, and emerging therapeutic modalities. Cell. 169 (6), 985–999. doi:10.1016/j.cell.2017.05.016
Oser, M. G., Sabet, A. H., Gao, W., Chakraborty, A. A., Schinzel, A. C., Jennings, R. B., et al. (2019). The KDM5A: RBP2 histone demethylase represses NOTCH signaling to sustain neuroendocrine differentiation and promote small cell lung cancer tumorigenesis. Genes. 33, 1718–1738. doi:10.1101/gad.328336.119
Park, S. B., Seo, K. W., So, A. Y., Seo, M. S., Yu, K. R., Kang, S. K., et al. (2012). SOX2 has a crucial role in the lineage determination and proliferation of mesenchymal stem cells through Dickkopf-1 and c-MYC. Cell. Death Differ. 19 (3), 534–545. doi:10.1038/cdd.2011.137
Parra, E. R., Villalobos, P., Behrens, C., Jiang, M., Pataer, A., Swisher, S. G., et al. (2018). Effect of neoadjuvant chemotherapy on the immune microenvironment in non-small cell lung carcinomas as determined by multiplex immunofluorescence and image analysis approaches. J. Immunother. Cancer 6 (1), 48. doi:10.1186/s40425-018-0368-0
Pauken, K. E., Sammons, M. A., Odorizzi, P. M., Manne, S., Godec, J., Khan, O., et al. (2016). Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 354 (6316), 1160–1165. doi:10.1126/science.aaf2807
Peng, H., Wu, X., Zhong, R., Yu, T., Cai, X., Liu, J., et al. (2021). Profiling tumor immune microenvironment of non-small cell lung cancer using multiplex immunofluorescence. Front. Immunol. 12, 750046. doi:10.3389/fimmu.2021.750046
Plaks, V., Kong, N., and Werb, Z. (2015). The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell. Stem Cell. 16 (3), 225–238. doi:10.1016/j.stem.2015.02.015
Rowbotham, S. P., Li, F., Dost, A. F. M., Louie, S. M., Marsh, B. P., Pessina, P., et al. (2018). H3K9 methyltransferases and demethylases control lung tumor-propagating cells and lung cancer progression. Nat. Commun. 9 (1), 4559. doi:10.1038/s41467-018-07077-1
Sabari, J. K., Lok, B. H., Laird, J. H., Poirier, J. T., and Rudin, C. M. (2017). Unravelling the biology of SCLC: Implications for therapy. Nat. Rev. Clin. Oncol. 14 (9), 549–561. doi:10.1038/nrclinonc.2017.71
Sanchez-Danes, A., and Blanpain, C. (2018). Deciphering the cells of origin of squamous cell carcinomas. Nat. Rev. Cancer 18 (9), 549–561. doi:10.1038/s41568-018-0024-5
Seidl, C., Panzitt, K., Bertsch, A., Brcic, L., Schein, S., Mack, M., et al. (2020). MicroRNA-182-5p regulates hedgehog signaling pathway and chemosensitivity of cisplatin-resistant lung adenocarcinoma cells via targeting GLI2. Cancer Lett. 469, 266–276. doi:10.1016/j.canlet.2019.10.044
Shi, Y. X., Wang, Y., Li, X., Zhang, W., Zhou, H. H., Yin, J. Y., et al. (2017). Genome-wide DNA methylation profiling reveals novel epigenetic signatures in squamous cell lung cancer. BMC Genomics 18 (1), 901. doi:10.1186/s12864-017-4223-3
Shibue, T., and Weinberg, R. A. (2017). EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 14 (10), 611–629. doi:10.1038/nrclinonc.2017.44
Siebel, C., and Lendahl, U. (2017). Notch signaling in development, tissue homeostasis, and disease. Physiol. Rev. 97 (4), 1235–1294. doi:10.1152/physrev.00005.2017
Soteriou, D., and Fuchs, Y. (2018). A matter of life and death: Stem cell survival in tissue regeneration and tumour formation. Nat. Rev. Cancer 18 (3), 187–201. doi:10.1038/nrc.2017.122
Su, S., Chen, J., Yao, H., Liu, J., Yu, S., Lao, L., et al. (2018). CD10(+)GPR77(+) Cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell. 172 (4), 841–856. doi:10.1016/j.cell.2018.01.009
Sutherland, K. D., Proost, N., Brouns, I., Adriaensen, D., Song, J. Y., and Berns, A. (2011). Cell of origin of small cell lung cancer: Inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell. 19 (6), 754–764. doi:10.1016/j.ccr.2011.04.019
