
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet. , 21 March 2023
Sec. Genetics of Common and Rare Diseases
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1098102
 Cristina Skrypnyk1,2*
Cristina Skrypnyk1,2* Aseel Ahmed Husain3
Aseel Ahmed Husain3 Hisham Y. Hassan4
Hisham Y. Hassan4 Jameel Ahmed5
Jameel Ahmed5 Abdulla Darwish6Latifa Almusalam6
Abdulla Darwish6Latifa Almusalam6 Noureddine Ben Khalaf7
Noureddine Ben Khalaf7 Fahad Al Qashar3
Fahad Al Qashar3Objective: Nemaline myopathies are a heterogeneous group of congenital myopathies caused by mutations in different genes associated with the structural and functional proteins of thin muscular filaments. Most patients have congenital onset characterized by hypotonia, respiratory issues, and abnormal deep tendon reflexes, which is a phenotype encountered in a wide spectrum of neuromuscular disorders. Whole-exome sequencing (WES) contributes to a faster diagnosis and facilitates genetic counseling.
Methods: Here, we report on two Arab patients from consanguineous families diagnosed with nemaline myopathy of different phenotype spectrum severities.
Results: Clinical assessment and particular prenatal history raised suspicion of neuromuscular disease. WES identified homozygous variants in NEB and KLHL40. Muscle biopsy and muscle magnetic resonance imaging studies linked the genetic testing results to the clinical phenotype. The novel variant in the NEB gene resulted in a classical type 2 nemaline myopathy, while the KLHL40 gene variant led to a severe phenotype of nemaline myopathy, type 8. Both patients were identified as having other gene variants with uncertain roles in their complex phenotypes.
Conclusions: This study enriches the phenotypic spectrum of nemaline myopathy caused by NEB and KLHL40 variants and highlights the importance of detailed prenatal, neonatal, and infancy assessments of muscular weakness associated with complex systemic features. Variants of uncertain significance in genes associated with nemaline myopathy may be correlated with the phenotype. Early, multidisciplinary intervention can improve the outcome in patients with mild forms of nemaline myopathies. WES is essential for clarifying complex clinical phenotypes encountered in patients from consanguineous families. Targeted carrier screening of extended family members would enable accurate genetic counseling and potential genetic prevention.
Nemaline myopathies (NMs) are a heterogeneous group of congenital myopathies caused by mutations in different genes associated with the structural and functional proteins of thin muscular filaments. The incidence of NM is estimated to be 1 in 30,000 births, with a carrier frequency of approximately 1/87 (Lehtokari et al., 2014). The spectrum of clinical phenotypes is very wide, ranging from severe neonatal forms to mild disorders in childhood (Sewry et al., 2019; Wang et al., 2020; Wen et al., 2020). NMs commonly show slow or no progression of symptoms, and only 16% of severe neonatal cases present with hypotonia, arthrogryposis, cardiomyopathy, and respiratory failure leading to early infant death (Cassandrini et al., 2017). Generalized weakness is part of the pediatric clinical phenotype of a wide spectrum of neuromuscular disorders and poses clinical diagnostic challenges. Whole-exome sequencing (WES) is now replacing more invasive procedures such as muscle and nerve biopsies and is helping clinicians reach a diagnosis faster (Mubaraki, 2021).
Nebulin is a giant protein of the skeletal sarcomeres that accounts for 4% of the total myofibrillar protein. Homozygous or compound heterozygous pathogenic variants in the nebulin gene (NEB) cause autosomal recessive nemaline myopathy type 2 (NEM2) (OMIM # 256030), which accounts for more than 50% of all NMs (Jin et al., 2014; Malfatti et al., 2014; Piga et al., 2016; Laitila et al., 2020; Laflamme et al., 2021). NEM2 appears in infancy with hypotonia and muscle weakness affecting the face, neck flexors, and proximal limb muscles. The most severe forms of NEM2 lead to death in the first 2 years of life (North and Ryan, 2002).
Nebulin and leiomodin 3, as striated skeletal muscle thin filaments, are stabilized by a kelch-like family member 40 protein detected in striated human fetal muscles, with the highest expression in adult skeletal muscles (Laitila and Wallgren-Pettersson, 2021). This protein maintains sarcomere integrity by modulating the regulatory circuits that control the expression of muscle-specific proteins (Cenik et al., 2015). Sarcomere dysfunction can cause secondary changes in the muscle’s ultrastructure, such as the nemaline rods, that further worsen muscle weaknesses (de Winter and Ottenheijm, 2017). Homozygous or compound heterozygous mutations in the kelch-like family member 40 gene (KLHL40) cause severe autosomal recessive nemaline myopathy type 8 (NEM8) (OMIM # 615348). Deficiencies in KLHL40 gene expression lead to marked reductions in nebulin and leiomodin 3 proteins, structural sarcomere irregularities, and a lethal loss of muscle function (Ravenscroft et al., 2013; Garg et al., 2014). KLHL40 variants are associated with severe forms of NMs that present with fetal akinesia or hypokinesia, contractures, fractures, respiratory failure, swallowing difficulties at birth, and early death in infancy (Kawase et al., 2015).
