 Yirao Chen1
Yirao Chen1 Lu Shen
Lu Shen Jifeng Guo
Jifeng Guo Qian Xu
Qian Xu- 1Department of Neurology, Xiangya Hospital, Central South University, Changsha, China
- 2Department of Neurology, Central Hospital, Bai Yin, China
- 3National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha, China
- 4Key Laboratory of Hunan Province in Neurodegenerative Disorders, Central South University, Changsha, China
- 5Hunan International Scientific and Technological Cooperation Base of Neurodegenerative and Neurogenetic Diseases, Changsha, China
- 6Engineering Research Center of Hunan Province in Cognitive Impairment Disorders, Central South University, Changsha, China
- 7Key Laboratory of Organ Injury, Aging and Regenerative Medicine of Hunan Province, Changsha, China
- 8Centre for Medical Genetics and Hunan Key Laboratory of Medical Genetics, School of Life Sciences, Central South University, Changsha, China
This study describes a patient with progressive myoclonic epilepsy-11 (EPM-11), which follows autosomal dominant inheritance caused by a novel SEMA6B variant. Most patients develop this disease during infancy or adolescence with action myoclonus, generalized tonic-clonic seizures (GTCS), and progressive neurological deterioration. No cases of adult-onset EPM-11 have been reported yet. Here, we present one case of adult-onset EPM-11 who experienced gait instability, seizures, and cognitive impairment, and harbored a novel missense variant, c.432C>G (p.C144W). Our findings provide a foundation for a better understanding of the phenotypic and genotypic profiles of EPM-11. Further functional studies are recommended to elucidate the pathogenesis of this disease.
Introduction
Progressive myoclonic epilepsies (PMEs) are a rare group of clinically and genetically heterogeneous disorders characterized by symptoms such as action myoclonus, GTCS, and progressive neurological deterioration (Andermann, 1990), typical onset is in childhood or adolescence. The concept of PMEs was first introduced by Herman Lundborg (Genton et al., 2016), who studied several Swedish families with a common ancestor in 1903 and noticed a particular form of epilepsy associated with progressive myoclonus, with varying degrees of severity. PMEs are typically inherited in an autosomal recessive manner, while a small number of patients have mitochondrial or autosomal dominant inheritance patterns (Franceschetti et al., 2014; Kälviäinen, 2015).
PMEs can be divided into two broad clinical groups. In the first group, the patients present with severe, treatment-resistant, and physically disabling myoclonus, tonic-clonic seizures, and ataxia, with intact cognitive skills (Berkovic et al., 1986). In the second group, the patients experience significant cognitive impairment and degeneration. In the early stages of PMEs, the clinical and electroencephalogram (EEG) characteristics may be similar to those of idiopathic generalized epilepsy syndromes, particularly juvenile myoclonic epilepsy. However, treatment failure, progressive aggravation of neurological symptoms, and EEG manifestations indicate PMEs (Shahwan et al., 2005). As for the management, classical anti-myoclonic agents, including valproate, and levetiracetam, often have limited lasting efficacy in patients with PME. Clonazepam is often helpful, but it typically leads to considerable sedation and tolerance over time. Zonisamide has good anti-epileptic effectiveness, sometimes with a long-lasting effect, and has been shown to improve inter-ictal myoclonus (Vossler et al., 2008; Herzog et al., 2021). Gene modification and enzyme replacement therapies may help improve the condition in the near future (Minassian, 2014).
In recent years, the clinical application of next-generation sequencing technology has led to the discovery of multiple gene mutations related to PME (such as GOSR2, ASAH1, KCTD7, TBC1D24, SCARB2, PRICKLE1, CARS2, and SERPINI) (Li et al., 2021). Pathogenic variants of semaphorin 6B (SEMA6B) can also cause EPM-11 [OMIM#618876], as demonstrated in a few case studies (Hamanaka et al., 2020; Courage et al., 2021; Herzog et al., 2021; Li et al., 2021; Shu et al., 2021; Xiaozhen et al., 2021; Duan et al., 2022). To date, 13 cases of SEMA6B-related PME have been reported, all of whom presented with seizures, while three presented with myoclonic seizures. Adult-onset EPM-11 has rarely been reported in the previous literature, and herein, we report one case of adult-onset EPM-11 who presented with late-onset gait instability, seizures, and cognitive impairment.
