 Yi Wu
Yi Wu Dan Wu
Dan Wu Yulong Lan
Yulong Lan Shaocong Lan
Shaocong Lan Duo Li1
Duo Li1 Hongwu Wang
Hongwu Wang Lian Ma
Lian Ma- 1Department of Pediatrics, Second Affiliated Hospital of Shantou University Medical College, Shantou, China
- 2Centre for Precision Health, School of Medical and Health Sciences, Edith Cowan University, Joondalup, WA, Australia
- 3Department of Cardiology, Second Affiliated Hospital of Shantou University Medical College, Shantou, China
- 4Department of clinical Medicine, Guangdong Medical University, Zhanjiang, China
- 5Department of Hematology and Oncology, Shenzhen Children’s Hospital of China Medical University, Shenzhen, China
- 6Shenzhen Public Service Platform of Molecular Medicine in Pediatric Hematology and Oncology, Shenzhen, China
- 7Department of Pediatrics, The Third Affiliated Hospital of Guangzhou Medical University (The Women and Children’s Hospital of Guangzhou Medical University), Guangzhou, China
Deletion in the Xp22.31 region is increasingly suggested to be involved in the etiology of epilepsy. Little is known regarding the genomic and clinical delineations of X-linked epilepsy in the Chinese population or the sex-stratified difference in epilepsy characteristics associated with deletions in the Xp22.31 region. In this study, we reported two siblings with a 1.69 Mb maternally inherited microdeletion at Xp22.31 involving the genes VCX3A, HDHD1, STS, VCX, VCX2, and PNPLA4 presenting with easily controlled focal epilepsy and language delay with mild ichthyosis in a Chinese family with a traceable 4-generation history of skin ichthyosis. Both brain magnetic resonance imaging results were normal, while EEG revealed epileptic abnormalities. We further performed an exhaustive literature search, documenting 25 patients with epilepsy with gene defects in Xp22.31, and summarized the epilepsy heterogeneities between sexes. Males harboring the Xp22.31 deletion mainly manifested with child-onset, easily controlled focal epilepsy accompanied by X-linked ichthyosis; the deletions were mostly X-linked recessive, with copy number variants (CNVs) in the classic region of deletion (863.38 kb–2 Mb). In contrast, epilepsy in females tended to be earlier-onset, and relatively refractory, with pathogenic CNV sizes varying over a larger range (859 kb–56.36 Mb); the alterations were infrequently inherited and almost combined with additional CNVs. A candidate region encompassing STS, HDHD1, and MIR4767 was the likely pathogenic epilepsy-associated region. This study filled in the knowledge gap regarding the genomic and clinical delineations of X-linked recessive epilepsy in the Chinese population and extends the understanding of the sex-specific characteristics of Xp22.31 deletion in regard to epilepsy.
Introduction
Epilepsy is a common neurological disturbance that manifests as recurrent and unprovoked seizures arising from largely unknown cellular and genetic mechanisms. Genetic contribution is increasingly emphasized in the etiology of epilepsies (Olson et al., 2014; EpiPM Consortium, 2015), notably copy number variants (CNVs) (Olson et al., 2014). Approximately 100 genes in CNV regions have been identified to be associated with epilepsy (EpiPM Consortium, 2015; Heyne et al., 2018), and rare sex chromosome CNVs have been identified in patients with epilepsy (Jabbari et al., 2018; Niestroj et al., 2020). However, evidence from case report studies has continuously suggested the involvement of Xp22.31 CNVs in epileptogenesis, for example, three case reports of X-linked familial focal epilepsy in the UK population (Mefford et al., 2011).
Although the specific causative genes for epilepsy and the functions of genes involved in this region are still elusive, CNVs in Xp22.31 have been suggested to play an important role in the genetics of epilepsy, albeit with variable penetrance (Mefford et al., 2011; Olson et al., 2014; Addis et al., 2018). Additionally, variants in the X chromosome lead to inconsistent disease phenotypes and prevalence rates between the sexes, owing to X-linked recessive inheritance and random X chromosome inactivation (XCI) in females, which protects females to a large extent (Fang et al., 2021). Furthermore, Xp22.3 is a domain of XCI escape and expresses genes such as STS with varying skewness, expanding the phenotypic diversity between the sexes (Meroni et al., 1996; Balaton and Brown, 2016; Balaton et al., 2018). Patients of both sexes with epilepsy have been found to have deletions in the Xp22.31 region (Olson et al., 2014; Addis et al., 2018; Gao et al., 2018); however, few studies have focused on the differences in variant size, source, and epilepsy patterns and types by sex.
Here, for the first time, we report two siblings with a maternally inherited microdeletion in a classic Xp22.31 region from a family with multifetal X-linked ichthyosis presenting with easily controlled focal epilepsy. Subsequently, we expanded the understanding of sex-specific characteristics of Xp22.31 deletion-associated epilepsy by reviewing the DECIPHER (Database of Chromosomal Imbalance and Phenotype in Humans Using Ensemble Resources) (Firth et al., 2009) and published articles. Together, we provided evidence for Xp22.31-microdeletion-related gene loss of function as an epilepsy risk factor using comparative genomic analysis.
Case description
The probands were referred to the Pediatric Clinic at the Second Affiliated Hospital of Shantou University Medical College (SUMC). Informed written consent was obtained from the children’s mother. This study was approved by the Research Ethics Committee of the Second Affiliated Hospital of SUMC (2020-4).
Proband 1
Proband 1 is currently 7 years and 8 months of age {height: 121.5 cm [−1.8 standard deviation (SD)], weight: 24.5 kg}. G1P1, at 38+ weeks of pregnancy, was born at term by C-section because of oligohydramnios detected by ultrasonic examination (birth weight: 2.8 kg). His mother noted no specific problems in the prenatal and perinatal periods. Intermittent daytime somnolence and nocturnal insomnia were problematic in infancy, but they returned to normal after age 3. The patient’s early developmental milestones were normal. His language development seemed delayed, as he could only say a few single words, e.g., “tea”,” mom”,” dad” at 1 year of age. He is presently in the first grade of primary school, and his academic performance is average.
