
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 20 April 2023
Sec. Cancer Genetics and Oncogenomics
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1066808
This article is part of the Research Topic Current Trends and Future Perspectives about Liquid Biopsy View all 6 articles
 Mengjing Zhao1,2
Mengjing Zhao1,2 Qingjuan Li1
Qingjuan Li1 Jing Yang2
Jing Yang2 Min Zhang2Xiaolan Liu2
Min Zhang2Xiaolan Liu2 Hongwei Zhang2Yunpeng Huang2
Hongwei Zhang2Yunpeng Huang2 Jing Li3Jiangping Bao2Jingfang Wang2Jun Du4
Jing Li3Jiangping Bao2Jingfang Wang2Jun Du4 Tao Guan2*Liping Su2*
Tao Guan2*Liping Su2*Background: Follicular lymphoma (FL), an indolent non-Hodgkin lymphoma (NHL), is generally incurable. Favourable prognosis and durable remission are crucial for FL patients. The genetic mutation spectrum provides novel biomarkers for determining the prognosis of FL patients, but its detection is easily affected by the collection of tumour tissue biopsies. In this study, we aimed to describe the mutational landscape of FL using circulating tumour DNA (ctDNA) samples and to explore the relationship between mutations and prognostic indicators of clinical outcome in patients with newly diagnosed follicular lymphoma and the prognostic value of such mutations.
Methods: A total of 28 patients with newly diagnosed FL were included in this study. A targeted NGS-based 59-gene panel was used to assess the ctDNA mutation profiles. Differences in clinical factors between patients carrying mutations and those without mutations were analysed. We also explored the relationship between gene mutation status, mean VAFs (variant allele frequencies) and clinical factors. The Kaplan‒Meier method was applied to analyse the overall survival (OS) and progression-free survival (PFS) of patients carrying mutations and those without mutations.
Results: ctDNA mutations were detectable in 21 (75%) patients. The most commonly mutated genes were CREBBP (54%, 15/28), KMT2D (50%, 14/28), STAT6 (29%, 8/28), CARD11 (18%, 5/28), PCLO (14%, 4/28), EP300 (14%, 4/28), BCL2 (11%, 3/28), and TNFAIP3 (11%, 3/28), with a mutation frequency of >10%. Patients with detectable ctDNA mutation tended to present with advanced Ann Arbor stage (III-IV) (p = 0.009), high FLIPI risk (3–5) (p = 0.023) and severe lymph node involvement (No. of involved areas ≥5) (p = 0.02). In addition, we found that the mean VAF was significantly higher in patients with advanced Ann Arbor stage, high-risk FLIPI, elevated lactate dehydrogenase (LDH: 0–248U/L), advanced pathology grade, bone marrow involvement (BMI) and lymph node involvement. Additionally, KMT2D, EP300, and STAT6 mutations were associated with inferior PFS (p < 0.05).
Conclusion: We described the ctDNA mutation landscapes in Chinese patients with newly diagnosed FL and found that ctDNA VAF means reflect tumour burden. Moreover, PFS was shorter in patients with KMT2D, EP300 and STAT6 mutations.
Follicular lymphoma (FL) is the second most common subtype of non-Hodgkin lymphoma (NHL), accounting for approximately 20% of all lymphomas in the United States and Western Europe (Teras et al., 2016; Freedman and Jacobsen, 2020). FL is a heterogeneous disease with variable outcomes that is associated with different genomic alterations (Pasqualucci et al., 2011). It has been concluded that FL is regulated by genes related to histone modifications, cell-signalling pathways, transcription factors and chromatin remodelling (Morin et al., 2011; Shih et al., 2012; Green, 2018). Understanding the genomic landscape of FL will be helpful for diagnosis, prognosis prediction and the development of precision therapy. Currently, tissue biopsy is the gold standard for pathological diagnosis and identification of genetic variants, but invasive biopsies have significant limitations. Tissue biopsies carry procedural risks and cannot account for spatial inter-and intratumor heterogeneity due to sampling from only one location in a single tumour lesion (Wang et al., 2016). Therefore, a non-invasive and reliable method is needed to better characterize genetic alterations.
Analysis of circulating tumour DNA (ctDNA) is the leading liquid biopsy approach in lymphomas and can potentially improve on the limitations of tissue biopsy (Costa & Schmitt, 2019). ctDNA is the DNA fragment derived from tumour cells, and it accounts for approximately 0.01% of cfDNA (cell-free DNA) (Schwarzenbach et al., 2011). Analysing somatic mutations in cell-free DNA (cfDNA) to identify ctDNA can provide the genetic profiles of tumour tissues. Hohaus S et al. first demonstrated that ctDNA is a quantified tumour-specific biomarker in NHLs and Hodgkin’s lymphoma (HL) by real-time PCR(Hohaus et al., 2009). It has been reported that ctDNA could be a novel and sensitive cancer biomarker that can be used for the diagnosis, follow-up of treatment and prognosis of various cancers (Melani and Roschewski, 2016; Roschewski et al., 2016; Cescon et al., 2020). Evidence has demonstrated that ctDNA can reflect tumour biopsies in regards to somatic mutations in diffuse large B cell lymphoma (DLBCL) because of the high concordance of mutation profiles between ctDNA samples and tissue samples, and the use of ctDNA can overcome the issue of intratumor spatial heterogeneity (Eskandari et al., 2019; Liu et al., 2020). However, there are few studies on the application of ctDNA in FL. Therefore, it is necessary to confirm the characteristics of the ctDNA mutation spectrum in Chinese FL patients and its clinical value. Moreover, along with the rapid advances in next-generation sequencing (NGS) technology, ctDNA provides new opportunities for a better understanding of the molecular mechanisms of FL.