We report two rare cases of nemaline myopathy with different clinical severity spectrums caused by homozygous variants in NEB and KLHL40, in two consanguineous Arab families (ClinVar accessions SCV003762114--SCV003762117). Clinical and laboratory data were collected from their medical records and analyzed. An extended literature review of previously reported cases with NEB and KLHL40 gene variants was conducted. The research protocol was approved by the appropriate research ethics committees (approval numbers: E016-PI-6/21 and BDF/R&REC/2022-659), and written consent for the publication of both cases, including the use of photographs, was obtained from the parents.
Patient 1, a boy aged 7 years and 10 months is the first child of a healthy and consanguineous Arab couple (first-degree paternal cousins) (Figure 1). He was born by natural delivery at 40 gestational weeks after a gestation with polyhydramnios, decreased, and weak fetal movements. His birth weight was 2.8 kg (15th percentile). Bilateral generalized arthrogryposis with elbow and knee flexion contractures, right foot talipes equinovarus, left foot vertical talipes, inguinal hernia, bilateral cryptorchidism, and hypotonia were observed at birth (Figure 2A). A brain magnetic resonance imaging (MRI) scan performed at 6 months of age was normal. His infancy was marked by poor feeding, food-swallowing difficulties, and recurrent respiratory infections. An early intensive physiotherapy program reduced upper limb arthrogryposis. The lower limb arthrogryposis and talipes required serial casts and surgical interventions that allowed him to walk independently at the age of 20 months.

FIGURE 1. Family pedigree of patient 1. NEB-nebulin gene variant c. 11969A>C; CREBBP: cyclic adenosine monophosphate response element binding protein gene variant c.5600G>A; NPC2: NPC intracellular cholesterol transporter 2 gene variant c.420-422delCTG. Proband (P) IV1 (purple): patient 1 was diagnosed with nemaline myopathy type 2 (NEB gene homozygous c.11969A>C) and heterozygous for CREBBP variant c.5600G>A; IV6 (red) diagnosed with Niemann-Pick disease type C2 (NPC2 gene homozygous c.420-422delCTG). (IV-2 & IV-3)* are dizygotic twins through pre-implantation genetic testing for monogenic disorders (PGT-M).
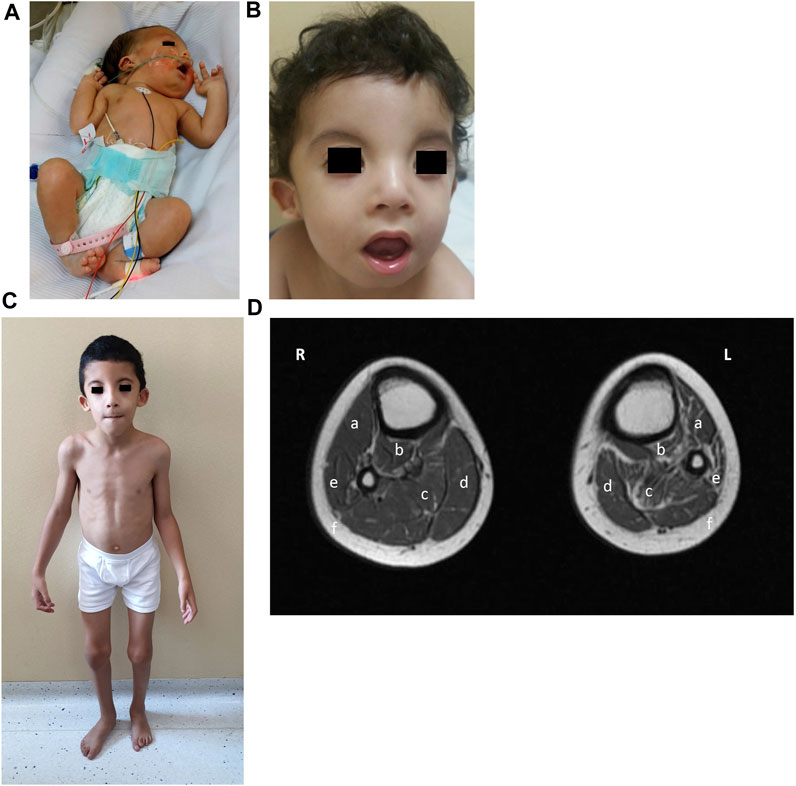
FIGURE 2. Phenotype of patient 1. (A) Clinical phenotype at the age of 2 days: bilateral generalized arthrogryposis with predominant bilateral elbow and knee flexion contractures, right foot talipes equinovarus, and left foot vertical talipes. (B) Clinical phenotype at the age of 18 months: wide forehead, bitemporal narrowing, hypertelorism, bilateral epicanthus, divergent strabismus, small depressed nasal bridge, small nose, short columella, anteverted nares, smooth and long filtrum, micrognathia, myopathic face appearance with facial muscle weakness, wide and opened mouth with an everted lower lip, high arched palate, small and anterior rotated ears with smooth helix and antihelix, and anterior and uplifted ear lobules. (C) Clinical phenotype at the age of 7 years and 10 months: myopathic facies with the same previous particularities, high-arched palate, overcrowded teeth, abnormal dental eruption, generalized thin build, hyperlordotic spine, mild elbow and knee contraction bilateral with hypotrophy of calf muscles, more marked on the left side, and overlapping second and third toes bilaterally. (D) MRI lower-limb (plain) (R-right, L-left): T1 axial of the calves showing mild atrophy of right gastrocnemius (c) and right soleus (d) muscles and subtle fatty infiltration; moderate atrophy of left anterior tibialis (a), posterior tibialis (b), gastrocnemius (c), and peroneal (e) muscles with prominent fatty infiltration; with a relative increase of the subcutaneous fat thickness in the left calf compared to the right one.