The patient’s first seizure occurred at the age of 58/12 years. He suddenly bowed, stared, and was unable to answer any questions while inside an elevator in the daytime. He recovered approximately 10 seconds later. Seizure attacks initially occurred every 3–4 days; later, seizures became more frequent, with almost 2-4 seizures daily. Each seizure lasted for ten to several tens of seconds, without notable provoking factors. The patient was admitted to the hospital and underwent 24-h electroencephalography (EEG), and abnormal brain waves were observed (Figure 1). No remarkable abnormality was found on brain magnetic resonance imaging (MRI). Griffiths Development Scales, Chinese (GDS-C) (Li et al., 2020) evaluated at the age of 78/12 years suggested that his personal-social ability and language ability lagged behind those of the same age, with an overall developmental quotient (DQ) of 92. The patient was diagnosed with “epilepsy” and treated with “carbamazepine (CBZ)” (50 mg BID for a week and subsequently 100 mg BID). Seizures were subsequently completely controlled within 1 month, and later, he was regularly reviewed by EEG, with no significant seizure wave found. He has taken CBZ for more than 2 years and gradually reduced doses.
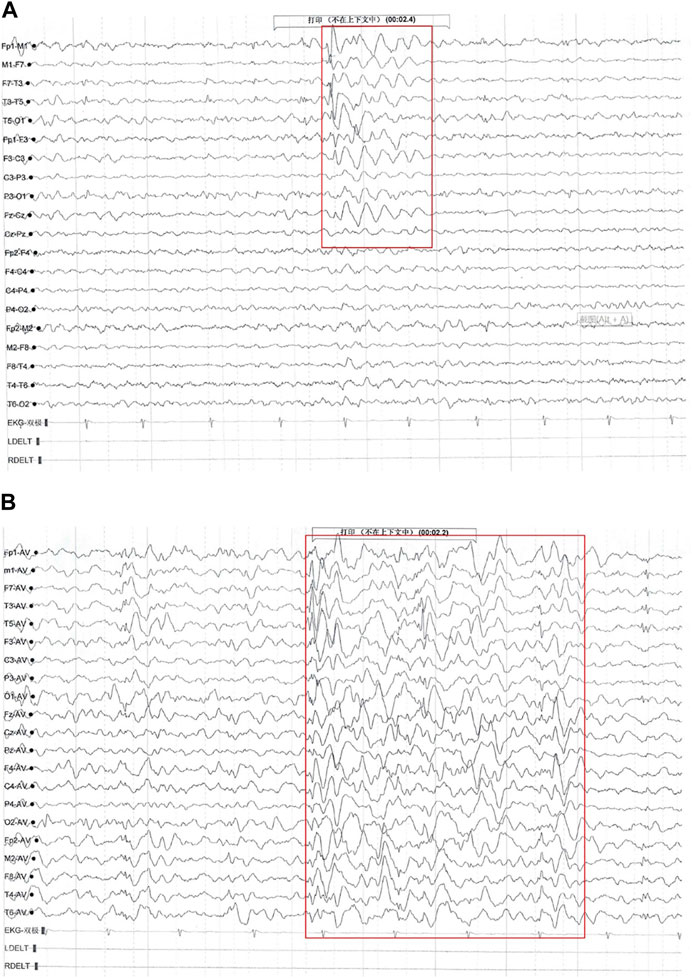
FIGURE 1. Electroencephalography (EEG) findings of Proband 1. Multiple paroxysmal events were recorded, with a clinical phenotype of sudden cessation of activity and daze. (A) During the interictal period, middle- and high-amplitude spine-slow waves were located in the bilateral temporal region, especially on the left side (EpiPM Consortium, 2015). (B) During the attack period, the low amplitude fast rhythm, obvious on the left, gradually evolved into a significant high amplitude spine-slow wave rhythm located in the bilateral temporal and frontal areas and Fz, and Cz showed and spread to the adjacent brain region (clinical representation: action stopped, daze).
Whole exome sequencing (WES) at the ages of 7 years and 6 months identified a 1.69 Mb maternally inherited hemizygous deletion on the X chromosome short arm (chr X: 6,451,786-8,138,073), affecting eight genes: VCX3A (OMIM 300533), HDHD1 (OMIM 306480), STS (OMIM 300747), VCX (OMIM 300229), PNPLA4 (OMIM 300102), MIR4767, MIR4767, and RPS27AP17. Details regarding WES were as follows: sheared blood DNA was prepared for Illumina paired-end sequencing and enriched for target regions with Agilent’s SureSelect Human All Exon V6 capture technology. The exome-captured library preparation was sequenced with the NovaSeq 6000 platform, reaching an average depth of >100X and Q30 > 90%. Reads were aligned to the GRCh37 human reference genome with BWA (v0.5.10). GATK Haplotype Caller v3.4 was used for variant calling and recalibration.]
Proband 2
Proband 2 is the youngest brother in this family. He is currently 310/12 years old. The birth history demonstrated no obvious special characteristics. No apparent feeding difficulties or sleep disorders were seen during infancy. Early milestones were mostly within normal limits.
His first seizure occurred at 38/12 years. In the morning, he was scolded by his father and in a bad mood. Approximately 10 minutes later, he suddenly tilted his head to one side, with his eyes open and turning upward, and could not talk or answer questions. The seizure lasted approximately half an hour; afterward, the patient became very drowsy and did not want to talk. A similar event occurred 1 hour later in the emergency room of a hospital, lasting for 15 min. Then, the patient was admitted to a pediatric department. A third event occurred 3 days later, with an approximately 3-min gaze described by a pediatrician. His body temperature was normal at the time, and he showed no signs of intercurrent illness.