Swanton, C., and Govindan, R. (2016). Clinical implications of genomic discoveries in lung cancer. N. Engl. J. Med. 374 (19), 1864–1873. doi:10.1056/NEJMra1504688
Tausendschon, M., Dehne, N., and Brune, B. (2011). Hypoxia causes epigenetic gene regulation in macrophages by attenuating Jumonji histone demethylase activity. Cytokine 53 (2), 256–262. doi:10.1016/j.cyto.2010.11.002
Teschendorff, A. E., and Feinberg, A. P. (2021). Statistical mechanics meets single-cell biology. Nat. Rev. Genet. 22 (7), 459–476. doi:10.1038/s41576-021-00341-z
Thai, A. A., Solomon, B. J., Sequist, L. V., Gainor, J. F., and Heist, R. S. (2021). Lung cancer. Lancet 398 (10299), 535–554. doi:10.1016/S0140-6736(21)00312-3
Wang, C., Yu, Q., Song, T., Wang, Z., Song, L., Yang, Y., et al. (2022). The heterogeneous immune landscape between lung adenocarcinoma and squamous carcinoma revealed by single-cell RNA sequencing. Signal Transduct. Target Ther. 7 (1), 289. doi:10.1038/s41392-022-01130-8
Wang, L., Li, H., Ren, Y., Zou, S., Fang, W., Jiang, X., et al. (2016). Targeting HDAC with a novel inhibitor effectively reverses paclitaxel resistance in non-small cell lung cancer via multiple mechanisms. Cell. Death Dis. 7 (1), e2063. doi:10.1038/cddis.2015.328
Wang, L., Liu, X., Ren, Y., Zhang, J., Chen, J., Zhou, W., et al. (2017). Cisplatin-enriching cancer stem cells confer multidrug resistance in non-small cell lung cancer via enhancing TRIB1/HDAC activity. Cell. Death Dis. 8 (4), e2746. doi:10.1038/cddis.2016.409
Wang, Q., Li, D. C., Li, Z. F., Liu, C. X., Xiao, Y. M., Zhang, B., et al. (2011a). Upregulation of miR-27a contributes to the malignant transformation of human bronchial epithelial cells induced by SV40 small T antigen. Oncogene 30 (36), 3875–3886. doi:10.1038/onc.2011.103
Wang, S. S., Liu, W., Ly, D., Xu, H., Qu, L., and Zhang, L. (2019). Tumor-infiltrating B cells: Their role and application in anti-tumor immunity in lung cancer. Cell. Mol. Immunol. 16 (1), 6–18. doi:10.1038/s41423-018-0027-x
Wang, W., Chen, D., and Zhu, K. (2018a). SOX2OT variant 7 contributes to the synergistic interaction between EGCG and Doxorubicin to kill osteosarcoma via autophagy and stemness inhibition. J. Exp. Clin. Cancer Res. 37 (1), 37. doi:10.1186/s13046-018-0689-3
Wang, W., Zheng, K., Pei, Y., and Zhang, X. (2018b). Histone demethylase JARID1B Is overexpressed in osteosarcoma and upregulates cyclin D1 expression via demethylation of H3K27me3. Oncol. Res. 26 (3), 373–384. doi:10.3727/096504017X14939809845080
Wang, Y., Liu, Y., Malek, S. N., Zheng, P., and Liu, Y. (2011b). Targeting HIF1α eliminates cancer stem cells in hematological malignancies. Cell. Stem Cell. 8 (4), 399–411. doi:10.1016/j.stem.2011.02.006
Wei, X., Chen, Y., Jiang, X., Peng, M., Liu, Y., Mo, Y., et al. (2021). Mechanisms of vasculogenic mimicry in hypoxic tumor microenvironments. Mol. Cancer 20 (1), 7. doi:10.1186/s12943-020-01288-1
Wu, Z. X., Teng, Q. X., Cai, C. Y., Wang, J. Q., Lei, Z. N., Yang, Y., et al. (2019). Tepotinib reverses ABCB1-mediated multidrug resistance in cancer cells. Biochem. Pharmacol. 166, 120–127. doi:10.1016/j.bcp.2019.05.015
Xie, W., Kagiampakis, I., Pan, L., Zhang, Y. W., Murphy, L., Tao, Y., et al. (2018). DNA Methylation patterns separate senescence from transformation potential and indicate cancer risk. Cancer Cell. 33 (2), 309–321. doi:10.1016/j.ccell.2018.01.008
Xu, X., Huang, L., Futtner, C., Schwab, B., Rampersad, R. R., Lu, Y., et al. (2014). The cell of origin and subtype of K-Ras-induced lung tumors are modified by Notch and Sox2. Genes. Dev. 28 (17), 1929–1939. doi:10.1101/gad.243717.114
