
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet., 02 February 2023
Sec. Genetics of Common and Rare Diseases
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1107460
 Joanna Kulikowska1*
Joanna Kulikowska1* Anna Jakubiuk-Tomaszuk2
Anna Jakubiuk-Tomaszuk2 Małgorzata Rydzanicz3Rafał Płoski3Jan Kochanowicz1
Małgorzata Rydzanicz3Rafał Płoski3Jan Kochanowicz1 Alina Kulakowska1
Alina Kulakowska1 Katarzyna Kapica-Topczewska1
Katarzyna Kapica-Topczewska1Variants in the ERCC4 gene have been described to be associated with the following autosomal recessive diseases: xeroderma pigmentosum group F (XPF), xeroderma pigmentosum type F/Cockayne syndrome (XPF/CS), Fanconi anemia complementation group Q (FANCQ), and XFE progeroid syndrome (XFEPS). In this paper, we present a case of a 53-year-old Caucasian female patient with rare variants in the ERCC4 gene. When she was 42 years old, falls and loss of balance occurred. At the age of 48, involuntary, uncoordinated movements of the upper limbs and head, tongue stereotypes (licking and extending movements), speech problems (dysarthria), memory deterioration, and hearing loss occurred. Since childhood, she has shown hypersensitivity to UV radiation. The neurological examination revealed chorea syndrome, cerebellar ataxia, dysarthria, and bilateral hearing loss. She has numerous pigmented lesions on the skin. Brain MRI demonstrated massive cortico-subcortical atrophy. The neuropsychological examination revealed dysfunctions in the executive domain in terms of attention, working memory, organizing, and planning activities. The genetic diagnostics was performed which excluded spinocerebellar ataxia types 1, 2, 3, 6, and 17, Huntington’s disease, and FMR1 premutation. In the genetic analysis of next-generation sequencing (NGS), two variants: c.2395C > T and c.1349G > A in the ERCC4 gene were identified in a heterozygote configuration. So far, a few cases of ERCC4 gene variants, which are associated with nucleotide excision repair pathways, have been described in connection with symptoms of cerebellar ataxia. In patients with ERCC4 biallelic variants, the adult neurological phenotype can sometimes be the first symptom and reason for access to genetic testing. The aforementioned case highlights the occurrence of rare genetic causes of progressive neurodegenerative diseases in adults, especially with the spectrum of autosomal recessive nucleotide excision repair pathway disorders (NERDs).
Chorea and cerebellar ataxia are rare causes of progressive neurodegeneration, most often of genetic etiology. After excluding secondary causes, like drug-induced dyskinesia or Sydenham chorea, genetic diagnosis must be conducted. In the differential diagnosis, the most common hereditary causes should be taken into consideration: Huntington’s disease, neuroacanthocytosis, spinocerebellar ataxia (SCA) types 1, 2, 3, 6, and 17, and others were found to be associated with chorea (Burgunder, 2022). Increasingly available genetic diagnostics, especially genomic sequencing, allows the detection of rare causes of cerebellar ataxia with chorea. ERCC4 gene variants have been described to be associated with the following autosomal recessive diseases: xeroderma pigmentosum group F (XPF) (Nabouli et al., 2021), xeroderma pigmentosum type F/Cockayne syndrome (XPF/CS) (Kashiyama et al., 2013), Fanconi anemia complementation group Q (FANCQ) (Bogliolo et al., 2013a), and XFE progeroid syndrome (XFEPS) (Mori et al., 2018). So far, a few cases of ERCC4 gene variants in connection with symptoms of cerebellar ataxia have been described. The role of nucleotide excision repair (NER) variants is highlighted in connection with adult-onset neurodegeneration (Cordts et al., 2022). Various gene variants can cause multiple phenotypes. We present a case of a patient with ERCC4 gene variants with the rare phenotype of progressive cerebellar ataxia, chorea, cognitive impairment, and sensory hearing loss with a history of skin photosensitivity.