The patient underwent a 5-h video-EEG during hospitalization, and no seizures occurred during the EEG test. The EEG showed that the background activity was normal, and many sharp waves were emitted in the left central area (only EEG report available). No obvious abnormality was found on the brain MRI. The patient was diagnosed with epilepsy and treated with valproic acid (VPA) (first 65 mg BID for 3 days and gradually increasing the dosage in accordance with the standard protocol, currently 130 mg BID). After taking VPA for 10 days, no similar symptoms were observed and have remained absent thus far. The EEG reexamined every 2 months thereafter was normal. Mild developmental and speech delays were also reported by a physiotherapist and a pediatrician, with GDS-C at the age of 310/12 years showing that his hearing and language and practical reasoning abilities lagged behind his peers; the total DQ score was 84. At this current age, the proband displayed moderate failure to thrive; height: 94 cm (−2 SD) and weight: 13 kg (−2 SD).
WES on blood DNA found the same maternally inherited deletion at Xp22.31 found in his elder brother.
Skin problems
Both boys had blatant ichthyosis since their early infancy. The abdominal wall and extremities showed epidermal dryness and scaling skin even in summer, consistent with ichthyosis (Figure 2), and deterioration was seen in cold weather or winter. However, the skin lesions did not appear severe when applying daily moisturizer to improve their symptoms.
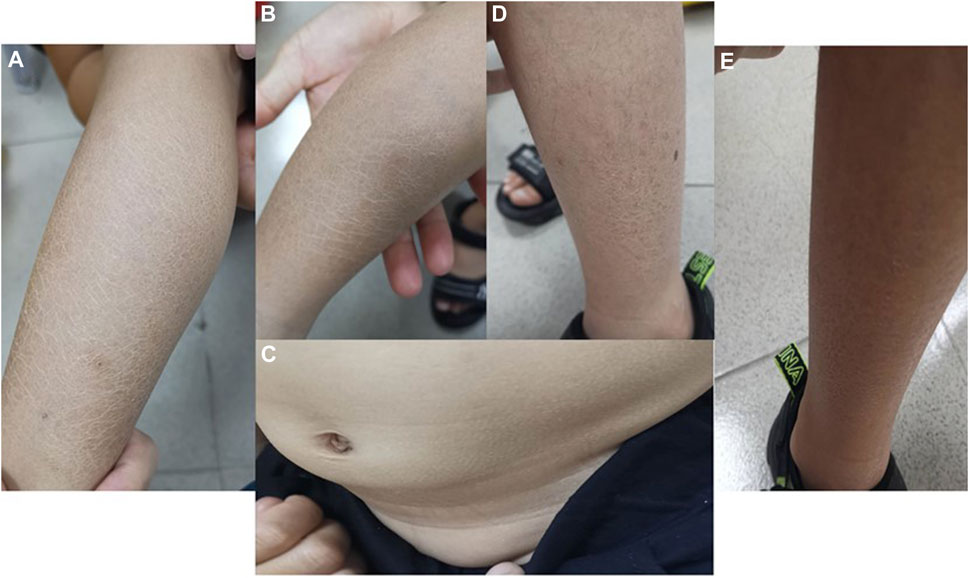
FIGURE 2. Photographs of ichthyosis on the skin (A, B) Photos of both lower limbs of Proband 1, with obviously dry and scaly skin in summer; (C) abdominal skin of Proband 1; (D, E) lateral skin of both lower limbs of Proband 2, with dry and scaly skin but no ulcer or break in summer.
Other Xp22.31 deletion-related comorbidities
No other comorbidities, including cryptorchidism, corneal opacity or cardiac arrhythmia, were noted in the two children after a detailed physical examination by a specialist. Nor were the apparent clinical presentations of ADHD and autism, despite a lack of professional scale evaluations for ADHD or autism for both children.
Family history
Both probands’ parents are of pure Chinese descent. The mother was 36 years old (height: 153 cm; weight: 51 kg), and the father was 40 years old (height: 170 cm; weight: 62 kg). Neither parent experienced epilepsy, skin abnormalities or other neurodevelopmental disorders. WES showed that their mother was an Xp22.31 microdeletion carrier, and their father did not carry any exon abnormalities.
The probands have a sister, the second child in the family, currently 6 years old. Her birth history and early developmental milestones were normal. Unlike her two brothers, her skin was smooth and delicate and good at communication. She was free of symptoms of epilepsy, speech delay and other neurological abnormalities. WES revealed her to be a carrier of the Xp22.31 microdeletion.
A maternal family history of epilepsy or intellectual disability (ID) was denied by the proband’s mother. However, ichthyosis has been present since the great grandfather’s generation (Figures 3, 4). A total of 6 males in 4 generations of the family manifested with ichthyosis; however, only the two probands additionally manifested with epilepsy.
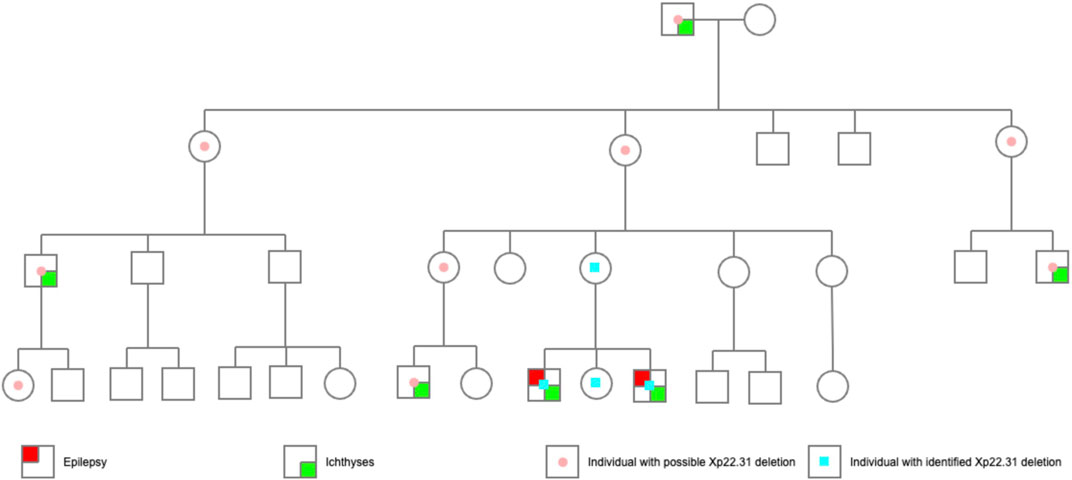
FIGURE 3. The pedigree of the probands. The probands’ great grandfather had obvious skin ichthyosis, whereas their great grandmother had normal skin; the second generation (their great grandfather’s 6 children) did not have the ichthyosis phenotype, but the males (the third generation) born to the women in this generation manifested a noticeable skin ichthyosis phenotype. Of the 10 children in the fourth generation, only three boys born to women in the third generation had an ichthyosis phenotype. Proband 1 and Proband 2 had epilepsy. Another boy, currently 2 years old, had no seizure attack thus far.