Case description
A 51-year-old Chinese man who presented with gait disturbance and GTCS was admitted to our neurology department. He first presented with clinical symptoms when he was 46 years old and experienced difficulty in walking steadily. The symptom slightly relieved after treatment with vitamins B1 and B12. His personality was dampened, and his memory deteriorated over time. He presented with generalized seizures at 50 years of age, which occurred twice a year. Physical examination revealed horizontal nystagmus, positive palmomental reflexes, postural and intention tremors, increased muscle tension in both lower extremities, tendon hyperreflexia, positive ankle clonus and heel-knee-shin tests of the right side, and dysmetria in the finger-to-nose test. Somatosensory evoked potential (SEP) showed giant evoked potentials in the bilateral upper limbs and enhanced C reflexes after stimulation of the median nerve bilaterally. A 24 h EEG showed bilaterally sharp waves over the fronto-centro-parietal electrodes, particularly over the right regions. Magnetic resonance imaging (MRI) revealed mild cortical and cerebellar atrophy (Figure 1A). The cognitive function was assessed using the Mini-Mental State Examination, with a score of 27, and the Montreal Cognitive Assessment, with a score of 23. We commenced treatment with levetiracetam (500 mg/d) and B vitamins. The patient had no seizures in 9 months, and his symptoms did not aggravate with follow-up.
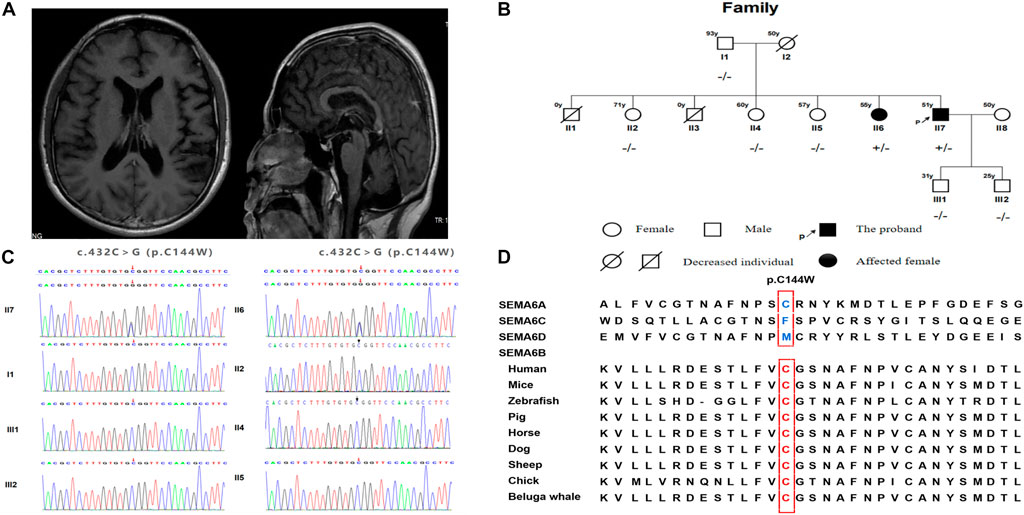
FIGURE 1. (A) Brain MRI of the proband. (B) SEMA6B gene sequencing of the family members. (C) Genetic pedigree of the family and corresponding individual genotypes. (D) The alignment of SEMA6B amino acid sequences.
There were no reports of consanguineous marriages within the patient’s family. Before the patient’s birth, his mother had two miscarriages. One of the proband’s sisters began having seizures, gait disturbances, and cognitive impairment in her 50s (Figure 1B). She experienced generalized seizures more frequently and had more severe cognitive impairment than the proband. She underwent antiepileptic treatment without any symptomatic improvement. Her condition worsened over time, and she had been bedridden for 2 years. Due to her cognitive function decline, she was unable to cooperate with us to complete the physical examination. However, her muscle tension was increased in all limbs, and the pathological signs were positive. The remaining members of the proband’s family were unaffected, while his mother died at 50 years of age in an accident. After obtaining consent, we performed further genetic analysis. We completed cytosine-adenine-guanine (CAG) repeat expansion detection and found no spinocerebellar ataxia (SCA)-related dynamic mutations; in contrast, whole exome sequencing (WES) detected a missense variant, c.432C>G (p.C144W), in exon six of SEMA6B in the proband. The proband’s sister carried the same heterozygous mutation (Figure 1C).