We present a case of a 53-year-old Caucasian female patient. When she was 42 years old, falls and loss of balance occurred. Furthermore, at the age of 48, progressive, involuntary, uncoordinated movements of the upper limbs and head, tongue stereotypes (licking and extending movements), speech problems (dysarthria), memory deterioration, and hearing loss occurred. Since childhood, she has shown skin and conjunctiva hypersensitivity to the sun. She underwent splenectomy due to spherocytosis at the age of 18 and cholecystectomy at the age of 20. Before the age of 40, she was diagnosed with premature ovarian failure. In vitro fertilization was performed with a donor oocyte. She gave birth to a female baby. There was no family history of genetic diseases and consanguineous parents. She pursued philological education. Initially, she had been working as a teacher of the Russian language and then in a library. Currently, she is living on a pension. The neurological examination revealed chorea syndrome, cerebellar ataxia, dysarthria, and bilateral hearing loss. She has numerous pigmented lesions on her brown-colored skin (Figure 1). Brain MRI demonstrated massive cortico-subcortical atrophy (Figures 2, 3 ). The consulting otolaryngologist diagnosed bilateral sensory hearing impairment. The electroneurographic examination did not reveal any signs of peripheral polyneuropathy. Laboratory tests showed the presence of macrocytic anemia, significant vitamin B12 deficiency, and high levels of homocysteine. The consulting hematologist recommended supplementation with vitamin B12, iron, and folic acid. Endoscopic examination showed atrophic gastropathy. At the age of 48, a neuropsychological examination was performed which revealed cognitive-behavioral disorders with a predominance of subcortical symptomatology. Additionally, a slowdown in the pace of work, periodic drops in the tension of free attention, difficulties in organizing complex visual materials, and deficits in learning and memory, mainly of an executive nature, were described. The neuropsychological examination performed at the age of 53 revealed dysfunctions in the executive domain in the field of attention (maintenance, selectivity, and divisibility), working memory, organizing, and planning activities (mainly activities on visual materials). There was variability in the severity of dysfunction in the cognitive and non-cognitive spheres. The clinical details are presented in Table 1. The genetic diagnostics was performed which excluded spinocerebellar ataxia types 1, 2, 3, 6, and 17, Huntington’s disease, and FMR1 premutation. Exome sequencing using the next-generation sequencing (NGS; SureSelectXT Human All Exome v7 (Agilent)) method was performed in further genetic diagnostics as previously described (Jakubiuk-Tomaszuk et al., 2019). Two variants in the ERCC4 gene, namely, missense c.2395C > T and a novel non-sense c.1349G > A, in the configuration of the heterozygote, were identified (Table 2). The presence of the c.1349G > A variant of the ERCC4 gene was found in a heterozygous state in the proband’s mother but excluded in the father, while the presence of the c.2395C > T variant of the ERCC4 gene was found in a heterozygous state in the proband’s father but excluded in the mother. It is consistent with in trans variant transmission in the autosomal recessive mode of inheritance. Moreover, in the proband, a heterozygous non-sense variant in the SPTB gene (hg38 chr14:064786837-C>T; NM_001355436.2:c.3128G>A (p.Trp1043Ter), rs1594775390) was identified (Figure 4). The c.3128G > A variant has 0 frequency in the gnomAD v3.1.2 database and was described in the ClinVar database as “pathogenic” and classified as “pathogenic” according to ACMG classification (total score 11, PVS1 very strong, PP5 moderate, and PM2 supporting). Variants in the SPTB gene, including the c.3128G > A variant identified in our patient, are associated with spherocytosis type 2, 8, and 9. The parents showed no clinical or laboratory signs of spherocytosis. So far, a rare phenotype of progressive spinocerebellar ataxia, chorea, cognitive impairment, and hearing loss with a history of skin photosensitivity has been described in single patients with ERCC4 gene variants.

FIGURE 1. Hyperpigmentation lesions on lower limbs.
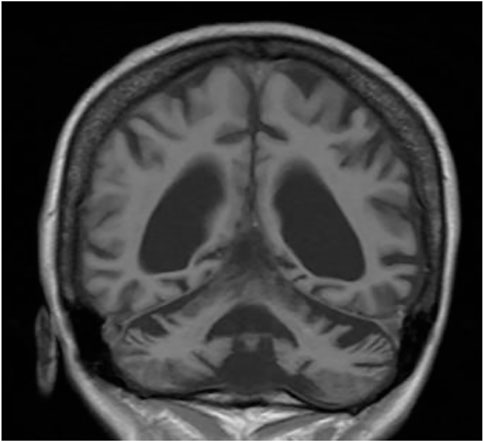
FIGURE 2. MRI—massive cortical and subcortical atrophy frontal section.
