 Chulaluk Kuptanon1†
Chulaluk Kuptanon1† Marie Morimoto2†
Marie Morimoto2† Elena-Raluca Nicoli2
Elena-Raluca Nicoli2 Joshi Stephen1David S. Yarnell1Heidi Dorward1
Joshi Stephen1David S. Yarnell1Heidi Dorward1 William Owen3
William Owen3 Suhag Parikh4Namik Yasar Ozbek5Baris Malbora6Carla Ciccone1Meral Gunay-Aygun1
Suhag Parikh4Namik Yasar Ozbek5Baris Malbora6Carla Ciccone1Meral Gunay-Aygun1 William A. Gahl1,2Wendy J. Introne1,7
William A. Gahl1,2Wendy J. Introne1,7 May Christine V. Malicdan1,2*
May Christine V. Malicdan1,2*- 1Human Biochemical Genetics Section, Medical Genetics Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD, United States
- 2National Institutes of Health Undiagnosed Diseases Program, National Institutes of Health Common Fund, Office of the Director, National Institutes of Health, Bethesda, MD, United States
- 3Children’s Hospital of The King’s Daughters, Norfolk, VA, United States
- 4Department of Pediatrics, School of Medicine, Emory University, Atlanta, GA, United States
- 5Division of Pediatric Hematology and Oncology, University of Yeni Yuzyil, Gaziosmanpasa Hospital, Istanbul, Türkiye
- 6Department of Pediatric Hematology/Oncology, Ankara City Hospital, The University of Health Sciences, Ankara, Türkiye
- 7Office of the Clinical Director, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD, United States
Introduction: Chediak-Higashi syndrome (CHS) is rare autosomal recessive disorder caused by bi-allelic variants in the Lysosomal Trafficking Regulator (LYST) gene. Diagnosis is established by the detection of pathogenic variants in LYST in combination with clinical evidence of disease. Conventional molecular genetic testing of LYST by genomic DNA (gDNA) Sanger sequencing detects the majority of pathogenic variants, but some remain undetected for several individuals clinically diagnosed with CHS. In this study, cDNA Sanger sequencing was pursued as a complementary method to identify variant alleles that are undetected by gDNA Sanger sequencing and to increase molecular diagnostic yield.
Methods: Six unrelated individuals with CHS were clinically evaluated and included in this study. gDNA Sanger sequencing and cDNA Sanger sequencing were performed to identify pathogenic LYST variants.
Results: Ten novel LYST alleles were identified, including eight nonsense or frameshift variants and two in-frame deletions. Six of these were identified by conventional gDNA Sanger sequencing; cDNA Sanger sequencing was required to identify the remaining variant alleles.
Conclusion: By utilizing cDNA sequencing as a complementary technique to identify LYST variants, a complete molecular diagnosis was obtained for all six CHS patients. In this small CHS cohort, the molecular diagnostic yield was increased, and canonical splice site variants identified from gDNA Sanger sequencing were validated by cDNA sequencing. The identification of novel LYST alleles will aid in diagnosing patients and these molecular diagnoses will also lead to genetic counseling, access to services and treatments and clinical trials in the future.
1 Introduction
Chediak-Higashi syndrome (CHS, OMIM 214500) is a rare autosomal recessive disorder characterized by partial oculocutaneous albinism, bleeding diathesis, recurrent infections, an increased risk of developing the hyperinflammatory condition called hemophagocytic lymphohistiocytosis (HLH), and neurologic impairment (Béguez César, 1943; Chediak, 1952; Higashi, 1954; Introne et al., 1999; Toro et al., 2009; Shirazi et al., 2019; Sharma et al., 2020). The presence of giant intracytoplasmic granules in leukocytes on peripheral blood smear is pathognomonic for CHS and establishes a clinical diagnosis (Toro et al., 2009). The disorder exhibits high variable expressivity, ranging from early onset classical CHS to later onset atypical CHS; those with atypical CHS often have subtle clinical features and are likely underdiagnosed (Weisfeld-Adams et al., 2013; Introne et al., 2016). Current treatment includes chemotherapy and continuation therapy for the HLH, followed by allogenic hematopoietic stem cell transplantation (HSCT) to treat the hematological and immunological dysfunction (Eapen et al., 2007; Toro et al., 2009; Umeda et al., 2016), and levodopa for patients who develop parkinsonism (Hauser et al., 2000; Toro et al., 2009; Bhambhani et al., 2013).
CHS has significant clinical overlap with several other genetic disorders, emphasizing the importance of a molecular diagnosis. Other disorders that include gray or silvery hair as observed in CHS include Griscelli syndrome (MIM 214450, MIM 607624, MIM 609227), Elejalde neuroectodermal melanolysosomal syndrome (MIM 256710), and oculocerebral syndrome with hypopigmentation (MIM 257800) (Gironi et al., 2019). There are also other disorders that have phenotypic overlap with one clinical aspect of CHS such as familial HLH or oculocutaneous albinism. Additionally, there are other multisystemic disorders with phenotypic overlap with several clinical features of CHS, such as Vici syndrome (MIM 242840) characterized by hypopigmentation, combined immunodeficiency, cardiomyopathy, cataracts, corpus callosum agenesis, microcephaly, and failure to thrive (Dafsari et al., 1993), and Hermansky-Pudlak syndrome (MIM 203300, MIM 608233, MIM 614072, MIM 614073, MIM 614074, MIM 614075, MIM 614076, MIM 614077, MIM 614171, MIM 617050, and MIM 619172) characterized by oculocutaneous albinism, a bleeding diathesis, neutropenia, granulomatous colitis, and pulmonary fibrosis (Huizing et al., 1993). These disorders can be distinguished from CHS due to differences in phenotypic presentation, the absence of giant intracytoplasmic granules in leukocytes, and distinct molecular causes.