In this study, targeted NGS was applied to draw a ctDNA mutational landscape in newly diagnosed FL patients. Then, we assessed the relationship between ctDNA mutations and clinical pathological features and the roles of ctDNA mutations, including mutation status and mean VAF, in the overall survival (OS) and progression-free survival (PFS) of these patients.
A total of 28 newly diagnosed FL patients were enrolled at Shanxi Cancer Hospital from December 2018 to June 2021. Patients were considered eligible if they were aged >18 years and had histologically confirmed FL according to the 2016 WHO Classification of Tumors of Hematopoietic and Lymphoid Tissue (Swerdlow et al., 2016). None of the patients had family history. The disease was staged based on the 2014 Lugano classification, and the FL international prognostic index (FLIPI) was applied for risk stratification. Bone marrow involvement was assessed by flow cytometry combined with IG rearrangement and PET-CT. All patients were treated with the R-CHOP regimen (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone). We retrospectively analysed the clinical data of these patients and followed them up to March 2022. The treatment response, including CR (complete response), PR (partial response), SD (stable disease) and PD (progressive disease), was assessed by CT/MRI and PET/CT according to local guidelines after two to four treatment cycles.
All study activities were approved by the Ethics Committee of Shanxi Cancer Hospital (Ethical approval No.202236), and informed consent was obtained in accordance with the Declaration of Helsinki.
Peripheral blood samples were collected before treatment using EDTA-containing tubes and centrifuged immediately at a speed of 820 g for 10 min to separate plasma, and then the plasma samples were centrifuged at 20,000 × g for 10 min. Next, cfDNA was extracted using a QIAamp Circulating Nucleic Acid Kit (QIAGEN, Germany) according to the manufacturer’s instructions. Subsequently, the mutation profiles of each cfDNA sample were detected using the targeted NGS-based 59-gene panel (Shanghai Rightongene Biotech Co., Ltd., Shanghai, China) with an Illumina NovaSeq 5,000 (2 × 150-paired-end reads) and the target regions for each gene assay are coding exons and splice sites. This method is based on our previous published research on DLBCL (Guan et al., 2022). Mutations were filtered out if the VAF ranged from 45% to 55% or 95%, as such VAFs were considered to indicate germline mutations. The VAF was defined as the number of mutant molecules at a specific nucleotide location over the total number of molecules present in the background at a specific given genomic location. The mean VAF was calculated as follows: Mean VAF = The sum of the VAF of all mutations/The number of mutations.
Categorical data were compared with Fisher’s exact or chi-squared test, and continuous data were compared using a two-tailed paired Mann‒Whitney U test. Progression-free survival (PFS) was defined as the period from the date of treatment onset to the date of progression, last follow-up, or death of any cause. Overall survival (OS) was calculated from the date of treatment onset to the date of death of any cause or last follow-up. Survival analysis was performed by the Kaplan‒Meier method, and survival differences between groups were analysed using the log-rank test. All statistical analyses were conducted with IBM SPSS software (26.0), and GraphPad Prism (9.0) was used for drawing. Probability values < 0.05 were considered significant.
A total of 28 newly diagnosed FL patients with valid targeted NGS data were included in this study, of which 21 patients (75%) carried ctDNA mutations. Eighty-six somatic mutations were detected, and the median mutation per sample was 4 (range from 1 to 8). The most frequently mutated genes in the whole cohort were CREBBP (54%, 15/28), KMT2D (50%, 14/28), STAT6 (29%, 8/28), CARD11 (18%, 5/28), PCLO (14%, 4/28), EP300 (14%, 4/28), BCL2 (11%, 3/28) and TNFAIP3 (11%, 3/28), with a mutation frequency of >10% (Figure 1). Additionally, pathway enrichment analysis for these mutations was performed, which showed that BCR/NF-κB signalling, epigenetic regulators and JAK/STAT signalling were universally activated in FL (Supplementary Figure S1).