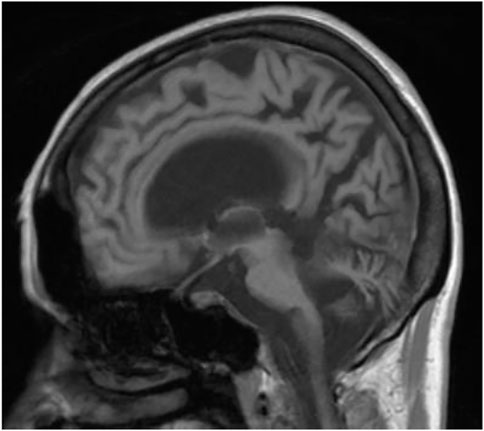
FIGURE 3. MRI—massive cortical and subcortical atrophy Sagittal section.
TABLE 1. Clinical details.

TABLE 2. Genetic diagnosis details.
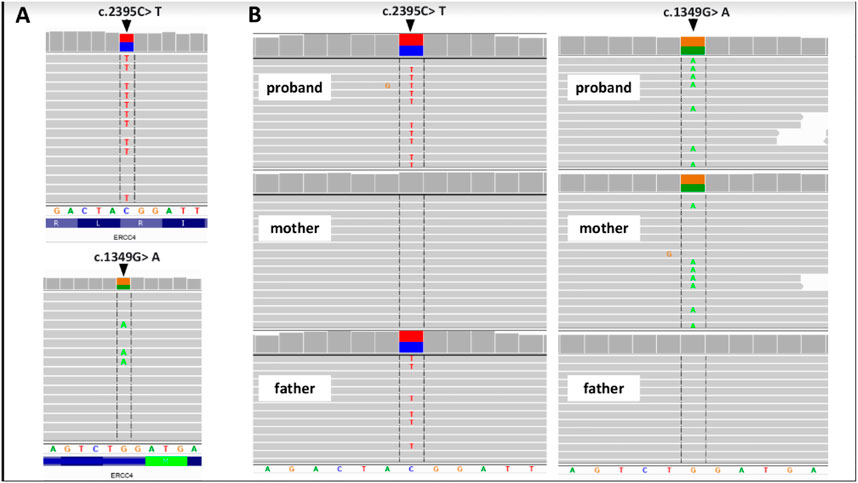
FIGURE 4. Genetic examination and family study of the ERCC4 gene. (A) Proband’s WES results and (B) results of family study performed by NGS-based amplicon deep sequencing. Integrative Genomics Viewer screenshots are presented.
We present a case of a Caucasian patient with chorea syndrome and cerebellar ataxia. The genetic diagnostics was performed which excluded spinocerebellar ataxia type 1, 2, 3, 6, and 17, Huntington’s disease, and FMR1 premutation. Exome sequencing using the NGS method revealed two variants, c.2395C > T and c.1349G > A, in the ERCC4 gene. NER is a mechanism responsible for DNA damage repair and occurs as a result of endogenous or exogenous factors (Spivak, 2015). NER impairment is associated with the occurrence of diseases such as XP and CS, and due to the possible occurrence of many variants of these genes, different phenotypes are described. Recently, attention has been paid to the development of adult-onset neurodegeneration in connection with damage to NER mechanisms, including the occurrence of ERCC4 variants (Cordts et al., 2022). All of them presented progressive symptoms of neurodegeneration beginning in adulthood: ataxia, chorea, and global brain atrophy. The concept of nucleotide excision repair disorders (NERDs) has recently been introduced to cover the spectrum of patients with adult-onset neurodegeneration with diagnosed NER impairment. Cordts et al., 2022 described 13 patients with NER gene variants who presented progressive neurological disorders, of which eight patients showed variants in the ERCC4 gene. In this study, it was estimated that among patients with ataxia, NER disorders occurred in 1% of patients and among patients with ataxia and cognitive disorders, NER disorders occurred in 8.6% (). ERCC4 variants have been described to be associated with autosomal recessive inheritance diseases: XP (Nabouli et al., 2021), FANCQ (Bogliolo et al., 2013b), CS (Kashiyama et al., 2013), and XFEPS (Mori et al., 2018). XP is a disease characterized by hypersensitivity to the sun, the presence of sun-induced skin lesions, and a high risk of skin cancer development, usually in the first decade of life (Nabouli et al., 2021). Niraj et al. reported cases of two patients with XPF who showed signs of cerebellar ataxia, chorea syndrome, hearing loss, and global atrophy on brain MRI. One of these patients had a history of basal cell carcinoma of the skin. In both patients described in this report, the c.2395C > T variant in the ERCC4 gene was identified (Shanbhag et al., 2018). Eight patients who carried variants in the ERCC4 gene were reported with adult-onset neurological deterioration. Seven patients harbored missense c.2395C > T variant in the heterozygous state (Cordts et al., 2022). In our case, the second variant c.1349G> A has not been reported previously. Since childhood, our patient has been highly hypersensitive to solar radiation, but the oncological history remains negative. Doi et al. (2018) described the coexistence of slowly progressive cerebellar ataxia, chorea syndrome, and mild hypersensitivity to solar radiation, in association with ERCC4 gene mutation. The neurological symptoms of XP occur in 20–30% of cases, mainly in the