The Lysosomal Trafficking Regulator (LYST) gene is the only known molecular cause of CHS and bi-allelic pathogenic variants in LYST establish a molecular diagnosis (Barbosa et al., 1996; Nagle et al., 1996; Toro et al., 2009). LYST has 53 exons within a 13,466 bp transcript spanning approximately 222 kb of genomic DNA and encodes a 429 kDa putative lysosomal trafficking protein. The key challenges to establishing a molecular diagnosis for individuals with CHS include the large size of LYST that can influence interpretation of variant pathogenicity, and the paucity of common pathogenic variants. Conventional molecular genetic testing of LYST by Sanger sequencing of genomic DNA (gDNA) detects the majority of pathogenic variants, but for some individuals clinically diagnosed with CHS, pathogenic variants remain undetected, likely due to large-scale indels and deep intronic variants or synonymous exonic variants that lead to aberrant splicing. Uniparental disomy leading to the inheritance of two copies of a pathogenic LYST variant from one parent has also been reported as a molecular mechanism for CHS (Dufourcq-Lagelouse et al., 1999; Manoli et al., 2010; Boluda-Navarro et al., 2021).
Several strategies have been used to sequence LYST. These strategies include conventional gDNA Sanger sequencing (Toro et al., 2009), cDNA Sanger sequencing (Boluda-Navarro et al., 2021), the protein truncation test followed by cDNA sequencing to screen for nonsense and frameshift variants and to identify substitutions and small indels in LYST (Certain et al., 2000), as well as next-generation sequencing approaches such as exome sequencing and genome sequencing, particularly if other candidate loci are being considered as part of the differential diagnosis (Jin et al., 2017).
In this study, we aimed to provide a complete molecular diagnosis for six individuals with CHS by utilizing both conventional gDNA Sanger sequencing and cDNA Sanger sequencing. cDNA Sanger sequencing was chosen as a complementary method of variant detection since patient LYST-expressing dermal fibroblasts were available and because this method has been shown to effectively detect variant alleles that may not be readily detectable by conventional gDNA Sanger sequencing (Certain et al., 2000; Boluda-Navarro et al., 2021). We hypothesize that this strategy will aid in the identification of variants in this technically challenging gene to better molecularly diagnose individuals with CHS in the future.
2 Materials and methods
2.1 Patients and clinical evaluation
Six patients clinically diagnosed with classical or atypical CHS were included in this study. These patients were enrolled over the course of 10–20 years in the Investigation of Chediak-Higashi Syndrome and Related Disorders protocol 00-HG-0153 (NCT00005917, clinicaltrials.gov) or the Diagnosis and Treatment of Patients with Inborn Errors of Metabolism or Other Genetic Disorders protocol 76-HG-0238 (NCT00369421, clinicaltrials.gov) approved by the National Human Genome Research Institute’s Institutional Review Board. Written informed consent was obtained prior to the study. Clinical evaluation included collecting the medical and family history, physical examination, complete blood count (CBC) blood test. Peripheral blood smear analysis and microscopic hair examination were used to assess the presence of giant inclusions in leukocytes and pigment clumping in hair shafts, pathognomonic features of CHS.
2.2 Primary cell culture
Primary dermal fibroblasts or melanocytes from CHS patients were cultured from forearm skin biopsies using standard protocols. Further details are presented in the Supplementary Data. Unaffected control primary fibroblasts and melanocytes were obtained from Coriell Institute for Medical Research and American Type Culture Collection. Details of the unaffected control primary cells used in the study are noted in Supplementary Table S1.
2.3 Immunofluorescence microscopy analysis
Indirect immunofluorescence detection of the late endosomal and lysosomal membrane marker LAMP3/CD63 was performed in cultured fibroblasts and melanocytes as previously described (Yarnell et al., 2020). Further details are presented in the Supplementary Data.
2.4 Gene expression analysis
Total RNA was extracted from cultured primary dermal fibroblasts or melanocytes using the RNeasy Mini Kit (Qiagen). Genomic DNA was removed by on-column DNase I digestion (Qiagen) during total RNA extraction. Reverse transcription was performed using the Omniscript Reverse Transcription Kit (Qiagen) and Oligo (dT)23 primers (O4387, Sigma-Aldrich). Quantitative PCR was performed using TaqMan gene expression assays according to the manufacturer’s specifications. Further details are presented in the Supplementary Data.
2.5 Genomic DNA Sanger sequencing
Genomic DNA (gDNA) was extracted from peripheral blood or cultured primary dermal fibroblasts using standard procedures. gDNA from cultured primary fibroblasts was used if the gDNA from peripheral blood was no longer available. Regions of the LYST (NM_000081.3) coding sequence (exons 3–53), including intron-exon boundaries, were amplified by PCR using the primers listed in Supplementary Table S2. The size and specificity of the PCR products were confirmed by agarose gel electrophoresis. Sanger sequencing was performed using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) and separated on the 3130xl Genetic Analyzer (Applied Biosystems). Chromatograms were analyzed using Sequencher v5.0 software (Gene Codes Corporation).
2.6 cDNA Sanger sequencing
Total RNA was extracted from cultured primary dermal fibroblasts or melanocytes using the RNeasy Mini Kit (Qiagen). cDNA was synthesized by reverse transcription with the High-Capacity RNA-to-cDNA Kit (Applied Biosystems). Regions of the LYST coding sequence (exons 3–53) were amplified by PCR using the primers listed in Supplementary Table S3. PCR amplicons were generated and confirmed by agarose gel electrophoresis prior to Sanger sequencing as described above.
2.7 Molecular cloning
PCR products were cloned into the pCR4-TOPO TA vector using the TOPO TA Cloning Kit (Invitrogen). Single clones were Sanger sequenced to individually analyze the cDNA alleles for which chromatograms obtained from the initial cDNA Sanger sequencing were difficult to interpret.