When seen in the genetic clinic at 18 months of age, the child’s growth parameters were: weight, 9 kg (3rd percentile); height, 81 cm (15th percentile), and head circumference, 46 cm (15th percentile). Particular dysmorphic craniofacial features were observed (Figure 2B), which included a wide forehead, bitemporal narrowing, hypertelorism, bilateral epicanthus, divergent strabismus, a small depressed nasal bridge, a small nose, a short columella, anteverted nares, a smooth and long philtrum, micrognathia, a myopathic facial appearance with facial muscle weakness, a wide and opened mouth with an everted lower lip, a high arched palate, small and anteriorly rotated ears with a smooth helix and antihelix, anterior and uplifted ear lobules, generalized muscle weakness, dropped shoulders, overlapping second and third toes bilaterally, dysphagia, generalized hypotonia, and motor and speech delays.
A Cytoscan HD SNP array (Affymetrix/Genome Diagnostics, Nijmegen, the Netherlands) reported a normal result of arr (1-22) x2, (XY) x1 but detected larger homozygous regions of 83.9 Mb (3% from the autosomal genome). The fukutin gene (FKTN) was suspected to be responsible for the phenotype, but its sequencing was normal. WES (targeted nucleotides covered ≥20X, 99.9%, Genome Diagnostics, Nijmegen, the Netherlands) reported a homozygous missense variant of uncertain significance in NEB, NM_001271208.1:c.11969A>C (p.(Asp3990Ala)), and a pathogenic heterozygous missense mutation in the cyclic adenosine monophosphate response element binding protein (CREBBP) gene, NM_004380.2:c.5600G>A (p.(Arg1867Gln). Both parents were confirmed as carriers of the NEB gene variant, while the CREBBP gene variant was identified in the healthy father.
Nemaline myopathy type 2 (NEM2) was considered as the diagnosis. A muscle biopsy was refused by the parents.
Patient 1 was fully able to speak by the age of 3 years and his muscle weakness improved under physiotherapy. His creatine kinase (CK) values also remained normal at serial re-evaluations. At the last visit to the genetic clinic at 7 years and 10 months of age (Figure 2C), his weight was 18 kg (1st percentile), his height was 117 cm (5th percentile), and his head circumference was 50 cm (5th percentile). The same particular myopathic facies with overcrowded teeth was noted again. His dysphagia had improved after intense occupational therapy. He showed winged scapulae bilaterally, a hyperlordotic spine, mild elbow and knee contraction bilaterally, and a generalized thin build with hypotrophy of the calf muscles, more marked on the left side. He was able to run but suffered frequent falls. Lower-limb muscle MRI performed at 7 years and 10 months of age revealed normal thigh muscles but moderate atrophy and fatty infiltration of the anterior, lateral, and posterior lower leg muscles. The left anterior and posterior tibialis, peroneal and gastrocnemius muscles showed predominant atrophy, with a relative increase in subcutaneous fat thickness (Figure 2D). Electrocardiography reported bradycardia, but a heart ultrasound was normal. Ophthalmology follow-up reported +3.5 D for both eyes, with divergent strabismus. He showed normal cognitive development and got good grades in school.
Patient 2 was a 10-week-old girl and the sixth child of a healthy, consanguineous Arab couple (first-degree paternal cousins) who had previously lost a pregnancy by second-trimester intrauterine fetal death and a child with severe congenital hypotonia and arthrogryposis by early neonatal death (Figure 3). The patient was born by natural delivery at 37 weeks of gestation after a pregnancy with significant polyhydramnios and decreased fetal movements. At birth her weight was 2.67 kg (30th percentile), her length was 47 cm (35th percentile), and her head circumference was 37 cm (99th percentile). She was apneic and bradycardic at birth and required active resuscitation, tracheal intubation, and neonatal intensive care unit admission. Her APGAR scores were 3, 4, and 5 points at 1, 5, and 10 min, respectively. On examination, her temperature was 36.3°C, her heart rate was 162 bpm, her respiratory rate was 52 breaths/min, and her oxygen saturation was 96% with FiO2 of 40%.