XPF (OMIM # 278760). This phenotype is often classified as CS. CS (OMIM # 278760) is characterized by microcephaly, significant sensitivity to the sun, and progressive neurodegeneration from the first months of life. The mean age at death is 12.5 years. Despite the similar genetic etiology, the differences in the clinical picture, including the age of the first symptoms, do not allow the patient to be diagnosed with CS. In-depth neurological diagnostics of the presented patient, apart from the highly developed chorea syndrome and cerebellar ataxia, showed significant cognitive impairment. In addition, bilateral sensory hearing loss was detected. Imaging diagnostics showed generalized brain atrophy. It is worth noting that the patient suffered from spherocytosis as a child, due to which she underwent splenectomy. Currently, she is diagnosed with macrocytic anemia due to vitamin B12 deficiency. FA (OMIM #615272) is characterized by bone marrow failure, congenital anomalies, and possible sun hypersensitivity, and the patient usually dies in childhood. Apart from the abnormalities mentioned previously, no symptoms of bone marrow failure have been found in our patient so far. It is worth emphasizing that the family history remained negative. Variants in the ERCC4 gene were found in the mother and father of the proband. In addition to increasing knowledge about NERD, the current literature does not provide a description of the coexistence of the two variants, c.2395 > T and c.1349G > A, in the ERCC4 gene. Only a few cases of cerebellar ataxia have been described so far in connection with the variants of the ERCC4 gene. The patient is aware of the genetic cause of her symptoms and has been informed about a very rare phenotype, which makes it impossible to attribute the symptoms to a specific genetic syndrome. However, due to the current cognitive impairment, information about the overall health condition is communicated to her husband. Adult-onset neurodegeneration, such as ataxia, chorea, cognitive impairment, and brain atrophy, should be considered a disrupted nucleotide excision repair mechanism, which has recently been referred to as NERD (Cordts et al., 2022). In patients with ERCC4 biallelic variants, the adult neurological phenotype can sometimes be the first symptoms and reason for access to genetic testing. The aforementioned case highlights the occurrence of rare genetic causes of progressive neurodegenerative diseases in adults, especially with the spectrum of autosomal NERD.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
JK: preparation, creation, and/or presentation of the published work, especially writing the initial draft, data analysis, and interpretation; AJ-T: data analysis and interpretation; MR: data analysis and interpretation; RP: conducting the research and investigation process, especially performing the experiments, and data/evidence collection; JK: critical revision of the article; AK: critical revision of the article; KK-T: final approval of the version to be published, idea formulation, and evolution of overarching research goals and aims.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Bogliolo, M., Schuster, B., Stoepker, C., Derkunt, B., Su, Y., Raams, A., et al. (2013). Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am. J. Hum. Genet. 92 (5), 800–806. doi:10.1016/J.AJHG.2013.04.002