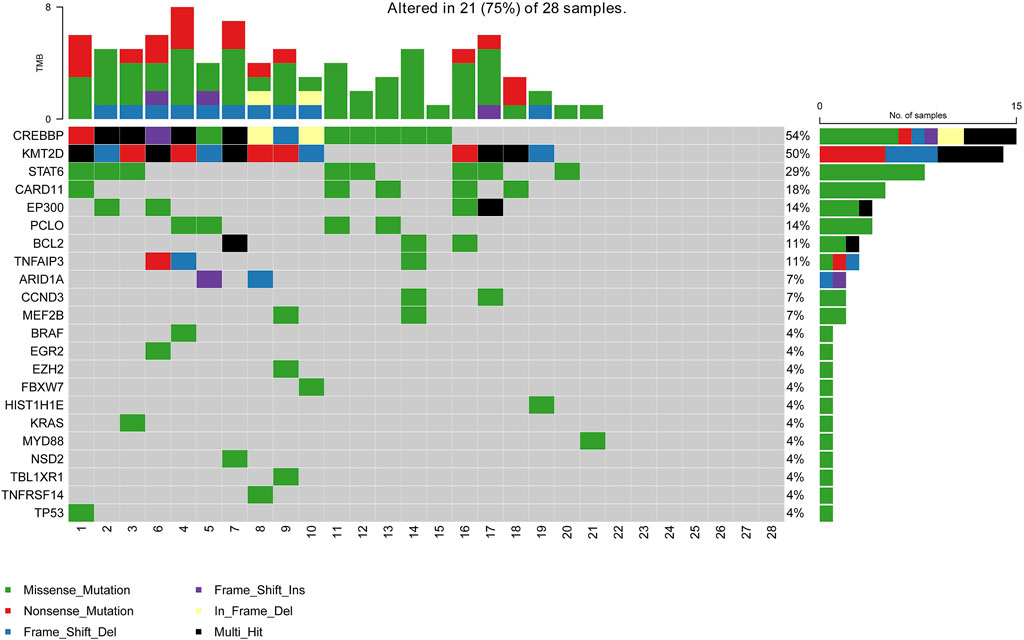
FIGURE 1. Mutation landscape of the ctDNA samples in newly diagnosed FL patients. The X-axis is the sample of individual patients. The upper histogram represents the number of mutated genes per sample. The Y-axis on the left shows the mutated genes, and the percentages on the right chart represent the mutation frequency of mutated genes.
Here, we analysed the relationship between prognostic indicators and ctDNA mutation status in newly diagnosed FL patients. Patients with detectable ctDNA mutations were significantly enriched in III-IV Ann Arbor stage (85.7% vs 28.6%, p = 0.009) and high-risk FLIPI (3–5) (52.45% vs 0, p = 0.023) and were inclined to have ≥5 lymph node-involved areas (81.0% vs 28.6%, p = 0.020) compared to patients without detected mutations in the plasma samples (Table 1). These results indicated that the ctDNA mutation status of newly diagnosed FL patients was closely associated with patient stage, FLIPI and number of lymph node-involved areas. It is worth noting that patients with ctDNA mutations were significantly enriched in lower pathological grades (grades 1–2) (81.0% vs 28.6%, p = 0.020).
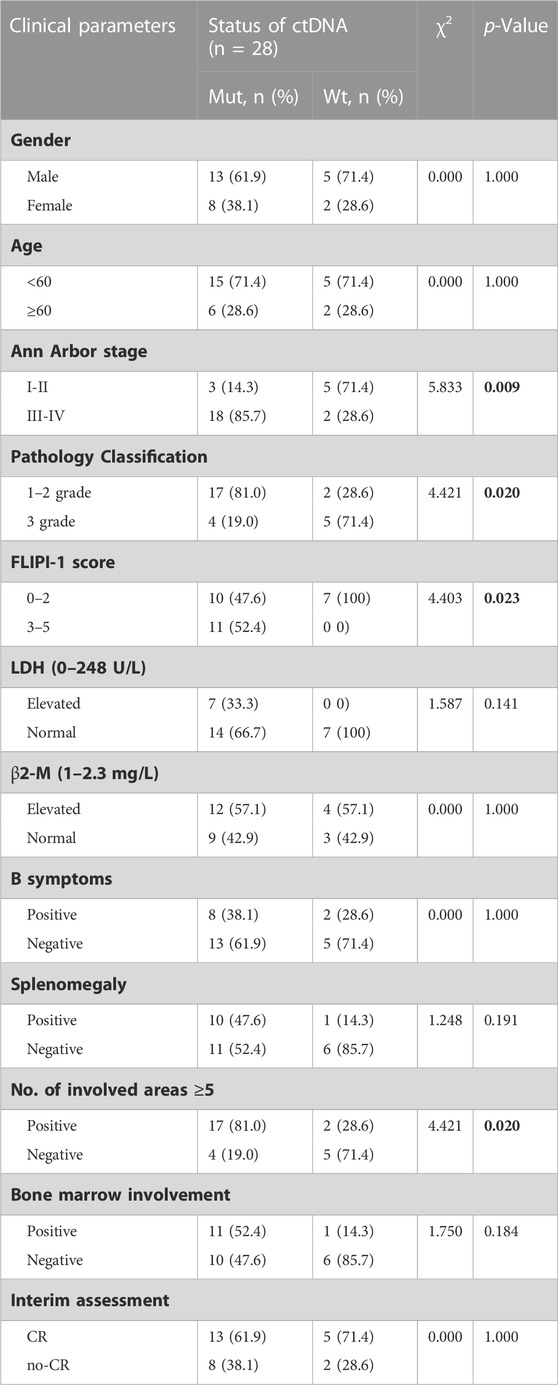
TABLE 1. The clinicopathologic features of FL patients with mutation or without mutation.
To further explore the clinical applications and value of ctDNA in newly diagnosed FL patients, we analysed the relationship between clinical indicators associated with prognosis and ctDNA levels. Here, we used mean VAFs (variant allele frequencies) to refer to mutation levels in ctDNA. As shown in Table 2, a comprehensive review and data analysis revealed that the VAF was closely correlated with poor clinical prognostic indicators, as a higher ctDNA mean VAF was associated with more advanced stage (p = 0.002; Figure 2A), high-risk FLIPI-1 (p = 0.015, Figure 2B) and elevated serum LDH levels (p = 0.003, Figure 2C). In addition, the ctDNA mean VAF was also significantly higher in patients with BMI (p = 0.011, Figure 2D) and severe lymph nodal involvement (p = 0.011, Figure 2E). However, the ctDNA mean VAF was significantly lower in grade 3 patients than in grade 1–2 patients (p = 0.019, Figure 2F). These results effectively suggest that ctDNA levels reflect tumour burden in newly diagnosed FL patients.