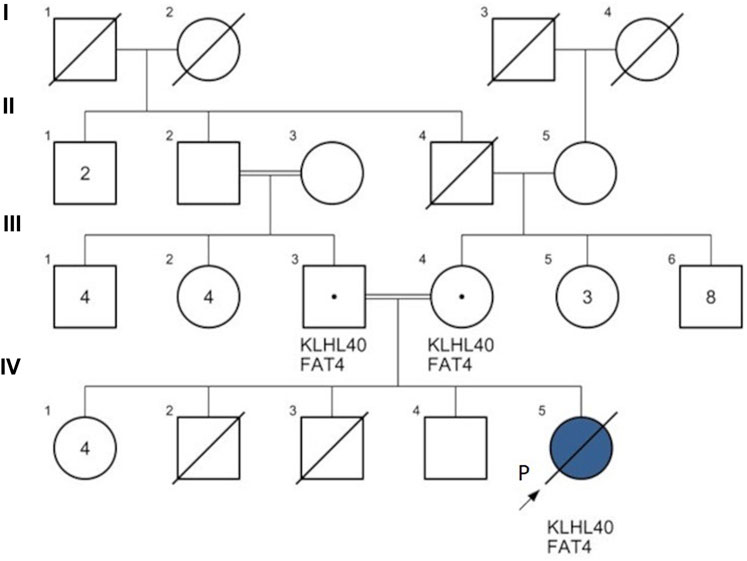
FIGURE 3. Family pedigree of patient 2. KLHL40: Kelch-like family member 40 gene variant c.1405G>A; FAT4: FAT atypical cadherin 4 gene variant c.13178A>T. Proband (P) IV5 (blue): patient 2 was diagnosed with nemaline myopathy type 8 (KLHL40 gene homozygous c.1405G>A), FAT4 gene homozygous c.13178A>T. The couple II-2 & II-3 are 2nd-degree relatives. IV-2: a full-term neonatal death after 24 h, unknown cause; IV-3: a 2nd-trimester intrauterine fetal death (IUFD) at 16 gestational weeks, unknown cause.
Physical examinations revealed severe hypotonia and diminished deep tendon reflexes (Figure 4A). Multiple congenital anomalies were observed, including craniofacial dysmorphism with narrowing of the bitemporal diameter, myopathic facies, exophthalmia, a bilateral sluggish pupillary reflex with bulbar muscle weakness, a bilateral epicanthal fold, a wide and depressed nasal bridge, a short nose, vertical nostrils, a long and smooth philtrum, thin lips, micrognathia, microtia, a short neck, a small chest and rib cage, arthrogryposis with joint stiffness of the upper and lower limbs, a right wrist with ulnar deviation, flexion of the left fingers, marked hip abduction, talipes equinovarus, right mid-shaft humeral and right proximal femoral dislocated non-communicated fractures (Figure 4B), and hypoplastic genitalia. The baby remained ventilator-dependent because of severe respiratory failure secondary to respiratory muscle weakness. Feeding was done via a nasogastric tube as there was no sucking reflex.
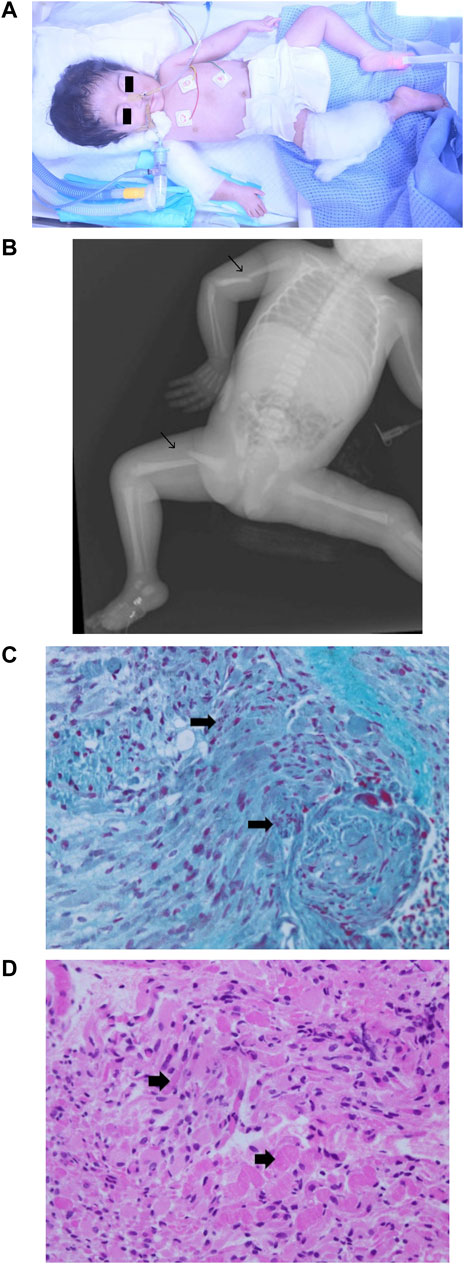
FIGURE 4. Phenotype of patient 2. (A) Clinical phenotype at the age of 1 day: craniofacial dysmorphism with narrowing of the bitemporal diameter, myopathic facies, exophthalmia, bilateral epicanthal fold, wide and depressed nasal bridge, short nose, vertical nostrils, long smooth philtrum, thin lips, micrognathia, microtia, short neck, small chest and rib cage, arthrogryposis with joint stiffness of the upper and lower limbs, right wrist with ulnar deviation, flexion of the left fingers, marked hip abduction, and talipes equinovarus. (B) X-ray skeletal survey: right mid-shaft humeral and right proximal femoral dislocated non-communicated fractures. (C) Muscle biopsy (left thigh) hematoxylin and eosin staining 40X: variation in muscle fiber size with degenerative changes. (D) Muscle biopsy (left thigh) Gomori trichrome stain 40X: degenerative muscle fibers containing red to purple rod inclusions in sarcoplasm.
The karyotype analysis was normal (46,XX). Other investigations also revealed normal levels of the muscle enzymes (CK, 266 IU/L; CK-MB, 29.4 IU/L) and normal muscle-specific receptor tyrosine kinase levels. At this point, a rare myopathy was suspected. WES (CentoXome Solo NGS-based CNV, targeted nucleotides covered ≥20X, 99.09%, Centogene, Rostock, Germany) reported two different variants of uncertain significance: a KLHL40 homozygous missense variant, NM_152393.3:c.1405G>A, p.(Gly469Ser), and a fat atypical cadherin 4 (FAT4) gene homozygous missense variant, NM_001291303.1:c.13178A>T, p.(Asp4393Val). The parents were confirmed to be double carriers for both genes variants.