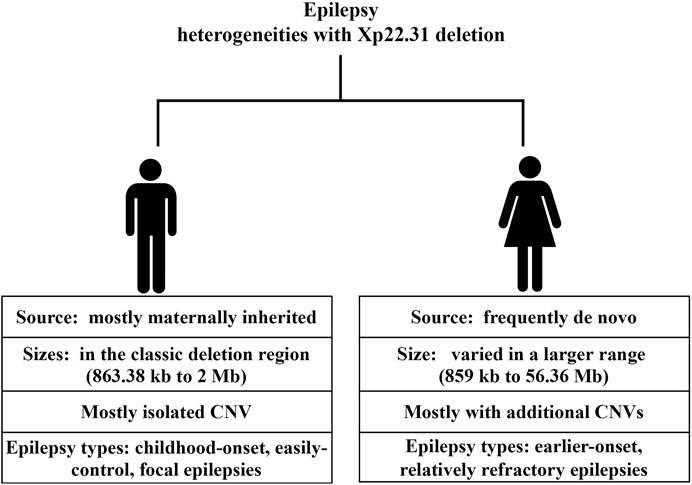
FIGURE 4. Summary of sex-specific epilepsy heterogeneities with Xp22.31 deletion.
Unfortunately, a detailed medical history collection, systematic examination or genetic testing of the rest of the family is unlikely, as another family adopted her mother at age ten, and she gradually lost contact with no desire to reconnect.
Characteristics of the reported epilepsy phenotypes with deletion in Xp22.31
We further searched the published studies and data from the DECIPHER database until Feb. 2022 and documented 23 patients with epilepsy and overlapping deletions in the Xp22.31 region ranging in size from 859 kb to 155.71 Mb (Supplementary eTable S2). Together with our 2 probands, there were 25 patients available for further examination. Of those, 14 were male, 8 were female, and 3 were of unknown sex. With regard to sex differences in variant sources, males were mostly maternally inherited (12/14), without any confirmed de novo change; in females, 4 were de novo (4/8), 2 were inherited (2/8), and the remaining 2 were of uncertain inheritance. The deletion sizes also differed greatly by sex. The majority (13/14) of males had isolated genetic changes, ranging from 863.38 kb to 2 Mb (mostly residing in the classic Xp22.31 deletion region). In contrast, females harbored deletion sizes that varied on a large scale (859 kb–56.36 Mb); three of them had isolated CNVs, and the rest (5/8) had additional CNVs in other chromosomes. Regarding the sex-associated epilepsy types, males tended to have focal or general epilepsy that was easily controlled with no or monotherapy with antiepileptic drugs. While data available for examining therapeutic effects were limited, females with Xp22.31 deletion tended to have early-onset and refractory epilepsy. Of 5 females with available data on seizure age or type (5/8), except for one patient with a de novo deletion of 1,688 kb at Xp22.31 who manifested with Rolandic epilepsy at age 6, the rest (4/8) had seizure types including infantile spasm (2/4) and generalized tonic-clonic seizures (GTC) (2/4). The epilepsy onset age was earlier in females than in males, with the earliest age of onset being 2 weeks. Notably, all 4 females with early-onset and refractory epilepsy harbored additional CNVs beyond the Xp22.31 deletion (4/4). Skin ichthyosis was notable in most of the included males rather than females. Other neurodevelopmental comorbidities associated with the Xp22.31 deletion included ID; among 14 males, 2 had uncertain status, 4 presented with ID, and the rest (8/14) had normal intelligence. In contrast, 4 females out of 8 had ID phenotypes.
CNVs in the deficit region of Xp22.31 of our probands encompassed genes including VCX3A, HDHD1, STS, VCX, PNPLA4, MIR4767, MIR651 and RPS27AP17 (Supplementary eTable S2). The minimum deletion region was 859 kb in females, though an additional 452 kb deletion in the Xq22.1 region and 33 kb deletion in 2p11.2 may confound the influences on epilepsy. In males, the minimum deletion region was 863.38 kb. The candidate region in Xp22.31, overlapping with the potentially pathogenic regions in all epilepsy cases, comprising STS, HDHD1, and MIR4767, is a risk factor for seizures.
Discussion
For the first time, we described two Chinese sibling boys with a maternally inherited deletion at Xp22.31, presenting with easily controlled focal epilepsy and mild ichthyosis from a family with a long history of XLI. Additional abnormalities included language delay in both probands and weak practical reasoning ability and growth retardation in Proband 2. Both probands possessed an ∼1.6 Mb microdeletion at Xp22.31. We further summarized the sex-specific differences in epilepsy characteristics in Xp22.31 deletion, focusing on variant source, size and epilepsy types, onset age and therapeutic experience, which are clinically informative. Furthermore, a candidate region encompassing three genes, STS, HDHD1, and MIR4767, should be the likely pathogenic epilepsy-associated region, despite a failure to further narrow down the region compared to Meyers’s study (2020) (Myers et al., 2020).