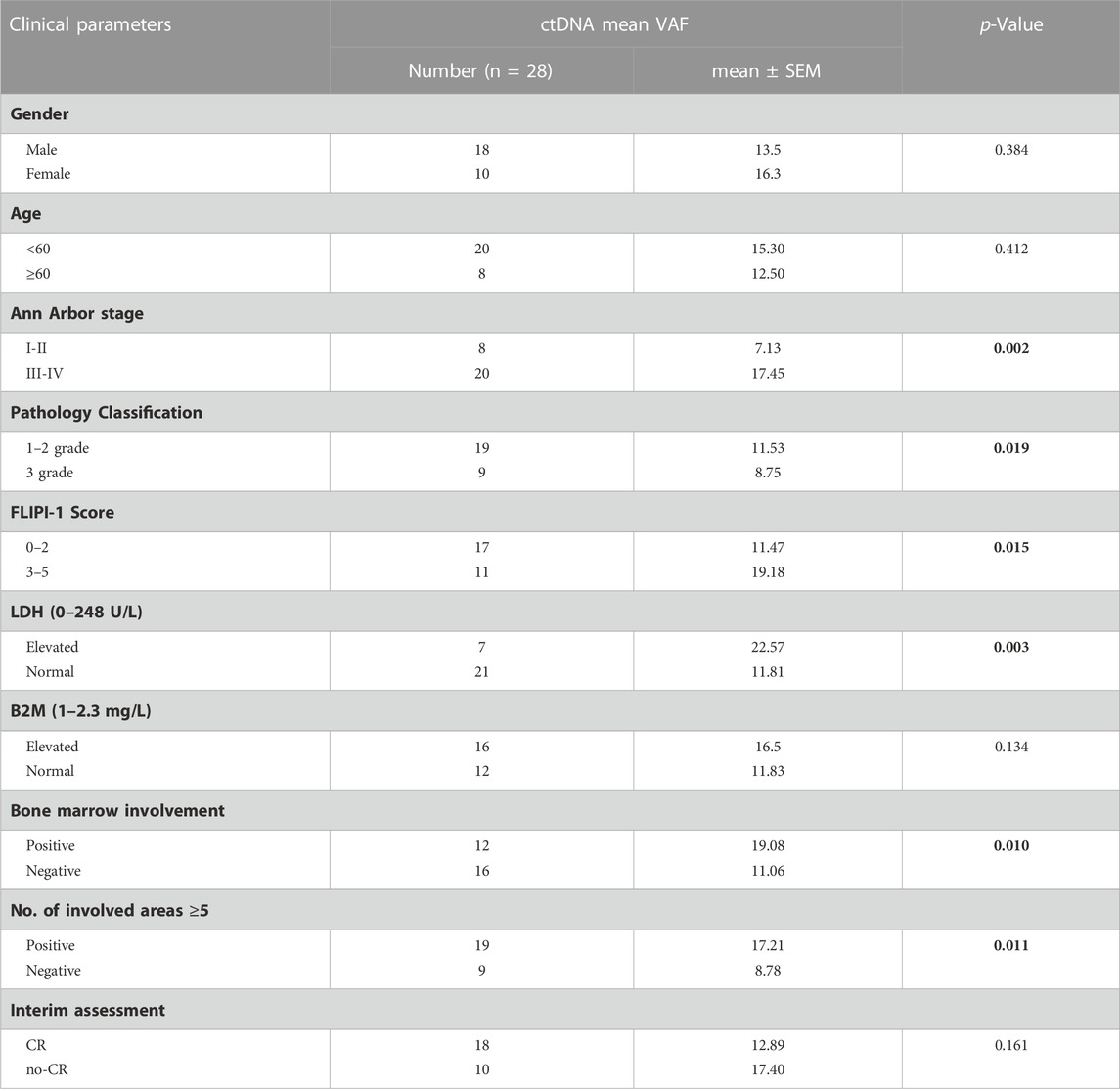
TABLE 2. Correlation between clinical indicators and the ctDNA levels.