2.8 ACMG-AMP classification
Variant pathogenicity classification was performed according to the standards and guidelines presented by the ACMG-AMP and the ACGS (Richards et al., 2015; Ellard et al., 2020). Further details are presented in the Supplementary Data.
2.9 Statistical analysis
Statistics were performed using GraphPad Prism 8 version 8.4.3 (GraphPad Software, San Diego, CA). A two-tailed Mann-Whitney test was performed for experiments comparing two sets of data. A p-value of less than 0.05 was considered statistically significant.
3 Results
3.1 Clinical findings
CHD3 presented with silvery hair, pale skin, and ocular albinism as well as a history of recurrent respiratory and skin infections and nose bleeds. He was clinically diagnosed with classical CHS at the age of 7 years. Abnormal pigment clumping was observed in his hair shafts (Figure 1A). He received a HSCT at age 9 years following HLH. Progressive neurological dysfunction with dementia was noted at 21 years of age.
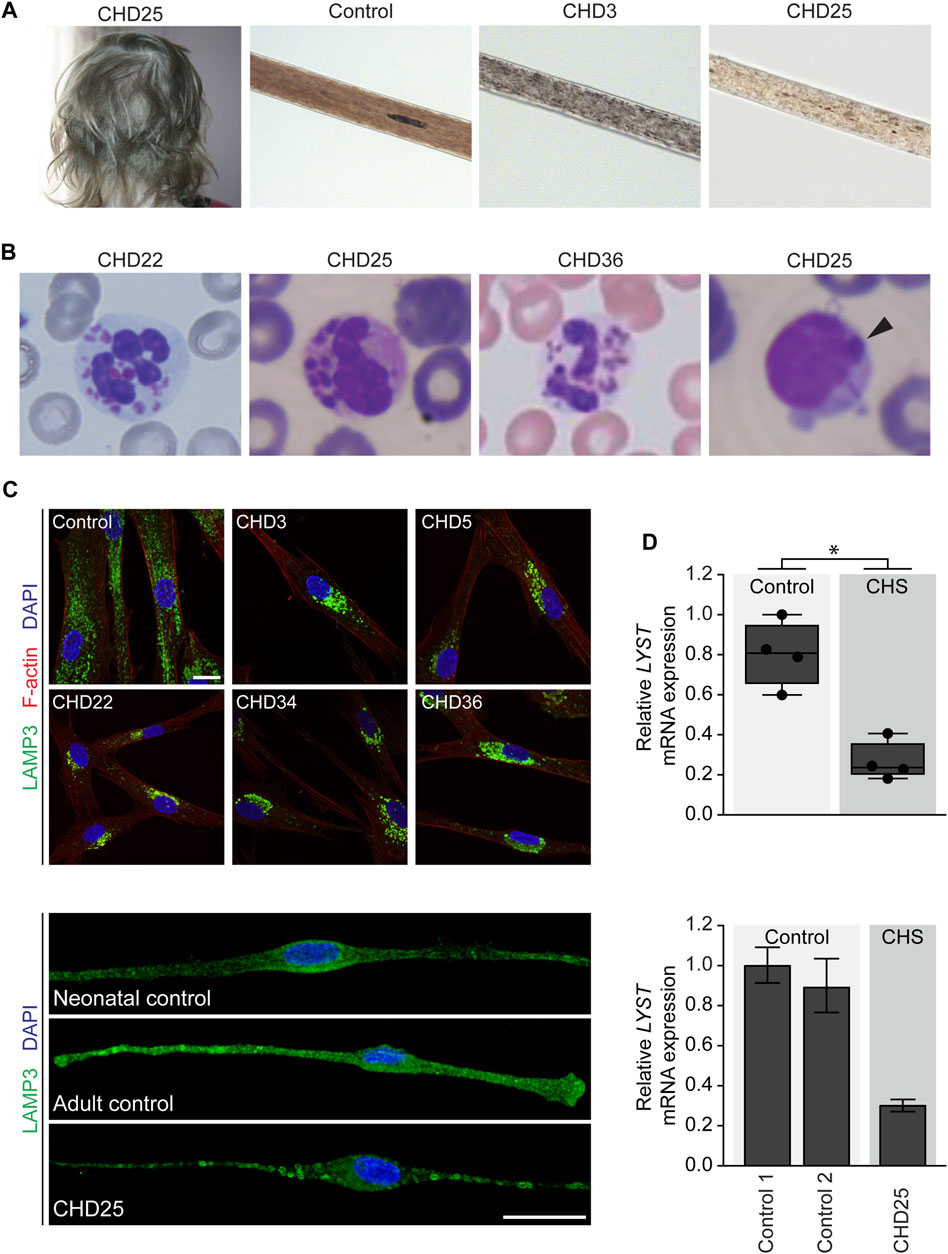
FIGURE 1. Clinical images of individuals with Chediak-Higashi syndrome (CHS) and cellular phenotype and LYST gene expression in primary cells from individuals with CHS (A) Partial oculocutaneous albinism often manifests as silvery hair and atypical pigment clumping of the hair shaft observed on light microscopy in individuals with CHS. (B) Peripheral blood smear demonstrating pathognomonic giant inclusions in polymorphonuclear leukocytes in CHD22, CHD25, and CHD36 and a single giant inclusion in a lymphocyte in CHD25 (arrowhead) (C) Primary fibroblasts (upper panel) and melanocytes (lower panel) were stained with LAMP3/CD63 antibodies to visualize lysosomal membranes. Primary fibroblasts were stained with phalloidin to visualize cell boundaries marked by F-actin. Lysosomes in unaffected control fibroblasts are distributed throughout the cell, while fibroblasts from individuals with CHS show enlarged lysosomes restricted to the perinuclear area. In melanocytes from CHD25, enlarged lysosomes were distributed throughout the cell compared to unaffected control melanocytes. Scale bar = 20 microns (D) Relative LYST mRNA expression in primary fibroblasts (upper panel) or melanocytes (lower panel). For the fibroblast expression data, the data are presented as a box and whisker plot relative to the highest LYST-expressing unaffected control with all individual mean data points represented. Group relative LYST expression differences were tested using a two-tailed Mann-Whitney test. For the melanocyte expression data, the data are presented as the mean of three technical replicates relative to the highest LYST-expressing unaffected control and error bars represent one standard deviation. Expression of HPRT1, POLR2A, and TBP were used as internal controls to normalize gene expression. Abbreviations: *, p < 0.05.