Consistent with our findings in the Chinese population, Xp22.31-linked epilepsy associated with XLI in other ethnicities/races manifested as an easily controlled epilepsy phenotype and normal brain MRI findings (Mefford et al., 2011; Olson et al., 2014; Myers et al., 2020). Interestingly, despite a long history of notable ichthyosis, no members of this family experienced seizure attacks other than the reported probands. We have several conjectures. First, X-linked recessive epilepsy likely displays incomplete penetrance (Mefford et al., 2011), as a minority (13%) of boys with CNVs in the most common disrupted regions had epilepsy with XLI (Rodrigo-Nicolás et al., 2018). A few literature reports and cohort studies have collectively implied that recurrent variants at the Xp22.31 region conferred risk for epilepsy but with variable penetrance (Olson et al., 2014; Addis et al., 2018; Choi et al., 2020). Second, it is possible that the size of CNVs in the same chromosomal region in the same family is not stable throughout inheritance. The mother may have inherited the STS deletion from her mother and her maternal grandfather, and then again, a new nonallelic homologous recombination (NAHR) event occurred in oogenesis to extend the STS deletion to the VCX gene (Van Esch et al., 2005). In Khelifa’s study, the size of the deletion in the healthy-phenotype mother differed from that in her son (Ben Khelifa et al., 2013). Mencarelli et al. also revealed an additional duplication involving the genes VCX, PNPLA4, and VCX2 at Xp22.3 in the proband relative to the mother (Mencarelli et al., 2008). Additionally, we could not completely exclude the involvement of environmental factors (e.g., toxin exposures during pregnancy) in epileptogenesis (Fukata and Fukata, 2017) that accounted for the occasional episode of seizures in the family clan.
It is essential to point out that current epilepsy–CNV associations have been identified through candidate locus screens. Accurate and efficient clinical consultation depends on the accumulation of clinical data. Although a male predominance in epilepsy phenotypes associated with Xp22.31 deletion was suggested in case reports (Rodrigo-Nicolás et al., 2018), we noted that both sexes with a deletion in Xp22.31 show epilepsy phenotypes. However, the types and severity of epilepsy and the source and size of variants differed greatly between sexes. Males who possess the Xp22.31 deletion mostly manifested with child-onset, easily controlled focal epilepsies that were X-linked recessive, accompanied by XLI. In contrast, except for one Rolandic epilepsy onset at age 6 years harboring a de novo deletion of 1,688 kb at Xp22.31 (7), epilepsy in females tended to be earlier-onset, relatively refractory and poor-prognosis, e.g., infantile spasm (Lux, 2013; Sahu, 2014) and GTC (Bresnahan et al., 2020), with pathogenic CNVs that were less likely to be inherited and combined with additional CNVs (4/4). The de novo variants and the combined additional CNVs may collectively account for the severity of seizure types. One Chinese girl with a de novo microdeletion at these same loci involving 4 genes (STS, VCX, PNPLA4) manifested a severe epilepsy phenotype (infantile spasms and hypotonic) and severe ID (Gao et al., 2018). De novo aberrations are known to cause worse outcomes than inherited changes (Watson et al., 2014). Previous findings have demonstrated that disease-causing de novo variants were associated with epilepsy types with the poorest prognosis (developmental and epileptic encephalopathies) (Allen et al., 2013). Additionally, we observed diverse combined neurodevelopmental abnormality phenotypes with epilepsy in Xp22.31 deletion in the review. The diverse manifestations of the aforementioned epilepsy phenotype, ID and speech delay are likely to support the incomplete penetrance property. Furthermore, the majority (8/13) of the males with CNV deletions from breakpoint STS and VCX-B manifested with focal epilepsy and ichthyosis but had normal cognition (Doherty et al., 2003), despite a deletion of a purported ID gene (VCX-3A). This finding supports mounting evidence that VCX-A is not an ID-associated gene (Cuevas-Covarrubias and González-Huerta, 2008; Mochel et al., 2008).
Notably, inherited deletions in Xp22.31 mostly contributed to epilepsy in males, whereas de novo deletions in Xp22.31 associated with severe epilepsy were observed only in females. Differing genetic backgrounds likely play a role in the contrasting enrichment patterns. We interpreted the lower susceptibility of males to complex refractory epilepsy as follows: de novo deletions in Xp22.31 are always lethal for males, and inherited deletions in Xp22.31 resulted in milder and weaker effects on females than on males. This finding is consistent with the phenomenon that some females, but not all, with the same X-linked deleterious allele are protected from its effects (Migeon, 2020).
There are several explanations underlying the diverse characteristics between sexes and among individuals. Lacking a second compensatory allele, males are naturally more frequently affected or have more severe phenotypes by X-linked pathogenic variants than females (Lyon, 1961). Phenotypes in females will be slightly milder than those in males, as females potentially have half the dose of pathogenic gene expression. Additionally, skewed XCI and XCI escape in the Xp22.31 region play an important role in the phenotypic heterogeneity of X-linked disorders (Tukiainen et al., 2017; Fang et al., 2021). Human genes that escape X inactivation are expressed at different levels in different individuals and in different tissues, as well as in different cells (Brown and Greally, 2003; Carrel and Willard, 2005; Migeon, 2020), suggesting remarkable expression heterogeneity among females (Zhang et al., 2013). As such, in females who possess the Xp22.31 deletion, the manifestation of phenotypes depends on whether the mutation is dominant, the expression level from the inactive X and the level of XCI skewing (Zhao et al., 2021). Indeed, skewed X-inactivation patterns due to X-chromosomal rearrangements have been reported to affect phenotypes of Xp22.3 defects (Suzuki et al., 2016; Ogushi et al., 2019). Additionally, the combined additional CNVs may contribute to the manifestation of phenotypes. Another possibility is ascertainment bias, whereby males with Xp22.31 deletions are picked up earlier than females due to their ichthyosis (only the most severely affected deletion-carrying females are likely to present), which may partly underlie the sex-specific phenotypic difference.