Discussion
PME has high genetic heterogeneity, and more than 40 genes are reportedly associated with this disorder. SEMA6B is one of the pathogenic genes that are related to PME and is located on chromosome 19p13; it contains 17 coding exons and a PPAR-binding site in the upstream sequence (Correa et al., 2001). SEMA6B is a member of the class-6 semaphorin family, which is involved in neural development, including neural crest cell migration, axon guidance, and cerebellar development (Andermatt et al., 2014). SEMA6B is highly expressed in multiple brain regions, including the cerebral cortex, cerebellar Purkinje cells, interneurons, and specific cell types, including excitatory and GABAergic inhibitory neurons (Hamanaka et al., 2020). Consequently, disruption of SEMA6B function in GABAergic neurons may contribute to epilepsy.
To the best of our knowledge, 13 cases of SEMA6B-associated PME, including the case described in the present study, have been reported. The first 12 had childhood-to-juvenile onset, and only our case presented as adult onset. The clinical characteristics and genetic detection of the EMP-11 patients are summarized in Table 1. Several reported cases identified patients harboring a truncating variant in the final exon of SEMA6B. Interestingly, a missense variant in exon 16 of SEMA6B (c.1834G > A/p. V612M) was found to be related to cerebellar hypoplasia, with symptoms including cerebellar ataxia and developmental delay (Aldinger et al., 2019). The SEMA6B c.432C>G (exon6) variant identified in this study is a novel mutation and is absent in the Genome Aggregation Database (gnomAD), Exome Aggregation Consortium (ExAC), and 1000 Genomes Project. This variant is predicted to be a damaging missense mutation by several missense prediction software packages, including Polythen2, MutationTaster, CADD, and ReVe, and p. C144W is highly conserved in other organisms (Figure 1D).
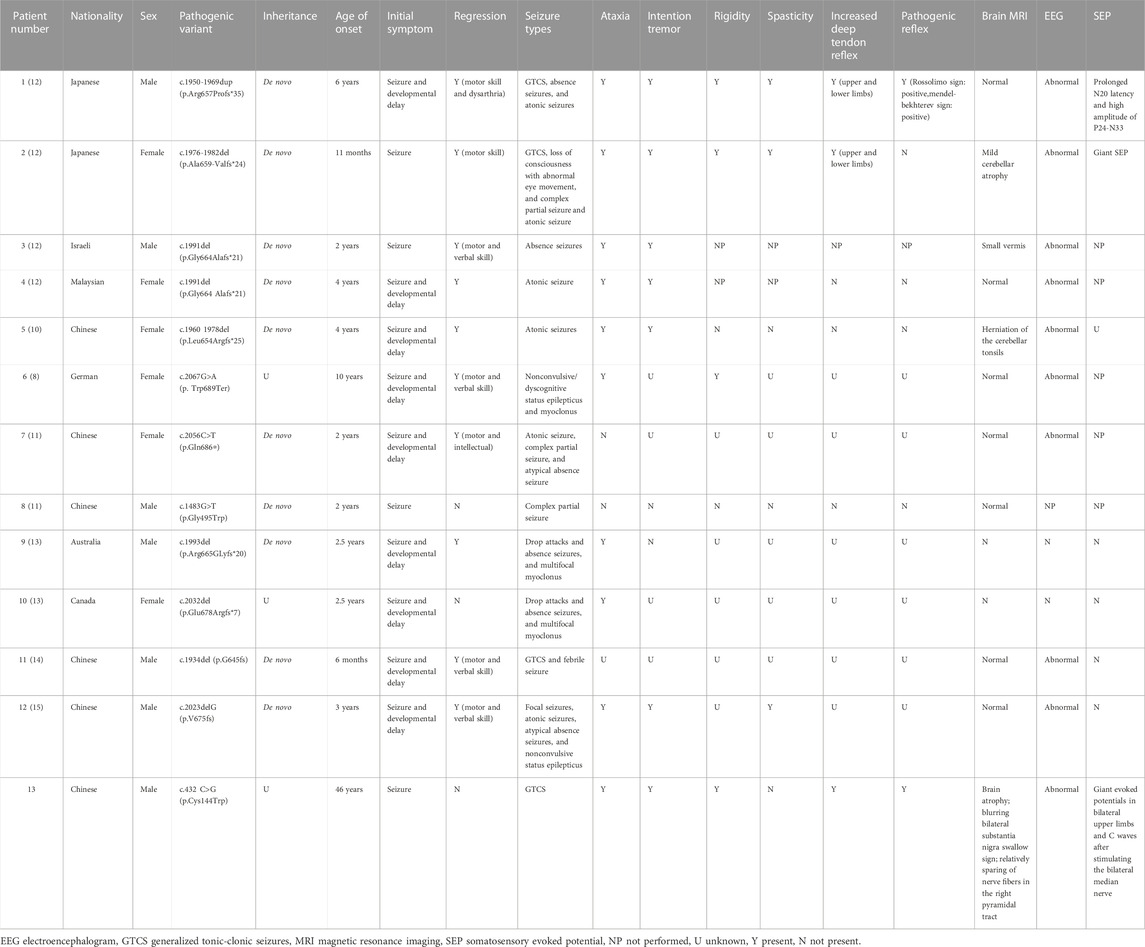
TABLE 1. Clinical features of the patient reported in this work and comparison with published cases of SEMA6B-related progressive myoclonic epilepsy.
In this family, the proband and his sister have similar phenotypes and carry the same variant, which is absent in other siblings. Therefore, we assume that this variant might have originated from their mother, who died in an accident. The mechanism of SEMA6B-related disease remains unclear. Previous studies have shown that missense and nonsense variants can lead to protein function problems, resulting in clinical symptoms such as epilepsy (Xiaozhen et al., 2021). Further functional studies will help us clarify the mechanism.
Conclusion