CHD5 has atypical CHS and was previously reported (Introne et al., 2016). She was diagnosed at the age of 19 years and presented with relatively mild disease manifesting as ocular albinism, gingival bleeding, and cutaneous methicillin-resistant Staphylococcus aureus (MRSA). She also had learning difficulties and a mood disorder. Pathognomonic giant inclusions were observed in her leukocytes and abnormal pigment clumping in her hair shafts (Introne et al., 2016).
CHD22 was hospitalized at 12 days of life due to omphalitis. She had recurrent sinopulmonary infections and otitis media, severely diminished platelet dense bodies, and patchy areas of hyper- and hypopigmentation of the skin. The presence of the pathognomonic giant cytoplasmic inclusions was confirmed by peripheral blood smear (Figure 1B), and she was clinically diagnosed with classical CHS at 1 year of age. She underwent HSCT at 1 year 8 months of age.
CHD25 was a 3.5-year-old female from Northern Turkey with a history of frequent bronchopulmonary infections who was admitted to the hospital due to persistent fever. Her mother and father were third degree relatives. She had one brother who was born at the 8th gestational month and died of an unknown cause in infancy. She also presented with silvery, hypopigmented hair, pale skin, and ocular albinism (Figure 1A). Peripheral blood smear confirmed giant cytoplasmic granules within the granulocytes, and lymphocytes showed a single giant inclusion (Figure 1B). She was clinically diagnosed with classical CHS. At presentation, she had evidence of recent Epstein-Barr Virus (EBV) infection and pancytopenia suggestive of HLH. Etoposide and methylprednisolone were started to treat her HLH. She had two posterior reversible leukoencephalopathy syndrome (PRES) attacks during the treatment and died due to respiratory failure during the second PRES attack, which involved the respiratory center of the pons.
CHD34 was diagnosed with pneumonia at birth and peripheral blood screening revealed giant granules pathognomonic for CHS. He was protected from infections until HSCT at 8 months of age and had no further infections or abnormal bleeding prior to transplant. He had a history of ocular albinism and patchy hyper- and hypopigmentation of the skin.
CHD36 had persistent thrush in the neonatal period that was unresponsive to nystatin and treated with two courses of fluconazole. She also had delayed separation of the umbilical cord and two episodes of respiratory infections. Upon referral to a dermatologist for an area of alopecia, she was noted to have silvery hair and patchy hypopigmentation of the skin, suggestive of CHS. Giant granules within the leukocytes were confirmed by blood smear (Figure 1B), and she was clinically diagnosed with classical CHS. She also had iris transillumination and platelet dense granule deficiency. She received HSCT at 10 months of age.
3.2 Cellular findings
To further support the clinical diagnosis of these six individuals, we pursued additional studies including assessment of lysosome morphology and intracellular localization by indirect immunofluorescence and relative LYST gene expression by quantitative PCR in cultured primary dermal fibroblasts or melanocytes. Indirect immunofluorescence staining of the late endosomal and lysosomal membrane marker LAMP3/CD63 in CHS fibroblasts and melanocytes revealed enlarged lysosomes (Figure 1C). Perinuclear localization of these lysosomes was apparent in CHS fibroblasts, while the enlarged lysosomes were located throughout CHD25 melanocytes. Quantitative PCR demonstrated reduced relative LYST mRNA expression in CHS fibroblasts and melanocytes compared to unaffected controls (Figure 1D, p-value = 0.0286 for fibroblast data). These cellular findings substantiate prior cellular findings reported in human CHS and in the beige mouse model (Perou et al., 1997; Westbroek et al., 2007) and support the clinical diagnosis of CHS in these six individuals.
3.3 Molecular genetic findings
To identify LYST variants in these six individuals with clinically confirmed CHS, conventional gDNA Sanger sequencing of the exonic regions of LYST was first performed. One or two variants were identified for CHD3, CHD5, CHD22, CHD34, and CHD36 (Figure 2A; Table 1). CHD3 was found to have a novel c.2413G>T variant predicted to lead to a p.(Glu805*) nonsense variant and a novel c.9925+1G>A variant predicted to affect the canonical splice donor site of intron 43. CHD5 was previously found to have a heterozygous c.7951G>T, p.(Val2651Phe) missense variant (Introne et al., 2016). CHD22 was found to have a novel c.1168del variant predicted to lead to a p.(Val390Cysfs*23) frameshift variant. CHD34 had a novel c.2016dup variant predicted to lead to a p.(Arg673Glnfs*12) frameshift variant. CHD36 had a novel c.2962C>T variant predicted to lead to a p.(Arg988*) nonsense variant and a novel c.9926-4_9936del variant predicted to affect the canonical splice acceptor site of intron 43. While no variants were identified for CHD25, PCR amplicons overlapping exons 39 and 40 failed to amplify, suggesting the possibility of a homozygous deletion in this region.