Variations in muscle fiber size with degenerative changes were visualized after hematoxylin and eosin staining of a skeletal muscle biopsy sample (Figure 4C). A high-power microscopic view after Gomori trichrome staining revealed red to purple rod inclusions in the sarcoplasm of degenerative muscle fibers (Figure 4D), linking the molecular genetic result to the muscular pathology features of NMs. The patient was diagnosed with nemaline myopathy type 8 (NEM8), developed severe respiratory failure, and passed away at the age of 10 weeks.
NMs usually present as early-onset proximal muscle weakness, and many cases are suspected prenatally. Prenatal ultrasonographic signs of NMs correlate with a poor prognosis and an increased risk of mortality. Fetal akinesia, polyhydramnios, arthrogryposis, and foot deformities are associated with NEB and KLHL40 gene variants (Lawlor, et al., 2011; Feingold-Zadok et al., 2017; Yeung et al., 2020; Yi et al., 2021). Both patients showed reduced fetal movement, polyhydramnios, neonatal arthrogryposis, and talipes, underlining again the importance of ultrasound imaging for the early detection of NMs. The earlier a rare myopathy is diagnosed, the easier it will be to quickly intervene and manage the condition, identify the recurrence risk, and initiate prevention.
The NEB gene variant, at exon 80, NM_001271208.1:c.11969A>C (p.(Asp3990Ala)) identified in patient 1 was assessed as a variant of unknown significance that led to the substitution of a strongly conserved nucleotide and amino acid residue. Nebulin has a fundamental role in skeletal muscle and variants affecting highly conserved regions of NEB may be regarded as pathogenic. To our knowledge, this specific variant has not been reported before in the literature or the Genome Aggregation Database (gnomAD) (https://gnomad.broadinstitute.org/); it is also not listed in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) or Varsome (https://varsome.com/variant/hg38/NEB). It may have a destabilizing effect on the protein, as predicted by Polyphen 2.0, leading to protein dysfunction. This variant is predicted as being deleterious by Combined Annotation Dependent Depletion (CADD), with a scaled score of 28.7 points (https://cadd.gs.washington.edu/snv). Considering the muscular phenotype of polyhydramnios, fetal hypokinesia, arthrogryposis, myopathic facies, neck flexor weakness, a nasal voice, dysarthria, dysphagia, and muscle weakness, we concluded that this homozygous missense NEB gene mutation was a likely candidate for the disease-causing variant seen in patient 1.
Classifying patients early based on their clinical phenotype is challenging, but disease progression during childhood enables the severity of their condition to be clarified (Lehtokari et al., 2014). At the age of 7 years and 10 months, patient 1 had good motor milestones and a slowly progressive, more distal lower limb myopathy corresponding to the classical NEM2 type. Lower-leg MRI showed findings already reported in this NEM2 type (Jungbluth et al., 2004; Gurgel-Giannetti et al., 2022); notably, the myopathy had completely spared the boy’s thigh muscles and predominantly led to atrophy of the left posterior and lateral calf muscles.
Nebulin is also present in the heart and brain, and cardiomyopathy is reported in about 13% of NEM2 patients (Amburgey et al., 2021). Electrocardiography showed bradycardia, but the cardiac ultrasound was reported to be normal for patient 1.
More than 200 NEB pathogenic variants are associated with a wide spectrum of variable phenotypes, from severe to mild forms, with associated learning and intellectual disabilities (variants in exons 27 and 143) (Jin et al., 2014; Lehtokari et al., 2014; Amburgey et al., 2021). No patients have been reported to have variants in exon 80 of the NEB gene. Our patient had a speech delay and mild learning difficulties as a toddler but experienced normal mental development and a slower myopathy progression.
Patient 1 was included in an intensive rehabilitation program and regularly followed up by a multidisciplinary medical team. He has benefited from physiotherapy and speech and behavioral therapies. His good evolution underlines the importance of both genetic diagnosis and early, sustained intervention.
His parents were confirmed carriers of the same NEB variant, and genetic counseling helped them to understand their recurrence risk and to make informed reproductive decisions. They opted for in vitro fertilization (IVF) and preimplantation genetic testing for the NEB gene variant (PGT-M), and their next pregnancy ended at term with two healthy male dizygotic twins. Multiple relatives later requested preconception carrier genetic testing, including for other rare genetic disorders reported in their extended consanguineous family, such as Niemann-Pick disease type 2C (Figure 1).
The KLHL40 gene variant, at exon 3, NM_152393.3:c.1405G>A (p.(Gly469Ser)), identified in our patient 2 led to the substitution of a conserved amino acid residue. This variant was reported before in ClinVar and Varsome as having uncertain significance for NEM8. It was assessed as likely pathological by ACMG, as probably damaging by the in silico prediction tool Polyphen 2.0 (https://genetics.bwh.harvard.edu/pph2/) with a score of 0.996 points (sensitivity: 0.55; specificity: 0.98), as disease-causing by Align GVGD (http://agvgd.hci.utah.edu/) and Mutation Taster (www.mutationtaster.org), and has a deleterious scaled CADD score of 31 points. The same variant was reported as a compound heterozygote with a KLHL40 gene variant c. 1498C>T in two NEM8 patients from Brazil, 17 months and 5 years of age, respectively, with a severe neonatal phenotype but later motor improvement (patients 29 and 30 in the study by Gurgel-Giannetti et al., 2022).