In conclusion, we reported a family with adult-onset PME-11 harboring a novel heterozygous missense variant. The new genetic variation reported here strengthens the gene–disease relationship. This finding may expand the consideration of the age of onset for EPM-11 and extend the mutational spectrum. However, further functional studies are required to better elucidate the pathogenesis of this disease.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Association of Xiangya Hospital of Central South University. The patients/participants provided their written informed consent to participate in this study.
Author contributions
QX conceived the study. YC drafted the manuscript. YC, XYang, XYan, LS, and QX participated in the clinical management of the patient. YC, XYan, LS, JG, and QX revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Natural Science Foundation of Hunan Province (reference number: 2021JJ31115), the National Natural Science Foundation of China (No. 82071437), and the Project Program of the National Clinical Research Center for Geriatric Disorders (Xiangya Hospital) (No. 2021KFJJ10).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aldinger, K. A., Timms, A. E., Thomson, Z., Mirzaa, G. M., Bennett, J. T., Rosenberg, A. B., et al. (2019). Redefining the etiologic landscape of cerebellar malformations. Am. J. Hum. Genet. 105 (3), 606–615. doi:10.1016/j.ajhg.2019.07.019
Andermatt, I., Wilson, N. H., Bergmann, T., Mauti, O., Gesemann, M., Sockanathan, S., et al. (2014). Semaphorin 6B acts as a receptor in post-crossing commissural axon guidance. Dev. Camb. Engl. 141 (19), 3709–3720. doi:10.1242/dev.112185
Berkovic, S. F., Andermann, F., Carpenter, S., and Wolfe, L. S. (1986). Progressive myoclonus epilepsies: Specific causes and diagnosis. N. Engl. J. Med. 315 (5), 296–305. doi:10.1056/NEJM198607313150506
Andermann (1990), Classification of progressive myoclonus epilepsies and related disorders. Marseille Consensus Group. Ann. neurology 28, 113, doi:10.1002/ana.410280129
Correa, R. G., Sasahara, R. M., Bengtson, M. H., Katayama, M. L., Salim, A. C., Brentani, M. M., et al. (2001). Human semaphorin 6B [(HSA)SEMA6B], a novel human class 6 semaphorin gene: Alternative splicing and all-trans-retinoic acid-dependent downregulation in glioblastoma cell lines. Genomics 73 (3), 343–348. doi:10.1006/geno.2001.6525
Courage, C., Oliver, K. L., Park, E. J., Cameron, J. M., Grabińska, K. A., Muona, M., et al. (2021). Progressive myoclonus epilepsies-Residual unsolved cases have marked genetic heterogeneity including dolichol-dependent protein glycosylation pathway genes. Am. J. Hum. Genet. 108 (4), 722–738. doi:10.1016/j.ajhg.2021.03.013
Duan, J., Chen, Y., Hu, Z., Ye, Y., Zhang, T., Li, C., et al. (2022). Non-convulsive status epilepticus in SEMA6B-related progressive myoclonic epilepsy: A case report with literature review. Front. Pediatr. 10, 859183. doi:10.3389/fped.2022.859183