FIGURE 2. LYST variant alleles in individuals with Chediak-Higashi syndrome (CHS) identified by gDNA and cDNA Sanger sequencing (A) Schematics of the LYST mRNA (NM_000081.3) and LYST protein (NP_000072.2) denoting the relative locations of the LYST variants identified by gDNA and cDNA Sanger sequencing. Previously reported variants (white), novel variants identified by gDNA Sanger sequencing (black), and novel variants identified by cDNA Sanger sequencing (red) are represented in the schematic and include frameshift (square),nonsense (diamond), missense (circle), and in-frame (triangle) variants. Functional domains of LYST include the WD repeats (light grey), the PH domain (medium grey), and the BEACH domain (dark grey), and the corresponding regions are color-coded in the LYST mRNA schematic. The LYST mRNA schematic is not drawn to scale, while the LYST protein schematic is drawn to scale (B) Chromatograms generated through cDNA Sanger sequencing of LYST, including the c.9785_9925del (exon 43 deletion) variant allele identified in CHD3, the c.11039_11195del (exon 51 deletion) variant allele identified in CHD5, the c.8359_8389del (partial exon 32 deletion) variant allele identified in CHD22, the homozygous c.9163_9560del (exon 39 and 40 deletion) variant allele identified in CHD25, the c.8152_8358del (exon 31 deletion) variant allele identified in CHD34, and the c.9926_10029del (partial exon 44 deletion) variant allele identified in CHD36. RT-PCR products were cloned to obtain sequence data for the c.11039_11195del variant allele. Abbreviations: BEACH domain, beige and Chediak-Higashi syndrome domain; LYST, lysosomal trafficking regulator; PH domain, pleckstrin homology domain.
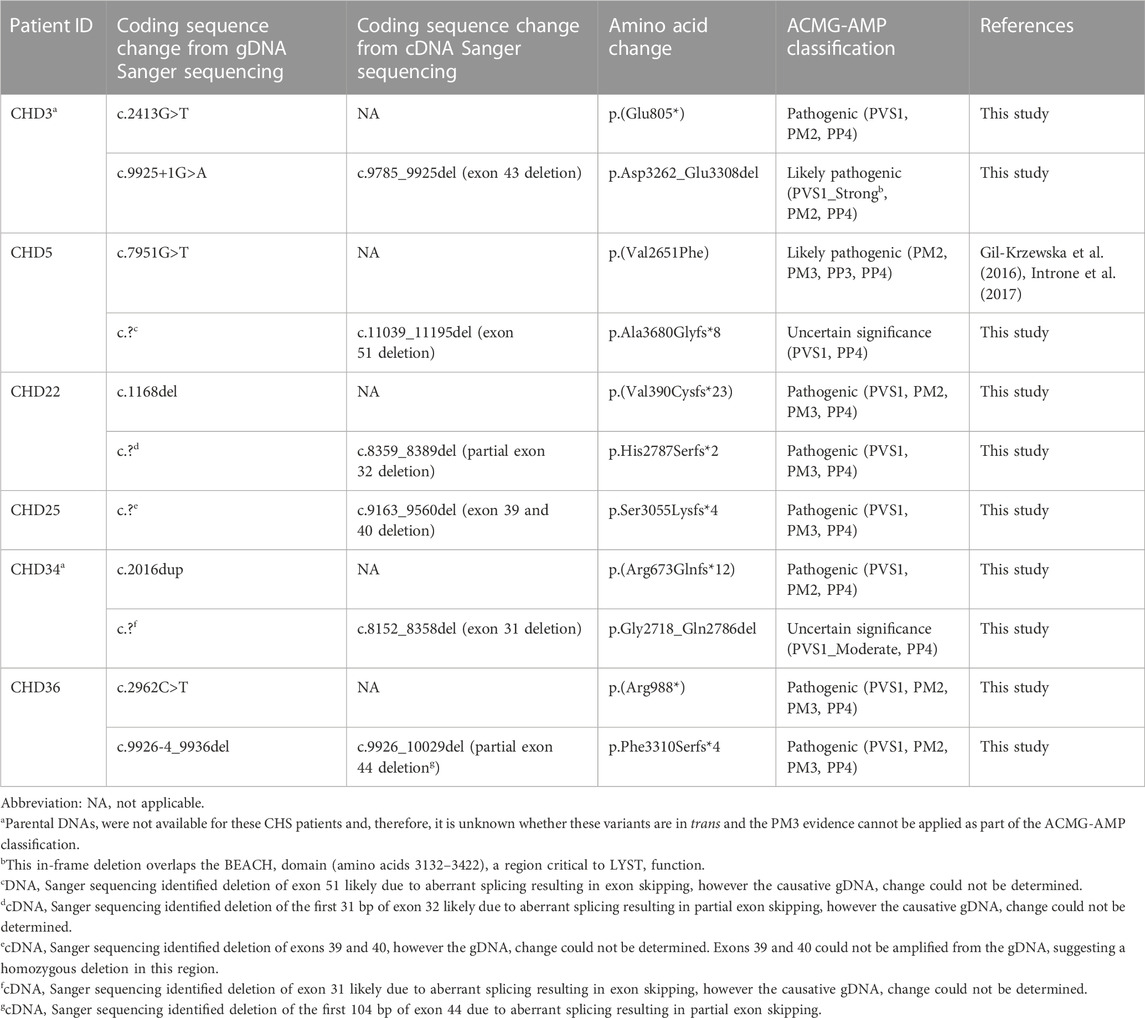
TABLE 1. Summary of variants identified in LYST (NM_000081.3, reference genome GRCh37/hg19) by gDNA and cDNA Sanger sequencing in six individuals with CHS.