Xu, X., Rock, J. R., Lu, Y., Futtner, C., Schwab, B., Guinney, J., et al. (2012). Evidence for type II cells as cells of origin of K-Ras-induced distal lung adenocarcinoma. Proc. Natl. Acad. Sci. U. S. A. 109 (13), 4910–4915. doi:10.1073/pnas.1112499109
Yang, L., Shi, P., Zhao, G., Xu, J., Peng, W., Zhang, J., et al. (2020). Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target Ther. 5 (1), 8. doi:10.1038/s41392-020-0110-5
Yu, W. K., Wang, Z., Fong, C. C., Liu, D., Yip, T. C., Au, S. K., et al. (2017). Chemoresistant lung cancer stem cells display high DNA repair capability to remove cisplatin-induced DNA damage. Br. J. Pharmacol. 174 (4), 302–313. doi:10.1111/bph.13690
Yu, Y., Schleich, K., Yue, B., Ji, S., Lohneis, P., Kemper, K., et al. (2018). Targeting the senescence-overriding cooperative activity of structurally unrelated H3K9 demethylases in melanoma. Cancer Cell. 33 (2), 322–336. doi:10.1016/j.ccell.2018.01.002
Zeng, Y., Li, N., Chen, R., Liu, W., Chen, T., Zhu, J., et al. (2020). Screening of hub genes associated with prognosis in non-small cell lung cancer by integrated bioinformatics analysis. Transl. Cancer Res. 9 (11), 7149–7164. doi:10.21037/tcr-20-1073
Zhang, K., Wang, J., Yang, L., Yuan, Y. C., Tong, T. R., Wu, J., et al. (2018). Targeting histone methyltransferase G9a inhibits growth and Wnt signaling pathway by epigenetically regulating HP1α and APC2 gene expression in non-small cell lung cancer. Mol. Cancer 17 (1), 153. doi:10.1186/s12943-018-0896-8
Zhang, L., Li, Y., Wang, Q., Chen, Z., Li, X., Wu, Z., et al. (2020). The PI3K subunits, P110α and P110β are potential targets for overcoming P-gp and BCRP-mediated MDR in cancer. Mol. Cancer 19 (1), 10. doi:10.1186/s12943-019-1112-1
Zhang, W., Zheng, X., Yu, Y., Zheng, L., Lan, J., Wu, Y., et al. (2022). Renal cell carcinoma-derived exosomes deliver lncARSR to induce macrophage polarization and promote tumor progression via STAT3 pathway. Int. J. Biol. Sci. 18 (8), 3209–3222. doi:10.7150/ijbs.70289
Zhang, Z., Yang, Y., and Zhang, X. (2017). MiR-770 inhibits tumorigenesis and EMT by targeting JMJD6 and regulating WNT/β-catenin pathway in non-small cell lung cancer. Life Sci. 188, 163–171. doi:10.1016/j.lfs.2017.09.002
Zhou, H. M., Zhang, J. G., Zhang, X., and Li, Q. (2021). Targeting cancer stem cells for reversing therapy resistance: Mechanism, signaling, and prospective agents. Signal Transduct. Target Ther. 6 (1), 62. doi:10.1038/s41392-020-00430-1
Zhu, M., Huang, Y., Bender, M. E., Girard, L., Kollipara, R., Eglenen-Polat, B., et al. (2021). Evasion of innate immunity contributes to small cell lung cancer progression and metastasis. Cancer Res. 81 (7), 1813–1826. doi:10.1158/0008-5472.CAN-20-2808
Keywords: epigenetics, stem cells, lung cancer, tumor microenvironment, oncogenesis, therapy resistance
Citation: Wu J, Feng J, Zhang Q, He Y, Xu C, Wang C and Li W (2023) Epigenetic regulation of stem cells in lung cancer oncogenesis and therapy resistance. Front. Genet. 14:1120815. doi: 10.3389/fgene.2023.1120815
Received: 10 December 2022; Accepted: 22 February 2023;
Published: 18 April 2023.
Edited by:
Pengxu Qian, Zhejiang University, ChinaReviewed by:
Pranabananda Dutta, Charles R. Drew University of Medicine and Science, United StatesCopyright © 2023 Wu, Feng, Zhang, He, Xu, Wang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weimin Li, d2VpbWkwMDNAc2N1LmVkdS5jbg==; Chengdi Wang, Y2hlbmdkaV93YW5nQHNjdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.