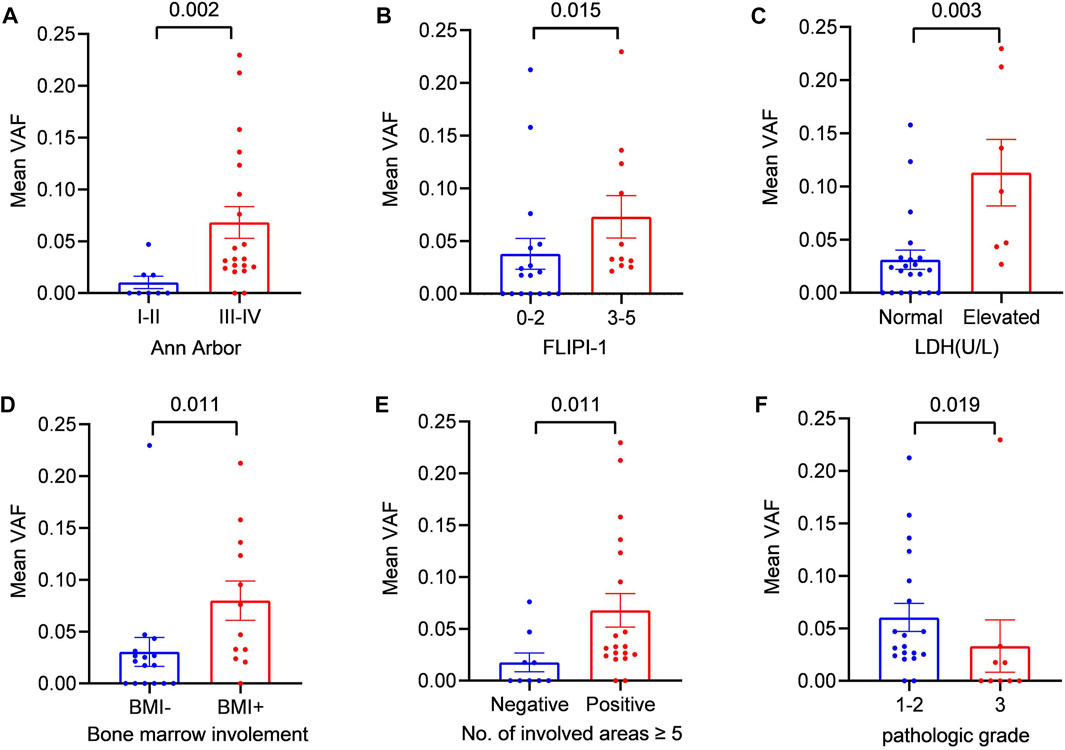
FIGURE 2. Relationship between the mean VAF and the clinical features of patients with newly diagnosed FL (A) Patients were divided into two groups according to Ann Arbor staging: the early-stage (I + II) group and the late-stage (III + IV) group (B) Statistical comparison between the low-FLIPI (0–2) group and the high-FLIPI (3–5) group was performed (C) Patients were divided into two groups according to LDH level: the elevated LDH group and the normal LDH group (ULN) (D) Patients were divided into two groups according to whether bone marrow was involved: BMI (+) and BMI (−) (E) Statistical comparison between the group with ≥5 lymph node-involved areas and the group with <5 lymph node-involved areas was performed. (F) Statistical comparison between the grade 1–2 cases and grade 3 cases was performed.
Then, we evaluated the impact of gene mutation status on OS and PFS in newly diagnosed FL patients using Kaplan‒Meier curves. We conducted an analysis of the survival of patients with mutated genes occurring in at least 4 cases (CREBBP, KMT2D, CARD11, STAT6, PCLO, and EP300). The results showed that KMT2D (p = 0.024) and STAT6 (p = 0.045) mutations were linked to significantly shorter PFS (Figures 3A, B). Similarly, in comparison with the patients without EP300 mutation (p = 0.044), the PFS was significantly shorter in the patients carrying a STAT6 mutation (Figure 3C). However, the difference in OS did not reach statistical significance. These results demonstrated that KMT2D, EP300 and STAT6 mutations predicted poor prognosis in patients with newly diagnosed FL. In addition, patients with detectable ctDNA mutations also showed a weak trend of shorter PFS than patients without ctDNA mutations. (p = 0.170) (Figure 3D).
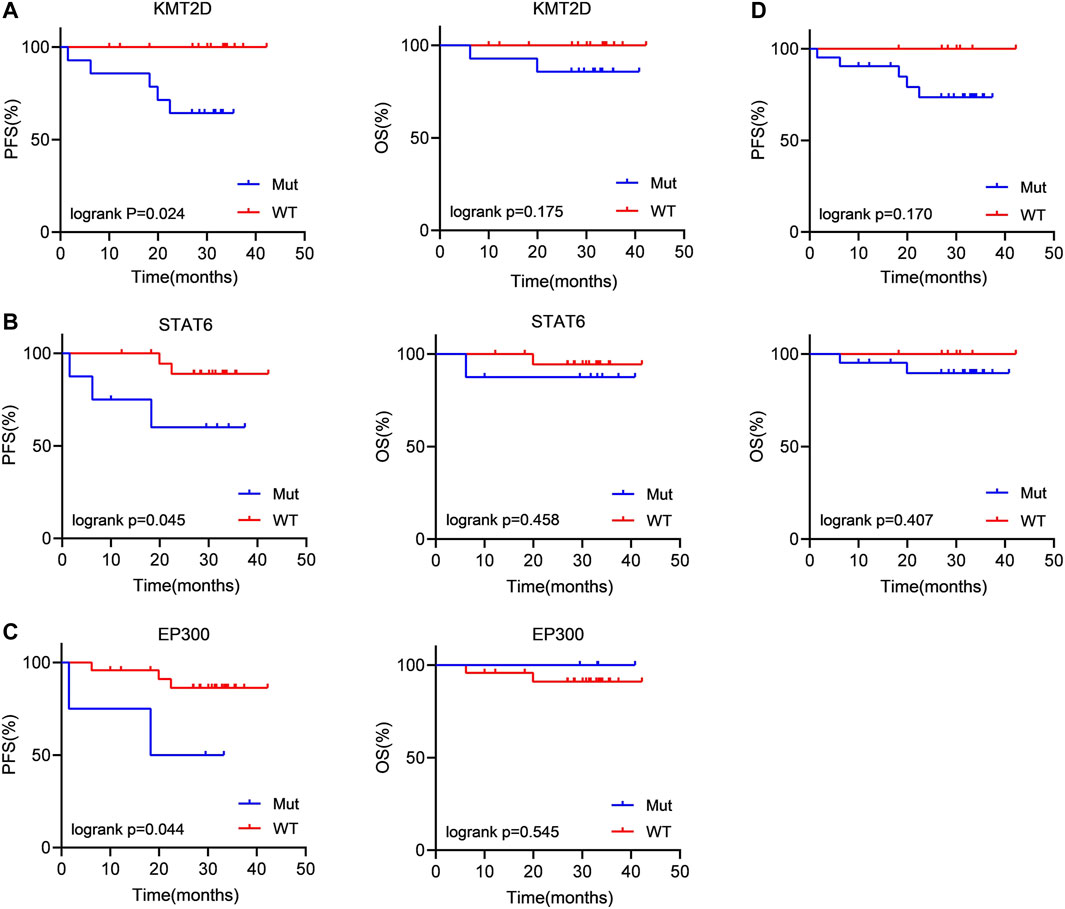
FIGURE 3. Patients carrying KMT2D, EP300 and STAT6 mutations tended to have a poor prognosis (A–C) Kaplan‒Meier curves were applied to compare the OS and PFS between patients carrying KMT2D, EP300 and STAT6 mutations and those without KMT2D, EP300 and STAT6 mutations. (D) Kaplan‒Meier curves were applied to compare the OS and PFS between patients with ctDNA mutation and without ctDNA mutation.