Moreover, it is important to note that the phenotypes may be immature and gradually develop during childhood. Males may have increased long-term risks of atrial fibrillation, leukemia, and end-stage renal disease, which need attention (Brcic et al., 2020). Females who possess genetic mutations or deletions in the Xp22.31 regions should be monitored for abnormal behavioral and psychiatric phenotypes (Cavenagh et al., 2019). Although the mother and her daughter appeared to be healthy carriers of this deletion, psychological evaluation is still needed, especially in the postpartum period (Cavenagh et al., 2019).
Genetic characterization of deleted and remaining sequences is an efficient way to map gene locations and functions associated with specific diseases (Monaco et al., 1985). Of the likely pathogenic genes, STS was supposed to be closely associated with the presence of epilepsy (Myers et al., 2020), as STS encodes a steroid sulfatase that hydrolyses neuro steroids that affect membrane potential and current conductance of the neuron, potentially controlling network excitability and seizure susceptibility (Ballabio et al., 1989a; Alperin and Shapiro, 1997; Kříž et al., 2008). Nevertheless, we must also acknowledge that STS deficiency alone is insufficient to cause seizures and that additional genetic or otherwise risk factors may be involved in the development of epilepsy. Additionally, HDHD1, encoding a pseudouridine-5′-phosphatase expressed at higher levels in the human brain, is also likely a possible gene candidate responsible for the seizure phenotype (Jenkins et al., 2004; Myers et al., 2020), as most of the epilepsy cases with Xp22.31 deletions harbored an HDHD1 deficiency in addition to STS (Supplementary eTable S2). Notably, the clinical abnormalities associated with Xp22.31 CNVs are thought to be influenced by contiguous gene syndromes, which manifest complex phenotypes resulting from the codeletion of adjacent genes (Ballabio et al., 1989b; Trent, 1993). Thus, further cytogenetic exploration to unravel not only the specific gene functions but also the interactions between genes and resulting gene function changes is warranted.
The limitations of this study should be noted. While we performed an exhaustive literature search on prior studies and the Decipher Database, the case review on epilepsy associated with Xp22.31 deletion is not all-inclusive. It is, however, the most comprehensive study thus far and provides enough data to highlight the variable epilepsy characteristics based on variant source, size, seizure type, onset age, and therapeutic effect between sexes. Additionally, we could not obtain the genetic background of the other members in the probands’ family despite the long family history of notable ichthyosis.
Conclusion
Our study demonstrates the significance of deletion at Xp22.31 in epilepsy and enriches the clinical phenotype profiles pertaining to the Xp22.31 microdeletion. Further investigation of the Xp22.31 region is warranted to unravel the pathophysiology of neurodevelopmental disorders.
Data availability statement
The datasets used and analyzed during the current study are available from the corresponding authors on reasonable request. Requests to access the datasets should be directed to HW, NDU4MjEzNDM3QHFxLmNvbQ==.
Ethics statement
The studies involving human participants were reviewed and approved by The Institutional Review Board of the Second Affiliated Hospital of Shantou University Medical College. Written informed consent to participate in this study was provided by the participants’ legal guardian. Written informed consent was obtained from the participants’ legal guardian for the publication of this case report.
Author contributions
YW, DW, HW, and LM were involved in the conception and design of the study. DW, YW, SL, and DL were responsible for drafting the manuscript. DW, ZZ, SL, and YL performed the statistical analysis. YW, DW, and LM obtained funding. LM and HW supervised the study. YW, DW, YL, LM, and HW had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Special Funds for Science and Technology in Guangdong Province (grant numbers: 202053-116 and 202053-121) and the Shantou Medical and Health Science and Technology Plan Project (grant number 190619105268874).
Acknowledgments
We thank the patients and their families who participated in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1025390/full#supplementary-material
References
Addis, L., Sproviero, W., Thomas, S. V., Caraballo, R. H., Newhouse, S. J., Gomez, K., et al. (2018). Identification of new risk factors for rolandic epilepsy: CNV at Xp22.31 and alterations at cholinergic synapses. J. Med. Genet. 55 (9), 607–616. doi:10.1136/jmedgenet-2018-105319
Allen, A. S., Berkovic, S. F., Cossette, P., Delanty, N., Dlugos, D., Eichler, E. E., et al. (2013). De novo mutations in epileptic encephalopathies. Nature 501 (7466), 217–221. doi:10.1038/nature12439
Alperin, E. S., and Shapiro, L. J. (1997). Characterization of point mutations in patients with X-linked ichthyosis. Effects on the structure and function of the steroid sulfatase protein. J. Biol. Chem. 272 (33), 20756–20763. doi:10.1074/jbc.272.33.20756