Bogliolo, M., Schuster, B., Stoepker, C., Derkunt, B., Su, Y., Raams, A., et al. (2013). Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am. J. Hum. Genet. 92 (5), 800–806. doi:10.1016/J.AJHG.2013.04.002
Burgunder, J. M. (2022). Chorea: An update on genetics. Eur. Neurol. 85 (5), 342–348. doi:10.1159/000526237
Cordts, I., Önder, D., Traschütz, A., Kobeleva, X., Karin, I., Minnerop, M., et al. (2022). Adult-onset neurodegeneration in nucleotide excision repair disorders (NERDND): Time to move beyond the skin. Mov. Disord. 37 (8), 1707–1718. doi:10.1002/MDS.29071
Doi, H., Koyano, S., Miyatake, S., Nakajima, S., Nakazawa, Y., Kunii, M., et al. (2018). Cerebellar ataxia-dominant phenotype in patients with ERCC4 mutations. J. Hum. Genet. 63 (4), 417–423. doi:10.1038/S10038-017-0408-5
Jakubiuk-Tomaszuk, A., Murcia Pienkowski, V., Zietkiewicz, S., Rydzanicz, M., Kosinska, J., Stawinski, P., et al. (2019). Syndromic chorioretinal coloboma associated with heterozygous de novo RARA mutation affecting an amino acid critical for retinoic acid interaction. Clin. Genet. 96 (4), 371–375. doi:10.1111/CGE.13611
Kashiyama, K., Nakazawa, Y., Pilz, D. T., Guo, C., Shimada, M., Sasaki, K., et al. (2013). Malfunction of nuclease ERCC1-XPF results in diverse clinical manifestations and causes Cockayne syndrome, xeroderma pigmentosum, and Fanconi anemia. Am. J. Hum. Genet. 92 (5), 807–819. doi:10.1016/J.AJHG.2013.04.007
Mori, T., Yousefzadeh, M. J., Faridounnia, M., Chong, J. X., Hisama, F. M., Hudgins, L., et al. (2018). ERCC4 variants identified in a cohort of patients with segmental progeroid syndromes. Hum. Mutat. 39 (2), 255–265. doi:10.1002/HUMU.23367
Nabouli, I., Chikhaoui, A., Othman, H., Elouej, S., Jones, M., Lagarde, A., et al. (2021). Case report: Identification of novel variants in ERCC4 and DDB2 genes in two Tunisian patients with atypical xeroderma pigmentosum phenotype. Front. Genet. 12, 650639. doi:10.3389/FGENE.2021.650639
Shanbhag, N. M., Geschwind, M. D., DiGiovanna, J. J., Groden, C., Godfrey, R., Yousefzadeh, M. J., et al. (2018). Neurodegeneration as the presenting symptom in 2 adults with xeroderma pigmentosum complementation group F. Neurol. Genet. 4 (3), e240. doi:10.1212/NXG.0000000000000240
Spivak, G. (2015). Nucleotide excision repair in humans. DNA Repair (Amst) 36, 13–18. doi:10.1016/J.DNAREP.2015.09.003
Vives-Corrons, J. L., Krishnevskaya, E., Rodriguez, I. H., and Ancochea, A. (2021). Characterization of hereditary red blood cell membranopathies using combined targeted next-generation sequencing and osmotic gradient ektacytometry. Int. J. Hematol. 113 (2), 163–174. doi:10.1007/S12185-020-03010-9
Keywords: ERCC4, gene variant, chorea, ataxia, brain atrophy, NER (nucleotide excision repair)
Citation: Kulikowska J, Jakubiuk-Tomaszuk A, Rydzanicz M, Płoski R, Kochanowicz J, Kulakowska A and Kapica-Topczewska K (2023) Case report: Variants in the ERCC4 gene as a rare cause of cerebellar ataxia with chorea. Front. Genet. 14:1107460. doi: 10.3389/fgene.2023.1107460
Received: 24 November 2022; Accepted: 18 January 2023;
Published: 02 February 2023.
Edited by:
Generoso Andria, University of Naples Federico II, ItalyReviewed by:
Alessandro Vaisfeld, University of Bologna, ItalyCopyright © 2023 Kulikowska, Jakubiuk-Tomaszuk, Rydzanicz, Płoski, Kochanowicz, Kulakowska and Kapica-Topczewska. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joanna Kulikowska, am9hbm5hYWt1bGlrb3dza2FAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.