Franceschetti, S., Michelucci, R., Canafoglia, L., Striano, P., Gambardella, A., Magaudda, A., et al. (2014). Progressive myoclonic epilepsies: Definitive and still undetermined causes. Neurology 82 (5), 405–411. doi:10.1212/WNL.0000000000000077
Genton, P., Striano, P., and Minassian, B. A. (2016). The history of progressive myoclonus epilepsies. Epileptic Disord. Int. epilepsy J. videotape 18 (S2), 3–10. doi:10.1684/epd.2016.0834
Hamanaka, K., Imagawa, E., Koshimizu, E., Miyatake, S., Tohyama, J., Yamagata, T., et al. (2020). De novo truncating variants in the last exon of SEMA6B cause progressive myoclonic epilepsy. Am. J. Hum. Genet. 106 (4), 549–558. doi:10.1016/j.ajhg.2020.02.011
Herzog, R., Hellenbroich, Y., Brüggemann, N., Lohmann, K., Grimmel, M., Haack, T. B., et al. (2021). Zonisamide-responsive myoclonus in SEMA6B-associated progressive myoclonic epilepsy. Ann. Clin. Transl. Neurology 8 (7), 1524–1527. doi:10.1002/acn3.51403
Kälviäinen, R. (2015). Progressive myoclonus epilepsies. Seminars Neurology 35 (03), 293–299. doi:10.1055/s-0035-1552620
Li, Q., Liu, M., Huang, D-P., Li, T., Huang, J., Jiang, P., et al. (2021). A de novo SEMA6B variant in a Chinese patient with progressive myoclonic epilepsy-11 and review of the literature. J. Mol. Neurosci. 71 (9), 1944–1950. doi:10.1007/s12031-021-01880-0
Minassian, B. A. (2014). The progressive myoclonus epilepsies. Prog. Brain Res. 213, 113–122. doi:10.1016/B978-0-444-63326-2.00006-5
Shahwan, A., Farrell, M., and Delanty, N. (2005). Progressive myoclonic epilepsies: A review of genetic and therapeutic aspects. Lancet Neurology 4 (4), 239–248. doi:10.1016/S1474-4422(05)70043-0
Shu, L., Xu, Y., Tian, Q., Chen, Y., Wang, Y., Xi, H., et al. (2021). A frameshift variant in the SEMA6B gene causes global developmental delay and febrile seizures. Neurosci. Bull. 37 (9), 1357–1360. doi:10.1007/s12264-021-00717-5
Vossler, D. G., Conry, J. A., and Murphy, J. V.ZNS-502/505 PME Study Group (2008). Zonisamide for the treatment of myoclonic seizures in progressive myoclonic epilepsy: An open-label study. Epileptic Disord. Int. epilepsy J. videotape 10 (1), 31–34. doi:10.1684/epd.2008.0168
Keywords: progressive myoclonic epilepsy, adult onset, SEMA6B, missense variant, case report
Citation: Chen Y, Yang X, Yan X, Shen L, Guo J and Xu Q (2023) A novel SEMA6B variant causes adult-onset progressive myoclonic epilepsy-11 in a Chinese family: A case report and literature review. Front. Genet. 14:1110310. doi: 10.3389/fgene.2023.1110310
Received: 30 November 2022; Accepted: 26 January 2023;
Published: 15 February 2023.
Edited by:
Chantal Depondt, Université libre de Bruxelles, BelgiumReviewed by:
Paul C. Marcogliese, University of Manitoba, CanadaJosef Finsterer, K.A. Rasmussen, Norway
Copyright © 2023 Chen, Yang, Yan, Shen, Guo and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qian Xu, eHl4dXFpYW4yMDE1QDE2My5jb20=