Balaton, B. P., and Brown, C. J. (2016). Escape artists of the X chromosome. Trends Genet. 32 (6), 348–359. doi:10.1016/j.tig.2016.03.007
Balaton, B. P., Dixon-McDougall, T., Peeters, S. B., and Brown, C. J. (2018). The eXceptional nature of the X chromosome. Hum. Mol. Genet. 27 (R2), R242–R249. doi:10.1093/hmg/ddy148
Ballabio, A., Bardoni, B., Carrozzo, R., Andria, G., Bick, D., Campbell, L., et al. (1989). Contiguous gene syndromes due to deletions in the distal short arm of the human X chromosome. Proc. Natl. Acad. Sci. U. S. A. 86 (24), 10001–10005. doi:10.1073/pnas.86.24.10001
Ballabio, A., Carrozzo, R., Parenti, G., Gil, A., Zollo, M., Persico, M. G., et al. (1989). Molecular heterogeneity of steroid sulfatase deficiency: A multicenter study on 57 unrelated patients, at DNA and protein levels. Genomics 4 (1), 36–40. doi:10.1016/0888-7543(89)90311-x
Ben Khelifa, H., Soyah, N., Ben-Abdallah-Bouhjar, I., Gritly, R., Sanlaville, D., Elghezal, H., et al. (2013). Xp22.3 interstitial deletion: A recognizable chromosomal abnormality encompassing VCX3A and STS genes in a patient with X-linked ichthyosis and mental retardation. Gene 527 (2), 578–583. doi:10.1016/j.gene.2013.06.018
Brcic, L., Underwood, J. F., Kendall, K. M., Caseras, X., Kirov, G., and Davies, W. (2020). Medical and neurobehavioural phenotypes in carriers of X-linked ichthyosis-associated genetic deletions in the UK Biobank. J. Med. Genet. 57 (10), 692–698. doi:10.1136/jmedgenet-2019-106676
Bresnahan, R., Panebianco, M., and Marson, A. G. (2020). Lamotrigine add-on therapy for drug-resistant generalised tonic-clonic seizures. Cochrane Database Syst. Rev. 7 (7), Cd007783. doi:10.1002/14651858.CD007783.pub3
Brown, C. J., and Greally, J. M. (2003). A stain upon the silence: genes escaping X inactivation. Trends Genet. 19 (8), 432–438. doi:10.1016/S0168-9525(03)00177-X
Carrel, L., and Willard, H. F. (2005). X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 434 (7031), 400–404. doi:10.1038/nature03479
Cavenagh, A., Chatterjee, S., and Davies, W. (2019). Behavioural and psychiatric phenotypes in female carriers of genetic mutations associated with X-linked ichthyosis. PLoS One 14 (2), e0212330. doi:10.1371/journal.pone.0212330
Choi, S., Mak, T., and O'Reilly, P. (2020). Tutorial: A guide to performing polygenic risk score analyses. Nat. Protoc. 15 (9), 2759–2772. doi:10.1038/s41596-020-0353-1
Cuevas-Covarrubias, S. A., and González-Huerta, L. M. (2008). Analysis of the VCX3A, VCX2 and VCX3B genes shows that VCX3A gene deletion is not sufficient to result in mental retardation in X-linked ichthyosis. Br. J. Dermatol 158 (3), 483–486. doi:10.1111/j.1365-2133.2007.08373.x
Doherty, M. J., Glass, I. A., Bennett, C. L., Cotter, P. D., Watson, N. F., Mitchell, A. L., et al. (2003). An Xp; Yq translocation causing a novel contiguous gene syndrome in brothers with generalized epilepsy, ichthyosis, and attention deficits. Epilepsia 44 (12), 1529–1535. doi:10.1111/j.0013-9580.2003.61702.x
EpiPM Consortium (2015). A roadmap for precision medicine in the epilepsies. Lancet Neurol. 14 (12), 1219–1228. doi:10.1016/S1474-4422(15)00199-4
Fang, H., Deng, X., and Disteche, C. (2021). X-factors in human disease: Impact of gene content and dosage regulation. Hum. Mol. Genet. 30, R285–R295. doi:10.1093/hmg/ddab221
Firth, H. V., Richards, S. M., Bevan, A. P., Clayton, S., Corpas, M., Rajan, D., et al. (2009). Decipher: Database of chromosomal imbalance and phenotype in humans using ensembl Resources. Am. J. Hum. Genet. 84 (4), 524–533. doi:10.1016/j.ajhg.2009.03.010
Fukata, Y., and Fukata, M. (2017). Epilepsy and synaptic proteins. Curr. Opin. Neurobiol. 45, 1–8. doi:10.1016/j.conb.2017.02.001
Gao, K., Zhang, Y., Zhang, L., Kong, W., Xie, H., Wang, J., et al. (2018). Large de novo microdeletion in epilepsy with intellectual and developmental Disabilities, with a Systems Biology analysis. Adv. Neurobiol. 21, 247–266. doi:10.1007/978-3-319-94593-4_9
Heyne, H. O., Singh, T., Stamberger, H., Abou Jamra, R., Caglayan, H., Craiu, D., et al. (2018). De novo variants in neurodevelopmental disorders with epilepsy. Nat. Genet. 50 (7), 1048–1053. doi:10.1038/s41588-018-0143-7
Jabbari, K., Bobbili, D. R., Lal, D., Reinthaler, E. M., Schubert, J., Wolking, S., et al. (2018). Rare gene deletions in genetic generalized and Rolandic epilepsies. PLoS One 13 (8), e0202022. doi:10.1371/journal.pone.0202022
Jenkins, C. M., Mancuso, D. J., Yan, W., Sims, H. F., Gibson, B., and Gross, R. W. (2004). Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J. Biol. Chem. 279 (47), 48968–48975. doi:10.1074/jbc.M407841200
Kříž, L., Bičíková, M., and Hampl, R. (2008). Roles of steroid sulfatase in brain and other tissues. Physiol. Res. 57 (5), 657–668. doi:10.33549/physiolres.931207
Li, P. Y., Fu, N. N., Li, Q. Y., Wang, G. F., Gao, L., and Zhang, X. (2020). The griffiths development scales-Chinese (gds-C): A reliable and valid neurodevelopmental assessment tool in children with ASD aged 3-8 years old in tianjin, China. Asian J. Psychiatr. 52, 102144. doi:10.1016/j.ajp.2020.102144
Lux, A. L. (2013). Latest American and European updates on infantile spasms. Curr. Neurol. Neurosci. Rep. 13 (3), 334. doi:10.1007/s11910-012-0334-z
Lyon, M. F. (1961). Gene action in the X-chromosome of the mouse (Mus musculus L). Nature 190, 372–373. doi:10.1038/190372a0