A similar KLHL40 variant appearing at the same position in exon 3, in a conserved functional domain, but with a different substitution, c.1405G>T, p(Gly469Cys), was found in a Japanese family with NEM8 (family 15 in the study by Ravenscroft et al. (2013)). Molecular modeling assessed this substitution as one that most likely destabilized the hydrophobic core of the kelch domain and reduced protein stability. It is highly probable that the KLHL40 gene variant, c.1405G>A, identified in our patient had the same impact and, to the best of our knowledge, this is the first time it has been reported in a family of Arab origin. The KLHL40 gene variant, c.1582G>A, is the common variant reported in Japanese, Kurdish, and Turkish patients with NEM8 (Seferian et al., 2016) but the variant, c.1516A>C, has only been reported in East Asian populations (Yeung et al., 2020). Considering the rarity of these cases, further studies might be needed to identify KLHL40 gene variants linked to NEM8 in the Arab population.
NEM8 is characterized by severe and distinctive fetal and neonatal features, and by an early death in the first months of life (Ravenscroft et al., 2013), with rare cases surviving into adolescence (Seferian et al., 2016). As previously reported (Kawase et al., 2015; Yi et al., 2021), our patient 2, also had fetal hypokinesia and polyhydramnios, a severe phenotype at birth with respiratory failure, long bone fractures, arthrogryposis, and hypotonia. Despite the early interventions, she passed away at the age of 10 weeks. The severity of patient 2’s clinical phenotype in association with the abnormal myofibrils and rod bodies identified by the muscular biopsy, supported the molecular findings and certified the diagnosis of a rare severe case of NEM8. When accepted by the family, a muscle biopsy can clarify the gene variant's impact on the structure and functionality of muscle fibers (Wang et al., 2020). The homozygous KLHL40 gene variant, c.1405G>A, may explain the intrauterine fetal death and the other neonatal fetal death previously recorded in this family.
A heterozygous missense variant was identified in exon 31 of the CREBBP gene, NM_004380.2:c.5600G>A (p.(Arg1867Gln)), in patient 1. This variant is classified as pathogenic by the American College of Medical Genetics and Genomics (ACMG), with a score of 17 points (https://varsome.com/variant/hg38/rs1131691326) and has a deleterious scaled CADD score of 28.9 points. Variants in exons 30 and 31 of the CREBBP gene are associated with Menke-Hennekam syndrome 1 (https://www.omim.org/entry/618332) (Menke et al., 2016; Menke et al., 2018). Two patients with craniofacial dysmorphism, intellectual disability, and the same CREBBP variant c.5600G>A, were previously reported, including a six-year-old girl [patient 8 in the study by Menke et al. (2016)] and a 57-year-old man [patient C16 in the study by Menke et al. (2018)]. Both patients had other genomic changes (del6p12.3 and dup9q34.3, respectively) with unclear influences on their phenotype. Some of their facial dysmorphic features were assessed as possibly associated with this CREBBP gene variant, including a depressed nasal bridge, a short nose, anteverted nares, a short columella, a long philtrum, and decreased elbow extension, which were present in our patient but were also reported previously in the NEM2 phenotype. Patient 1 had a speech delay but not a developmental delay. Segregation analysis studies were conducted on the parents and other relatives (their siblings and cousins). The patient’s father has the same CREBBP variant, but none of the signs reported in the literature; therefore, it remains uncertain whether this is a case of non-penetrance and whether this variant has any influence on our patient’s phenotype.
Patient 2 was reported to also be homozygous for a missense variant of uncertain significance in the FAT4 gene, c.13178A>T, (p.(Asp4393Val)), which was not reported in gnomAD but has been predicted as damaging by the in silico tool, Mutation Taster. Pathogenic variants in the FAT4 gene are associated with autosomal recessive Van Maldergem syndrome type 2, and Hennekam lymphangiectasia-lymphedema syndrome type 2. Hypotonia, respiratory difficulties, and talipes are also features of Van Maldergem syndrome. A typically periventricular nodular heterotopia linked to this syndrome could not be ruled out; therefore, the impact of this variant on our patient’s phenotype, if any, is uncertain.
The professional care and guidance received led to an excellent collaboration with the families of both patients. Genetic counseling facilitated an understanding of complex genetic pathologies, clarifying the impact of consanguinity and the importance of prevention. The counseling helped both families to cope with this rare disease diagnosis and to make informed decisions going forward, regarding adequate management and future offspring. Getting another healthy pregnancy with the help of IVF and PGT-M for the NEB gene variant brought a lot of joy to the first family, and both families have become strong advocates of the importance of preconception genetic counseling in the context of consanguinity and a history of affected children.