To further identify LYST alleles, cDNA Sanger sequencing of the coding regions of LYST was performed. Four additional variant alleles were identified, and cDNA-level consequences were revealed for the two splice site variants (Figure 2; Table 1). CHD5 was found to have a novel c.11039_11195del variant allele (deletion of exon 51) that is predicted to lead to a p.Ala3680Glyfs*8 frameshift variant; CHD22 had a novel c.8359_8389del variant allele (deletion of the first 31 bp of exon 32) predicted to lead to a p.His2787Serfs*2 frameshift variant. CHD25 had a novel homozygous c.9163_9560del variant allele (deletion of exons 39 and 40) that is predicted to lead to a p.Ser3055Lysfs*4 frameshift variant. Finally, CHD34 was found to have a novel c.8152_8358del variant allele (deletion of exon 31) that is predicted to lead to a p.Gly2718_Gln2786 in-frame deletion. Variants at the level of gDNA could not be determined. For the canonical splice site variants in CHD3 and CHD36, the c.9925+1G>A variant led to c.9785_9925del (deletion of exon 43), confirming an in-frame variant (p.Asp3262_Glu3308del), and the c.9926-4_9936del variant led to c.9926_10029del (partial deletion of exon 44), confirming a frameshift variant (p.Phe3310Serfs*4). These findings establish a molecular diagnosis of CHS for all these individuals.
4 Discussion
In this study, we performed detailed clinical phenotyping and gDNA Sanger sequencing complemented by cDNA Sanger sequencing to establish a complete clinical and molecular diagnosis for six individuals with CHS. We report ten novel LYST alleles that may aid the molecular diagnosis of patients in the future; four of these variant alleles were undetectable by conventional gDNA Sanger sequencing and subsequently identified by cDNA Sanger sequencing.
Pathogenic variants have been identified throughout the LYST gene with no hotspots or hypermutated regions, and the majority of reported pathogenic variants in LYST have been nonsense or frameshift variants (personal communication) (Toro et al., 2009; Sanchez-Guiu et al., 2014). Eight of the ten novel variants reported in this study are predicted to lead to a nonsense or frameshift variant, while the remaining two novel variants are predicted to lead to an in-frame deletion (Figure 2; Table 1). Individuals with classical CHS often have bi-allelic nonsense or frameshift variants, while individuals with atypical CHS often have at least one missense variant, although exceptions have been reported (personal communication) (Toro et al., 2009; Sanchez-Guiu et al., 2014). Although CHD34 presented with classical CHS, one of his variants leads to an in-frame deletion of exon 31 (p.Asp3262_Glu3308del) and, while there are no known functional domains in this region of LYST, this region may represent a critical domain important for LYST function. Interestingly, CHD3 also presented with classical CHS and, in addition to a nonsense variant, CHD3 had an in-frame deletion within the BEACH domain (p.Gly2718_Gln2786del) that is critical for LYST function.
cDNA Sanger sequencing in combination with conventional gDNA Sanger sequencing offers several advantages. First, cDNA Sanger sequencing can detect variant alleles that are not readily detectable by conventional gDNA Sanger sequencing. While establishing a clinical diagnosis is valuable, the confirmation of a molecular diagnosis is critical for optimal patient care, genetic counseling for the individual and their family members, and eligibility in future clinical trials. Second, cDNA Sanger sequencing is relatively easy to incorporate into the molecular genetic testing workflow, requiring only routine cell culture and total RNA extraction techniques. An important consideration is patient sample archiving of LYST-expressing cell types, including dermal fibroblasts, melanocytes, NK cells, and lymphoblastoid cell lines, for the preparation of total RNA. Archiving cells from each parent and all available family members would also be useful for variant phasing and segregation studies.
A key limitation of this study was that we were unable to identify the cause of the full or partial exon skipping at the gDNA level for some alleles. While efforts were made to sequence nearby intronic regions and identify exonic synonymous variants that might lead to the aberrant splicing, proving the consequence of such variants requires further functional validation. With the decreasing costs of genome sequencing and targeted next-generation sequencing, these are becoming viable options for the identification of deep intronic variants leading to splicing changes in LYST. Furthermore, only cDNA from each of the six patients was analyzed in this study due to sample availability. While we were unable to assess cDNA from cells derived from family members due to sample availability, cDNA Sanger sequencing could be performed to segregate LYST alleles in a family even if the variant at the level of gDNA is unknown.
In this study, the integration of thorough clinical phenotyping and conventional gDNA Sanger sequencing in combination with cDNA Sanger sequencing increased the mutational spectrum of CHS. Based on our experience from this study, we provide the following recommendations for molecular genetic testing of the LYST gene in CHS. Because of the increasing access to next-generation sequencing and its rapid turnaround time, we recommend the utilization of clinical genome sequencing to include copy number variant analysis for patients in which CHS is suspected. When available, RNA sequencing using lymphoblastoid cell lines and melanocytes could help delineate the resulting splice site abnormalities from intronic variants. For standard clinical practice using exome sequencing and when clinical genome sequencing is not available, targeted gDNA Sanger sequencing and copy number detection is recommended for patients highly suspected to have clinical CHS, followed by RNA sequencing (as above) or cDNA Sanger sequencing if only one or no variants are detected by gDNA Sanger sequencing. It will also be interesting to pursue efforts on long-read sequencing technologies to identify deletions in these large genes and to phase variants in the absence of parental samples in the future. Overall, the identification of disease-causing variants will aid in the future diagnosis and care of individuals with CHS.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by National Human Genome Research Institute IRB. The patients/participants provided their written informed consent to participate in this study.
Author contributions
CK performed the cDNA sequencing and cell culture experiments, interpreted the data, and wrote the initial draft. MM extensively revised the manuscript, performed qPCR and statistical analysis, confirmed variants and variant nomenclature, and generated the figures. ERN helped conceptualize the approach for finding mutations, performed immunofluorescent studies, and organized the sample cohort list. JS helped conceptualize the approach for finding mutations, designed primers, performed cloning, and interpreted data. DSY and HD assisted in the immunofluorescent studies in cells and imaging. WO, SP, NYO, BM, and MG-A provided clinical consult, helped in interpreting clinical data, and contributed to manuscript writing. CC assisted in overall organization of samples and provided assistance in cell culture. WAG provided project direction and funding, and assisted in interpreting data. WJI provided clinical consult and expertise, interpreted clinical data, and provided supervision. MCVM provided overall project design, supervised personnel, interpreted molecular data, assisted in interpreting clinical data, and finalized the manuscript. All authors contributed to manuscript writing.