This study independently reported the prognostic value of ctDNA and the use of ctDNA to evaluate tumour burden in newly diagnosed FL patients. In detail, we analysed the characteristics of genetic mutation profiles and the clinical value of ctDNA from plasma samples in 28 newly diagnosed FL patients. Our results confirmed the relevance of high ctDNA levels and ctDNA mutation status during pretreatment in regards to Ann Arbor stage, FLIPI-1, LDH level, bone marrow involvement, pathology classification and lymph nodal involvement. In addition, we found that KMT2D, EP300 and STAT6 mutations indicated a poor prognosis in patients with FL.
We applied targeted NGS of 59 lymphoma-related genes and analysed the plasma ctDNA mutation characteristics and found that CREBBP (54%, 15/28) is the most commonly mutated gene in newly diagnosed FL patients. In fact, that was also observed in a study of the tumour tissue mutation landscape of follicular lymphoma patients (Shin et al., 2019). Moreover, the report of the tumour tissue mutation landscape in follicular lymphoma patients maintained high consistency with ctDNA mutation profiles in our study (Bastos-Oreiro et al., 2021). These results suggest that the mutation spectrum detected in plasma ctDNA samples of FL patients can reliably reflect the gene mutation information obtained from tumour tissue biopsy. However, there are also some mutated genes detected in ctDNA that are not found in tumour biopsy, which may be due to the heterogeneity of FL disease and the spatial limitations of tumour biopsy sampling. Unfortunately, neither solid nor liquid tumour biopsies can capture the complete genetic profile of the tumour, but ctDNA can be used as a supplement to bring additional mutational information because ctDNA overcomes tumour heterogeneity and contains molecular information that might be missed by examining a traditional single tumour sample.
CtDNA VAF, which is defined as the number of mutated molecules over the total number of molecules at a specific location in the genome, is closely associated with clinical features and prognosis and is considered a new biomarker of tumour burden (Arisi et al., 2022; van Velzen et al., 2022). In the present study, we found that the mean VAF was significantly increased in patients with advanced Ann Arbor stage, high-risk FLIPI, elevated serum LDH levels, bone marrow involvement, higher pathological grade and severe lymph nodal involvement. Our results suggest that ctDNA levels reflect overall tumour burden.
Moreover, patients with advanced Ann Arbor stage, high-risk FLIPI and severe lymph nodal involvement are more prone to ctDNA mutations. An unexpected finding is that ctDNA mutation generally tends to occur in FL patients with pathological grades of 1–2 rather than 3. Thus, we considered the following two aspects: 1). The 59-gene panel may not be sufficient to cover all mutated genes of FL; 2). In a recent study, gene expression profiling pointed out significant differences separating grade 1 to 2 cases from both grade 3A and grade 3B cases (Horn et al., 2018). Laurent, C et al. suggested that the high-grade FL cohort showed less frequent CREBBP mutations than grade 1–2 cases (Laurent et al., 2021). This further demonstrates the value of plasma genotyping for capturing clinically relevant tumour heterogeneity.
Dynamic ctDNA monitoring has been associated with PFS in DLBCL patients, but it is still under investigation in FL. Here, we analysed the association between ctDNA mutation status and prognosis in newly diagnosed FL patients. We found that FL patients with detectable ctDNA mutations tended to have significantly shorter PFS than those without detectable ctDNA mutations, but the difference was not statistically significant (p = 0.17), and we considered that the sample sizes in our study were relatively small, resulting in limited statistical power. According to previous studies, KMT2D is the most commonly mutated gene in patients with newly diagnosed FL (Morin et al., 2011), and it was also found that KMT2D mutation acts to “accelerate the mutation” and promote invasive tumour cell subpopulation formation during FL development (Green et al., 2013). Moreover, our results showed that patients with KMT2D mutations had shorter progression-free survival (PFS). Inactivating mutation of EP300 frequently occurs in DLBCL and FL, the two most common B-cell lymphomas (Pasqualucci et al., 2011). EP300 mutation has been reported to be closely related to inferior PFS (Pastore et al., 2015), which is consistent with our analysis. In addition, patients carrying STAT6 mutations had significantly shorter PFS than those without STAT6 mutations. These results suggest that KMT2D, EP300 and STAT6 mutations have a strong predictive value in the prognosis of pretreatment FL patients.