Homozygous variants of the NEB2 gene, c.11969A>C, and the KLHL40 gene, c.1405G>A, were correlated with the phenotypes of the two patients presented here. This study enriches our knowledge about the phenotypic spectrum of nemaline myopathy caused by variants in NEB and KLHL40 and highlights the importance of detailed prenatal, neonatal, and infancy assessments of muscular weakness associated with complex systemic features. Variants of uncertain significance in genes associated with nemaline myopathies may also contribute to the phenotype. Early and multidisciplinary intervention can improve the outcome, especially in patients with mild forms of the disease. WES is essential for clarifying complex clinical phenotypes encountered in patients from consanguineous families. Targeted carrier screening of extended family members facilitates accurate genetic counseling and enables genetic prevention.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
The studies involving human participants were reviewed and approved by the Ethics Committee of Arabian Gulf University (E016-PI-6/21) and of Bahrain Defence Force Hospital, Royal Medical Services (BDF/R&REC/2022-659). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
CS and FA designed the project and oversaw clinical data collection, CS, HYH, and NBK interpreted the genetic testing results, JA interpreted the MRI data, LA and AD performed the muscle biopsy analysis, CS, AAH, and FA conducted the bibliography search, and all authors contributed to the manuscript preparation.
We thank the teams from Genome Diagnostics Nijmegen, the Netherlands, and Centogene, Rostock, Germany, for performing the genetic testing; we are pleased to thank both families for their generous collaboration.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
NMs, nemaline myopathies; NEM2, nemaline myopathy type2; NEM8, nemaline myopathy type 8.
Amburgey, K., Acker, M., Saeed, S., Amin, R., Beggs, A. H., Bönnemann, C. G., et al. (2021). A cross-sectional study of nemaline myopathy. Neurology 96 (10), e1425–e1436. doi:10.1212/WNL.0000000000011458
Cassandrini, D., Trovato, R., Rubegni, A., Lenzi, S., Fiorillo, C., Baldacci, J., et al. (2017). Congenital myopathies: Clinical phenotypes and new diagnostic tools. Ital. J. Pediatr. 43 (1), 101. doi:10.1186/s13052-017-0419-z
Cenik, B. K., Garg, A., McAnally, J. R., Shelton, J. M., Richardson, J. A., Bassel-Duby, R., et al. (2015). Severe myopathy in mice lacking the MEF2/SRF-dependent gene leiomodin-3. J. Clin. Invest. 125 (4), 1569–1578. doi:10.1172/JCI80115
de Winter, J. M., and Ottenheijm, C. A. C. (2017). Sarcomere dysfunction in nemaline myopathy. J. Neuromuscul. Dis. 4 (2), 99–113. doi:10.3233/JND-160200
Feingold-Zadok, M., Chitayat, D., Chong, K., Injeyan, M., Shannon, P., Chapmann, D., et al. (2017). Mutations in the NEB gene cause fetal akinesia/arthrogryposis multiplex congenita. Prenat. Diagn 37 (2), 144–150. doi:10.1002/pd.4977
Garg, A., O'Rourke, J., Long, C., Doering, J., Ravenscroft, G., Bezprozvannaya, S., et al. (2014). KLHL40 deficiency destabilizes thin filament proteins and promotes nemaline myopathy. J. Clin. Invest. 124 (8), 3529–3539. doi:10.1172/JCI74994
Gurgel-Giannetti, J., Souza, L. S., Yamamoto, G. L., Belisario, M., Lazar, M., Campos, W., et al. (2022). Nemaline myopathy in Brazilian patients: Molecular and clinical characterization. Int. J. Mol. Sci. 23 (19), 11995. doi:10.3390/ijms231911995
Jin, H. S., Lee, J. B., Kim, K., Lee, K. Y., Choi, V. N., Kim, J. S., et al. (2014). Identification of the rare compound heterozygous variants in the NEB gene in a Korean family with intellectual disability, epilepsy and early-childhood-onset generalized muscle weakness. J. Hum. Genet. 59 (12), 643–647. doi:10.1038/jhg.2014.87
Jungbluth, H., Sewry, C. A., Counsell, S., Allsop, J., Chattopadhyay, A., Mercuri, E., et al. (2004). Magnetic resonance imaging of muscle in nemaline myopathy. Neuromuscul. Disord. 14 (12), 779–784. doi:10.1016/j.nmd.2004.08.005
Kawase, K., Nishino, I., Sugimoto, M., Togawa, T., Sugiura, T., Kouwaki, M., et al. (2015). Nemaline myopathy with KLHL40 mutation presenting as congenital totally locked-in state. Brain Dev. 37 (9), 887–890. doi:10.1016/j.braindev.2015.02.002
Laflamme, N., Lace, B., Thonta Setty, S., Rioux, N., Labrie, Y., Droit, A., et al. (2021). A homozygous deep intronic mutation alters the splicing of nebulin gene in a patient with nemaline myopathy. Front. Neurol. 12, 660113. doi:10.3389/fneur.2021.660113
Laitila, J., and Wallgren-Pettersson, C. (2021). Recent advances in nemaline myopathy. Neuromuscul. Disord. 31 (10), 955–967. doi:10.1016/j.nmd.2021.07.012