Funding
This study was supported by the Intramural Research Program of the NIH National Human Genome Research Institute (Bethesda, MD).
Acknowledgments
The authors would like to express their gratitude to the patients and their families for participating in this study. This study was supported by the Intramural Research Program of the NIH National Human Genome Research Institute (Bethesda, MD). The authors thank Karen Simon for sharing her technical expertise in Sanger sequencing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1072784/full#supplementary-material
Abbreviations
CBC, complete blood count; CHS, Chediak-Higashi syndrome; EBV, Epstein-Barr virus; HLH, hemophagocytic lymphohistiocytosis; HSCT, hematopoietic stem cell transplantation; LYST, Lysosomal Trafficking Regulator; MRSA, methicillin-resistant Staphylococcus aureus; PRES, posterior reversible leukoencephalopathy syndrome.
References
Barbosa, M. D., Nguyen, Q. A., Tchernev, V. T., Ashley, J. A., Detter, J. C., Blaydes, S. M., et al. (1996). Identification of the homologous beige and Chediak-Higashi syndrome genes. Nature 382 (6588), 262–265. doi:10.1038/382262a0
Béguez César, A. (1943). Neutropenia crónica maligna familiar con granulaciones atípicas de los leucocitos. Bol. Soc. Cuba. Pediatr. 15 (12), 900–922.
Bhambhani, V., Introne, W. J., Lungu, C., Cullinane, A., and Toro, C. (2013). Chediak-Higashi syndrome presenting as young-onset levodopa-responsive parkinsonism. Mov. Disord. 28 (2), 127–129. doi:10.1002/mds.25386
Boluda-Navarro, M., Ibanez, M., Liquori, A., Franco-Jarava, C., Martinez-Gallo, M., Rodriguez-Vega, H., et al. (2021). Case report: Partial uniparental disomy unmasks a novel recessive mutation in the LYST gene in a patient with a severe phenotype of Chediak-Higashi syndrome. Front. Immunol. 12, 625591. doi:10.3389/fimmu.2021.625591
Certain, S., Barrat, F., Pastural, E., Le Deist, F., Goyo-Rivas, J., Jabado, N., et al. (2000). Protein truncation test of LYST reveals heterogenous mutations in patients with Chediak-Higashi syndrome. Blood 95 (3), 979–983.
Chediak, M. M. (1952). New leukocyte anomaly of constitutional and familial character. Rev. Hematol. 7 (3), 362–367.
Dafsari, H. S., Ebrahimi-Fakhari, D., Saffari, A., Deneubourg, C., Fanto, M., and Jungbluth, H. (1993). “EPG5-related disorder,” in GeneReviews((R)). Editors M. P. Adam, D. B. Everman, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, and L. J. H. Bean (Seattle (WA): University of Washington).
Dufourcq-Lagelouse, R., Lambert, N., Duval, M., Viot, G., Vilmer, E., Fischer, A., et al. (1999). Chediak-Higashi syndrome associated with maternal uniparental isodisomy of chromosome 1. Eur. J. Hum. Genet. 7 (6), 633–637. doi:10.1038/sj.ejhg.5200355
Eapen, M., DeLaat, C. A., Baker, K. S., Cairo, M. S., Cowan, M. J., Kurtzberg, J., et al. (2007). Hematopoietic cell transplantation for Chediak-Higashi syndrome. Bone Marrow Transpl. 39 (7), 411–415. doi:10.1038/sj.bmt.1705600
Ellard, S., Baple, E. L., Callaway, A., Berry, I., Forrester, N., Turnbull, C., et al. (2020). ACGS best practice guidelines for variant classification in rare disease 2020. Available at: https://www.acgs.uk.com/media/11631/uk-practice-guidelines-for-variant-classification-v4-01-2020.pdf (Accessed October 6, 2022).
Gil-Krzewska, A., Wood, S. M., Murakami, Y., Nguyen, V., Chiang, S. C. C., Cullinane, A. R., et al. (2016). Chediak-Higashi syndrome: Lysosomal trafficking regulator domains regulate exocytosis of lytic granules but not cytokine secretion by natural killer cells. J. Allergy Clin. Immunol. 137 (4), 1165–1177. doi:10.1016/j.jaci.2015.08.039
Gironi, L. C., Zottarelli, F., Savoldi, G., Notarangelo, L. D., Basso, M. E., Ferrero, I., et al. (2019). Congenital hypopigmentary disorders with multiorgan impairment: A case report and an overview on gray hair syndromes. Med. Kaunas. 55 (3), 78. doi:10.3390/medicina55030078
Hauser, R. A., Friedlander, J., Baker, M. J., Thomas, J., and Zuckerman, K. S. (2000). Adult Chediak-Higashi parkinsonian syndrome with dystonia. Mov. Disord. 15 (4), 705–708. doi:10.1002/1531-8257(200007)15:4<705::aid-mds1016>3.0.co;2-b
Higashi, O. (1954). Congenital gigantism of peroxidase granules; the first case ever reported of qualitative abnormity of peroxidase. Tohoku J. Exp. Med. 59 (3), 315–332. doi:10.1620/tjem.59.315
Huizing, M., Malicdan, M. C. V., Gochuico, B. R., and Gahl, W. A. (1993). “Hermansky-pudlak syndrome,” in GeneReviews((R)). Editors M. P. Adam, D. B. Everman, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, and L. J. H. Bean (Seattle (WA): University of Washington).