In fact, ctDNA mutations and their possible clinical applications have been initially explored in FL. Nagy, A et al. applied ddPCR to analyse the mutation and prognostic value of EZH2 in plasma samples from 4 patients with FL (Nagy et al., 2020), and Shin, S et al. discovered CREBBP mutations in plasma cfDNA using targeted deep sequencing in two follicular lymphoma patients. However, most current studies are limited to analysing single-gene mutations using liquid biopsy in FL, or other mutation signatures of ctDNA have only been characterized in a very small cohort. An important limitation of our study was the small number of cases that we were able to collect, but the clinical relevance of ctDNA was well displayed in our independent study.
In conclusion, our data demonstrated the feasibility of ctDNA targeted-NGS in newly diagnosed FL patients and confirmed that ctDNA mutation status and mean VAF can reflect tumour burden and are related to shorter PFS. Given the clinical relevance of the reported results, further development and validation of our findings in large patient cohorts will be needed.
The data presented in the study are deposited in the National Genomics Data Center repository (https://ngdc.cncb.ac.cn/gsa-human/s/e5a7JHq7), and accession number is PRJCA015152.
The studies involving human participants were reviewed and approved by Ethics Committee of Shanxi Cancer Hospital. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
LS contributed to conception and design of the study. QL organized the database. MiZ and XL performed the statistical analysis. MeZ and TG wrote the first draft of the manuscript. JB, HZ, and JW wrote sections of the manuscript. JL and YH followed up the patients. JD and JY participated in the revision of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
This study was supported by the foundation of Key Laboratory Construction Project Supported by Health Commission of Shanxi Province (Grant No. 2020SYS11), Shanxi Province Health Research Project Supported by Health Commission of Shanxi Province (Grant No. 2022121), and Research Project Supported by Shanxi Scholarship Council of China (Grant No. 2020-194).
We thank Shanghai Rightongene Biotechnology Co., Ltd. (Shanghai, China) for the assistance in targeted sequencing and mutation analysis.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1066808/full#supplementary-material
Arisi, M. F., Dotan, E., and Fernandez, S. V. (2022). Circulating tumor DNA in precision oncology and its applications in colorectal cancer. Int. J. Mol. Sci. 23 (8), 4441. doi:10.3390/ijms23084441
Bastos-Oreiro, M., Suárez-González, J., Andrés-Zayas, C., Carrión, N. C., Moreno, S., Carbonell, D., et al. (2021). Incorporation of next-generation sequencing in clinical practice using solid and liquid biopsy for patients with non-Hodgkin's lymphoma. Sci. Rep. 11 (1), 22815. doi:10.1038/s41598-021-02362-4
Cescon, D. W., Bratman, S. V., Chan, S. M., and Siu, L. L. (2020). Circulating tumor DNA and liquid biopsy in oncology. Nat. Cancer 1 (3), 276–290. doi:10.1038/s43018-020-0043-5
Costa, J. L., and Schmitt, F. C. (2019). Liquid biopsy: A new tool in oncology. Acta Cytol. 63 (6), 448. doi:10.1159/000501355
Eskandari, M., Manoochehrabadi, S., Pashaiefar, H., Zaimy, M. A., and Ahmadvand, M. (2019). Clinical significance of cell-free DNA as a prognostic biomarker in patients with diffuse large B-cell lymphoma. Blood Res. 54 (2), 114–119. doi:10.5045/br.2019.54.2.114
Freedman, A., and Jacobsen, E. (2020). Follicular lymphoma: 2020 update on diagnosis and management. Am. J. Hematol. 95 (3), 316–327. doi:10.1002/ajh.25696
Green, M. R. (2018). Chromatin modifying gene mutations in follicular lymphoma. Blood 131 (6), 595–604. doi:10.1182/blood-2017-08-737361
Green, M. R., Gentles, A. J., Nair, R. V., Irish, J. M., Kihira, S., Liu, C. L., et al. (2013). Hierarchy in somatic mutations arising during genomic evolution and progression of follicular lymphoma. Blood 121 (9), 1604–1611. doi:10.1182/blood-2012-09-457283
Guan, T., Zhang, M., Liu, X., Li, J., Xin, B., Ren, Y., et al. (2022). Circulating tumor DNA mutation profile is associated with the prognosis and treatment response of Chinese patients with newly diagnosed diffuse large B-cell lymphoma. Front. Oncol. 12, 1003957. doi:10.3389/fonc.2022.1003957
Hohaus, S., Giachelia, M., Massini, G., Mansueto, G., Vannata, B., Bozzoli, V., et al. (2009). Cell-free circulating DNA in Hodgkin's and non-Hodgkin's lymphomas. Ann. Oncol. 20 (8), 1408–1413. doi:10.1093/annonc/mdp006
Horn, H., Kohler, C., Witzig, R., Kreuz, M., Leich, E., Klapper, W., et al. (2018). Gene expression profiling reveals a close relationship between follicular lymphoma grade 3A and 3B, but distinct profiles of follicular lymphoma grade 1 and 2. Haematol. (Roma) 103 (7), 1182–1190. doi:10.3324/haematol.2017.181024
Laurent, C., Adelaide, J., Guille, A., Tesson, B., Gat, E., Evrard, S., et al. (2021). High-grade follicular lymphomas exhibit clinicopathologic, cytogenetic, and molecular diversity extending beyond grades 3A and 3B. Am. J. Surg. Pathol. 45 (10), 1324–1336. doi:10.1097/PAS.0000000000001726
Liu, H., Yang, C., Zhao, X., Le, J., Wu, G., Wei, J., et al. (2020). Genotyping on ctDNA identifies shifts in mutation spectrum between newly diagnosed and relapse/refractory DLBCL. Onco Targets Ther. 13, 10797–10806. doi:10.2147/OTT.S275334
Melani, C., and Roschewski, M. (2016). Molecular monitoring of cell-free circulating tumor DNA in non-hodgkin lymphoma[J]. Oncol. Willist. Park) 30 (8), 731–738.