Mefford, H. C., Yendle, S. C., Hsu, C., Cook, J., Geraghty, E., McMahon, J. M., et al. (2011). Rare copy number variants are an important cause of epileptic encephalopathies. Ann. Neurol. 70 (6), 974–985. doi:10.1002/ana.22645
Mencarelli, M. A., Katzaki, E., Papa, F. T., Sampieri, K., Caselli, R., Uliana, V., et al. (2008). Private inherited microdeletion/microduplications: Implications in clinical practice. Eur. J. Med. Genet. 51 (5), 409–416. doi:10.1016/j.ejmg.2008.06.003
Meroni, G., Franco, B., Archidiacono, N., Messali, S., Andolfi, G., Rocchi, M., et al. (1996). Characterization of a cluster of sulfatase genes on Xp22.3 suggests gene duplications in an ancestral pseudoautosomal region. Hum. Mol. Genet. 5 (4), 423–431. doi:10.1093/hmg/5.4.423
Migeon, B. R. (2020). X-Linked diseases: Susceptible females. Genet. Med. 22 (7), 1156–1174. doi:10.1038/s41436-020-0779-4
Mochel, F., Missirian, C., Reynaud, R., and Moncla, A. (2008). Normal intelligence and social interactions in a male patient despite the deletion of NLGN4X and the VCX genes. Eur. J. Med. Genet. 51 (1), 68–73. doi:10.1016/j.ejmg.2007.11.002
Monaco, A. P., Bertelson, C. J., Middlesworth, W., Colletti, C. A., Aldridge, J., Fischbeck, K. H., et al. (1985). Detection of deletions spanning the Duchenne muscular dystrophy locus using a tightly linked DNA segment. Nature 316 (6031), 842–845. doi:10.1038/316842a0
Myers, K. A., Simard-Tremblay, E., and Saint-Martin, C. (2020). X-linked familial focal epilepsy associated with Xp22.31 deletion. Pediatr. Neurol. 108, 113–116. doi:10.1016/j.pediatrneurol.2020.02.008
Niestroj, L. M., Perez-Palma, E., Howrigan, D. P., Zhou, Y., Cheng, F., Saarentaus, E., et al. (2020). Epilepsy subtype-specific copy number burden observed in a genome-wide study of 17 458 subjects. Brain 143 (7), 2106–2118. doi:10.1093/brain/awaa171
Ogushi, K., Hattori, A., Suzuki, E., Shima, H., Izawa, M., Yagasaki, H., et al. (2019). DNA methylation status of SHOX-flanking CpG islands in healthy individuals and short stature patients with pseudoautosomal copy number variations. Cytogenet Genome Res. 158 (2), 56–62. doi:10.1159/000500468
Olson, H., Shen, Y., Avallone, J., Sheidley, B. R., Pinsky, R., Bergin, A. M., et al. (2014). Copy number variation plays an important role in clinical epilepsy. Ann. Neurol. 75 (6), 943–958. doi:10.1002/ana.24178
Rodrigo-Nicolás, B., Bueno-Martínez, E., Martín-Santiago, A., Cañueto, J., Vicente, A., Torrelo, A., et al. (2018). Evidence of the high prevalence of neurological disorders in nonsyndromic X-linked recessive ichthyosis: A retrospective case series. Br. J. Dermatol 179 (4), 933–939. doi:10.1111/bjd.16826
Sahu, J. K. (2014). Infantile spasms-evidence based medical management. Indian J. Pediatr. 81 (10), 1052–1055. doi:10.1007/s12098-014-1499-z
Suzuki, E., Shima, H., Toki, M., Hanew, K., Matsubara, K., Kurahashi, H., et al. (2016). Complex X-chromosomal rearrangements in two women with ovarian dysfunction: Implications of chromothripsis/chromoanasynthesis-dependent and -independent origins of complex genomic alterations. Cytogenet Genome Res. 150 (2), 86–92. doi:10.1159/000455026
Trent, R. J. (1993). Contiguous gene syndromes and small chromosome abnormalities. Med. J. Aust. 159 (2), 128–131. doi:10.5694/j.1326-5377.1993.tb137753.x
Tukiainen, T., Villani, A. C., Yen, A., Rivas, M. A., Marshall, J. L., Satija, R., et al. (2017). Landscape of X chromosome inactivation across human tissues. Nature 550 (7675), 244–248. doi:10.1038/nature24265
Van Esch, H., Hollanders, K., Badisco, L., Melotte, C., Van Hummelen, P., Vermeesch, J. R., et al. (2005). Deletion of VCX-A due to NAHR plays a major role in the occurrence of mental retardation in patients with X-linked ichthyosis. Hum. Mol. Genet. 14 (13), 1795–1803. doi:10.1093/hmg/ddi186
Watson, C., Marques-Bonet, T., Sharp, A., and Mefford, H. (2014). The genetics of microdeletion and microduplication syndromes: An update. Annu. Rev. genomics Hum. Genet. 15, 215–244. doi:10.1146/annurev-genom-091212-153408
Zhang, Y., Castillo-Morales, A., Jiang, M., Zhu, Y., Hu, L., Urrutia, A. O., et al. (2013). Genes that escape X-inactivation in humans have high intraspecific variability in expression, are associated with mental impairment but are not slow evolving. Mol. Biol. Evol. 30 (12), 2588–2601. doi:10.1093/molbev/mst148
Keywords: Xp22.31, epilepsy, microdeletion, copy number variations, X-linked ichthyosis, sex-specific
Citation: Wu Y, Wu D, Lan Y, Lan S, Li D, Zheng Z, Wang H and Ma L (2023) Case report: Sex-specific characteristics of epilepsy phenotypes associated with Xp22.31 deletion: a case report and review. Front. Genet. 14:1025390. doi: 10.3389/fgene.2023.1025390
Received: 22 August 2022; Accepted: 23 May 2023;
Published: 06 June 2023.
Edited by:
Corrado Romano, University of Catania, ItalyReviewed by:
William Davies, Cardiff University, United KingdomChristopher McGraw, Massachusetts General Hospital and Harvard Medical School, United States
Copyright © 2023 Wu, Wu, Lan, Lan, Li, Zheng, Wang and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongwu Wang, NDU4MjEzNDM3QHFxLmNvbQ==; Lian Ma, bWFsaWFuODk2NUBzaW5hLmNvbQ==
†These authors have contributed equally to this work and share first authorship