Introne, W., Boissy, R. E., and Gahl, W. A. (1999). Clinical, molecular, and cell biological aspects of Chediak-Higashi syndrome. Mol. Genet. Metab. 68 (2), 283–303. doi:10.1006/mgme.1999.2927
Introne, W. J., Westbroek, W., Cullinane, A. R., Groden, C. A., Bhambhani, V., Golas, G. A., et al. (2016). Neurologic involvement in patients with atypical Chediak-Higashi disease. Neurology 86 (14), 1320–1328. doi:10.1212/WNL.0000000000002551
Introne, W. J., Westbroek, W., Groden, C. A., Bhambhani, V., Golas, G. A., Baker, E. H., et al. (2017). Neurologic involvement in patients with atypical Chediak-Higashi disease. Neurology 88 (7), e57–e65. doi:10.1212/WNL.0000000000003622
Jin, Y., Zhang, L., Wang, S., Chen, F., Gu, Y., Hong, E., et al. (2017). Whole genome sequencing identifies novel compound heterozygous lysosomal trafficking regulator gene mutations associated with autosomal recessive Chediak-Higashi syndrome. Sci. Rep. 7, 41308. doi:10.1038/srep41308
Manoli, I., Golas, G., Westbroek, W., Vilboux, T., Markello, T. C., Introne, W., et al. (2010). Chediak-Higashi syndrome with early developmental delay resulting from paternal heterodisomy of chromosome 1. Am. J. Med. Genet. A 152A (6), 1474–1483. doi:10.1002/ajmg.a.33389
Nagle, D. L., Karim, M. A., Woolf, E. A., Holmgren, L., Bork, P., Misumi, D. J., et al. (1996). Identification and mutation analysis of the complete gene for Chediak-Higashi syndrome. Nat. Genet. 14 (3), 307–311. doi:10.1038/ng1196-307
Perou, C. M., Leslie, J. D., Green, W., Li, L., Ward, D. M., and Kaplan, J. (1997). The Beige/Chediak-Higashi syndrome gene encodes a widely expressed cytosolic protein. J. Biol. Chem. 272 (47), 29790–29794. doi:10.1074/jbc.272.47.29790
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Sanchez-Guiu, I., Anton, A. I., Garcia-Barbera, N., Navarro-Fernandez, J., Martinez, C., Fuster, J. L., et al. (2014). Chediak-Higashi syndrome: Description of two novel homozygous missense mutations causing divergent clinical phenotype. Eur. J. Haematol. 92 (1), 49–58. doi:10.1111/ejh.12203
Sharma, P., Nicoli, E. R., Serra-Vinardell, J., Morimoto, M., Toro, C., Malicdan, M. C. V., et al. (2020). Chediak-higashi syndrome: A review of the past, present, and future. Drug Discov. Today Dis. Models 31, 31–36. doi:10.1016/j.ddmod.2019.10.008
Shirazi, T. N., Snow, J., Ham, L., Raglan, G. B., Wiggs, E. A., Summers, A. C., et al. (2019). The neuropsychological phenotype of Chediak-Higashi disease. Orphanet J. Rare Dis. 14 (1), 101. doi:10.1186/s13023-019-1049-x
Toro, C., Nicoli, E. R., Malicdan, M. C., Adams, D. R., and Introne, W. J. (2009). “Chediak-Higashi syndrome,” in GeneReviews. Editors M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. W. Grippet al. (Seattle: University of Washington), 1993–2020. [Internet].
Umeda, K., Adachi, S., Horikoshi, Y., Imai, K., Terui, K., Endo, M., et al. (2016). Allogeneic hematopoietic stem cell transplantation for Chediak-Higashi syndrome. Pediatr. Transpl. 20 (2), 271–275. doi:10.1111/petr.12626
Weisfeld-Adams, J. D., Mehta, L., Rucker, J. C., Dembitzer, F. R., Szporn, A., Lublin, F. D., et al. (2013). Atypical Chediak-Higashi syndrome with attenuated phenotype: Three adult siblings homozygous for a novel LYST deletion and with neurodegenerative disease. Orphanet J. Rare Dis. 8, 46. doi:10.1186/1750-1172-8-46
Westbroek, W., Adams, D., Huizing, M., Koshoffer, A., Dorward, H., Tinloy, B., et al. (2007). Cellular defects in Chediak-Higashi syndrome correlate with the molecular genotype and clinical phenotype. J. Invest. Dermatol 127 (11), 2674–2677. doi:10.1038/sj.jid.5700899
Keywords: LYST, rare disorders, monogenic diseases, oculocutaneous albinism, splicing abnormalities, personalized medicine, molecular diagnosis
Citation: Kuptanon C, Morimoto M, Nicoli E-R, Stephen J, Yarnell DS, Dorward H, Owen W, Parikh S, Ozbek NY, Malbora B, Ciccone C, Gunay-Aygun M, Gahl WA, Introne WJ and Malicdan MCV (2023) cDNA sequencing increases the molecular diagnostic yield in Chediak-Higashi syndrome. Front. Genet. 14:1072784. doi: 10.3389/fgene.2023.1072784
Received: 17 October 2022; Accepted: 22 February 2023;
Published: 08 March 2023.
Edited by:
Gaurav K. Varshney, Oklahoma Medical Research Foundation, United StatesReviewed by:
Nima Parvaneh, Tehran University of Medical Sciences, IranJenny Barker, Nationwide Children’s Hospital, United States
Copyright © 2023 Kuptanon, Morimoto, Nicoli, Stephen, Yarnell, Dorward, Owen, Parikh, Ozbek, Malbora, Ciccone, Gunay-Aygun, Gahl, Introne and Malicdan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: May Christine V. Malicdan, bWF5Y2hyaXN0aW5lLm1hbGljZGFuQG5paC5nb3Y=
†These authors have contributed equally to this work