Morin, R. D., Mendez-Lago, M., Mungall, A. J., Goya, R., Mungall, K. L., Corbett, R. D., et al. (2011). Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 476 (7360), 298–303. doi:10.1038/nature10351
Nagy, A., Batai, B., Balogh, A., Illes, S., Mikala, G., Nagy, N., et al. (2020). Quantitative analysis and monitoring of EZH2 mutations using liquid biopsy in follicular lymphoma. Genes (Basel) 11 (7), 785. doi:10.3390/genes11070785
Pasqualucci, L., Dominguez-Sola, D., Chiarenza, A., Fabbri, G., Grunn, A., Trifonov, V., et al. (2011). Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 471 (7337), 189–195. doi:10.1038/nature09730
Pastore, A., Jurinovic, V., Kridel, R., Hoster, E., Staiger, A. M., Szczepanowski, M., et al. (2015). Integration of gene mutations in risk prognostication for patients receiving first-line immunochemotherapy for follicular lymphoma: A retrospective analysis of a prospective clinical trial and validation in a population-based registry. Lancet Oncol. 16 (9), 1111–1122. doi:10.1016/S1470-2045(15)00169-2
Roschewski, M., Staudt, L. M., and Wilson, W. H. (2016). Dynamic monitoring of circulating tumor DNA in non-Hodgkin lymphoma. Blood 127 (25), 3127–3132. doi:10.1182/blood-2016-03-635219
Schwarzenbach, H., Hoon, D. S., and Pantel, K. (2011). Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 11 (6), 426–437. doi:10.1038/nrc3066
Shih, A. H., Abdel-Wahab, O., Patel, J. P., and Levine, R. L. (2012). The role of mutations in epigenetic regulators in myeloid malignancies. Nat. Rev. Cancer 12 (9), 599–612. doi:10.1038/nrc3343
Shin, S. H., Kim, Y. J., Lee, D., Cho, D., Ko, Y. H., Cho, J., et al. (2019). Analysis of circulating tumor DNA by targeted ultra-deep sequencing across various non-Hodgkin lymphoma subtypes. Leuk. Lymphoma 60 (9), 2237–2246. doi:10.1080/10428194.2019.1573998
Swerdlow, S. H., Campo, E., Pileri, S. A., Harris, N. L., Stein, H., Siebert, R., et al. (2016). The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 127 (20), 2375–2390. doi:10.1182/blood-2016-01-643569
Teras, L. R., Desantis, C. E., Cerhan, J. R., Morton, L. M., Jemal, A., and Flowers, C. R. (2016). 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J. Clin. 66 (6), 443–459. doi:10.3322/caac.21357
van Velzen, M., Creemers, A., van den Ende, T., Schokker, S., Krausz, S., and Reinten, R. J. (2022). Circulating tumor DNA predicts outcome in metastatic gastroesophageal cancer[J]. Gastric Cancer 25, 906. doi:10.1007/s10120-022-01313-w
Wang, Y., Tian, P. W., Wang, W. Y., Wang, K., Zhang, Z., Chen, B. J., et al. (2016). Noninvasive genotyping and monitoring of anaplastic lymphoma kinase (ALK) rearranged non-small cell lung cancer by capture-based next-generation sequencing. Oncotarget 7 (40), 65208–65217. doi:10.18632/oncotarget.11569
Keywords: follicular lymphoma, circulating tumour DNA, targeted next-generation sequencing, mutation, prognosis
Citation: Zhao M, Li Q, Yang J, Zhang M, Liu X, Zhang H, Huang Y, Li J, Bao J, Wang J, Du J, Guan T and Su L (2023) Application of circulating tumour DNA in terms of prognosis prediction in Chinese follicular lymphoma patients. Front. Genet. 14:1066808. doi: 10.3389/fgene.2023.1066808
Received: 11 October 2022; Accepted: 27 March 2023;
Published: 20 April 2023.
Edited by:
Elisa Frullanti, University of Siena, ItalyReviewed by:
Rina Kansal, University at Buffalo, United StatesCopyright © 2023 Zhao, Li, Yang, Zhang, Liu, Zhang, Huang, Li, Bao, Wang, Du, Guan and Su. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tao Guan, dGFvZ3VhbjMzMUBnbWFpbC5jb20=; Liping Su, c3VscDIwMDVAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.