Laitila, J. M., McNamara, E. L., Wingate, C. D., Goullee, H., Ross, J. A., Taylor, R. L., et al. (2020). Nebulin nemaline myopathy recapitulated in a compound heterozygous mouse model with both a missense and a nonsense mutation in Neb. Acta Neuropathol. Commun. 8 (1), 18. doi:10.1186/s40478-020-0893-1
Lawlor, M. W., Ottenheijm, C. A., Lehtokari, V. L., Cho, K., Pelin, K., Wallgren-Pettersson, C., et al. (2011). Novel mutations in NEB cause abnormal nebulin expression and markedly impaired muscle force generation in severe nemaline myopathy. Skelet. Muscle 1 (1), 23. doi:10.1186/2044-5040-1-23
Lehtokari, V. L., Kiiski, K., Sandaradura, S. A., Laporte, J., Repo, P., Frey, J. A., et al. (2014). Mutation update: The spectra of nebulin variants and associated myopathies. Hum. Mutat. 35 (12), 1418–1426. doi:10.1002/humu.22693
Malfatti, E., Lehtokari, V. L., Böhm, J., De Winter, J. M., Schäffer, U., Estournet, B., et al. (2014). Muscle histopathology in nebulin-related nemaline myopathy: Ultrastrastructural findings correlated to disease severity and genotype. Acta Neuropathol. Commun. 2, 44. doi:10.1186/2051-5960-2-44
Menke, L. A., Gardeitchik, T., Hammond, P., Heimdal, K. R., Houge, G., Hufnagel, S. B., et al. (2018). Further delineation of an entity caused by CREBBP and EP300 mutations but not resembling Rubinstein-Taybi syndrome. Am. J. Med. Genet. A 176 (4), 862–876. doi:10.1002/ajmg.a.38626
Menke, L. A., van Belzen, M. J., Alders, M., Cristofoli, F., Ehmke, N., Fergelot, P., et al. (2016). CREBBP mutations in individuals without Rubinstein-Taybi syndrome phenotype. Am. J. Med. Genet. A 170 (10), 2681–2693. doi:10.1002/ajmg.a.37800
Mubaraki, A. A. (2021). Nemaline myopathy: A case report. Case Rep. Neurol. 13 (2), 499–503. doi:10.1159/000517898
North, K. N., and Ryan, M. M. (2002). “Nemaline myopathy – retired chapter, for historical reference only,” in GeneReviews® [internet]. Editors M. P. Adam, D. B. Everman, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, L. J. H. Beanet al. (Seattle (WA): University of Washington, Seattle), 1993–2022. [updated 2015 Jun 11].
Piga, D., Magri, F., Ronchi, D., Corti, S., Cassandrini, D., Mercuri, E., et al. (2016). New mutations in NEB gene discovered by targeted next-generation sequencing in nemaline myopathy Italian patients. J. Mol. Neurosci. 59 (3), 351–359. doi:10.1007/s12031-016-0739-2
Ravenscroft, G., Miyatake, S., Lehtokari, V. L., Todd, E. J., Vornanen, P., Yau, K. S., et al. (2013). Mutations in KLHL40 are a frequent cause of severe autosomal-recessive nemaline myopathy. Am. J. Hum. Genet. 93 (1), 6–18. doi:10.1016/j.ajhg.2013.05.004
Seferian, A. M., Malfatti, E., Bosson, C., Pelletier, L., Taytard, J., Forin, V., et al. (2016). Mild clinical presentation in KLHL40-related nemaline myopathy (NEM 8). Neuromuscul. Disord. 26 (10), 712–716. doi:10.1016/j.nmd.2016.07.011
Sewry, C. A., Laitila, J. M., and Wallgren-Pettersson, C. (2019). Nemaline myopathies: A current view. J. Muscle Res. Cell. Motil. 40 (2), 111–126. doi:10.1007/s10974-019-09519-9
Wang, Q., Hu, Z., Chang, X., Yu, M., Xie, Z., Lv, H., et al. (2020). Mutational and clinical spectrum in a cohort of Chinese patients with hereditary nemaline myopathy. Clin. Genet. 97 (6), 878–889. doi:10.1111/cge.13745
Wen, Q., Chang, X., and Guo, J. (2020). A childhood-onset nemaline myopathy caused by novel heterozygote variants in the nebulin gene with literature review. Acta Neurol. Belg 120 (6), 1351–1360. doi:10.1007/s13760-019-01230-3
Yeung, K. S., Yu, F. N. Y., Fung, C. W., Wong, S., Lee, H. H. C., Fung, S. T. H., et al. (2020). The KLHL40 c.1516A>C is a Chinese-specific founder mutation causing nemaline myopathy 8: Report of six patients with pre- and postnatal phenotypes. Mol. Genet. Genomic Med. 8 (7), e1229. doi:10.1002/mgg3.1229
Keywords: congenital, myopathy, nemaline, WES, NEB, KLHL40
Citation: Skrypnyk C, Husain AA, Hassan HY, Ahmed J, Darwish A, Almusalam L, Ben Khalaf N and Al Qashar F (2023) Case report: Homozygous variants of NEB and KLHL40 in two Arab patients with nemaline myopathy. Front. Genet. 14:1098102. doi: 10.3389/fgene.2023.1098102
Received: 14 November 2022; Accepted: 27 February 2023;
Published: 21 March 2023.
Edited by:
Fan Jin, Zhejiang University, ChinaReviewed by:
Baiba Lace, Centre hospitalier de l’Université Laval, CanadaCopyright © 2023 Skrypnyk, Husain, Hassan, Ahmed, Darwish, Almusalam, Ben Khalaf and Al Qashar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristina Skrypnyk, Y3Jpc3RpbmFzQGFndS5lZHUuYmg=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.