 María Carolina Manotas1†
María Carolina Manotas1† Ana Lucía Rivera1Ana Milena Gómez1Patricia Abisambra2Gonzalo Guevara1Vilma Medina1Sandra Tapiero1Antonio Huertas1Julián Riaño-Moreno1Juan Carlos Mejía1Angélica María Gonzalez-Clavijo1Mireya Tapiero-García1
Ana Lucía Rivera1Ana Milena Gómez1Patricia Abisambra2Gonzalo Guevara1Vilma Medina1Sandra Tapiero1Antonio Huertas1Julián Riaño-Moreno1Juan Carlos Mejía1Angélica María Gonzalez-Clavijo1Mireya Tapiero-García1 Andrés Arturo Cuéllar-Cuéllar1
Andrés Arturo Cuéllar-Cuéllar1 Luis Felipe Fierro-Maya1
Luis Felipe Fierro-Maya1 María Carolina Sanabria-Salas1,3*†
María Carolina Sanabria-Salas1,3*†- 1Medical Subdirection, Instituto Nacional de Cancerología, Bogotá, Colombia
- 2Department of Medicine, Universidad del Norte, Barranquilla, Colombia
- 3Subdirection of Research, Instituto Nacional de Cancerología, Bogotá, Colombia
Pheochromocytomas (PCCs) and paragangliomas (PGLs) (known as PPGL in combination) are rare neuroendocrine tumors of the adrenal medulla and extra-adrenal ganglia. About 40% of the patients with PPGL have a hereditary predisposition. Here we present a case-series of 19 unrelated Colombian patients with a clinical diagnosis of PPGL tumors that underwent germline genetic testing as part of the Hereditary Cancer Program developed at the Instituto Nacional de Cancerología, Colombia (INC-C), the largest reference cancer center in the country. Ten of 19 patients (52.63%) were identified as carriers of a pathogenic/likely pathogenic (P/LP) germline variant in a known susceptibility gene. The majority of the P/LP variants were in the SDHB gene (9/10): one corresponded to a nonsense variant c.268C>T (p.Arg90*) and eight cases were found to be carriers of a recurrent CNV consisting of a large deletion of one copy of exon 1, explaining 42% (8/19) of all the affected cases. Only one additional case was found to be a carrier of a missense mutation in the VHL gene: c.355T>C (p.Phe119Leu). Our study highlights the major role of SDHB in Colombian patients with a clinical diagnosis of PGL/PCC tumors and supports the recommendation of including the analysis of large deletions/duplications of the SDHB gene as part of the genetic counselling to improve the detection rate of hereditary cases and their clinical care.
1 Introduction
Pheochromocytoma (PCC) was first described by Fränkel in 1886; since then, important discoveries and advances have been made in relation to diagnosis, genetics, and management (Wiseman et al., 2019). PCC is a rare neuroendocrine tumor, which originates from the chromaffin cells of the adrenal medulla, and is related to paraganglioma (PGL), which originates from extra-adrenal chromaffin cells (Aygun and Uludag, 2020); both are histologically identical (Berends et al., 2018). Chromaffin cells of the adrenal medulla are major components of the autonomic nervous system that releases catecholamines into the blood stream; interestingly, recent studies have uncovered new cellular origin of chromaffin cells from Schwann cell precursors, and understanding these differentiation pathways may help to comprehend the origin of these tumors and improve their clinical management (Furlan et al., 2017; Kastriti et al., 2019). Most PCCs and PGLs are secreting or functioning endocrine tumors, and their clinical presentation is associated with excessive secretion of catecholamines (epinephrine, norepinephrine, and dopamine) and their metabolites (Aygun and Uludag, 2020). PCCs and PGLs, in combination, are called PPGL (Boyd et al., 2019). Sympathetic PGLs cause excess catecholamines, while parasympathetic PGLs are mostly non-secreting. Symptoms of PCC/PGL are due to mass effects or catecholamine hypersecretion (e.g., sustained or paroxysmal elevations in blood pressure, headache, profuse sweating, intense palpitations, paleness, and apprehension or anxiety) (Else et al., 2018). Hypertension is present in 80–90% of the patients, while approximately 25% of the patients present with the classically defined symptom triad of headache, sweating, and palpitations (Benn et al., 2015; Aygun and Uludag, 2020). Diagnosis is typically confirmed by determination of plasma and urinary fractionated catecholamines and metanephrines, being the determination of plasma free metanephrines the test of choice (Lenders et al., 2002). Non-hormone-producing PGLs are found in the head and neck area, and less frequently in the thorax; symptoms related to this type of tumors are due to pressure on surrounding nerves and include hearing loss, pulsatile tinnitus, cough, hoarseness, dysphagia, facial paralysis, pain or abnormal tongue motility (Benn et al., 2015). Tumor location is determined based on imaging studies, such as magnetic resonance imaging, computed tomography, positron-emission tomography, and/or scintigraphy (Oleaga and Goñi, 2008).
PPGLs have an incidence between 2 and 8/1,000,000 inhabitants; they occur most frequently between 40 and 50 years and mostly in women (55%) (Farrugia et al., 2017; Garcia-Carbonero et al., 2021). However, 20% of the cases are pediatric with a peak of presentation between 11 and 13 years of age (Aygun and Uludag, 2020; Jain et al., 2020). At the Instituto Nacional de Cancerología, Colombia (INC-C), in 2019, three new cases of malignant PCC and one case of malignant PGL with unknown primary location were reported (Anuario estadístico 2019, 2021). In 2020 COVID-19 affected, the number of new cases was similar with three new cases of malignant PCC and two cases of malignant PGL with unknown primary location (Anuario estadístico 2020, 2021). The proportion of women was 4/5 in 2020, while in 2019 the proportion of women was 2/4.
PPGLs can occur sporadically or as part of different hereditary tumor syndromes (Welander et al., 2011; Fishbein, 2016; Aygun and Uludag, 2020). Several studies have reported that 30–40% of the cases are caused by germline mutations (Neumann et al., 2002; Welander et al., 2011; Gimenez-Roqueplo et al., 2012; Dahia, 2014; Favier et al., 2015; Fishbein, 2016; Aygun and Uludag, 2020). The syndromes associated with inherited PGL/PCC are characterized by a predisposition to the development of PGLs distributed along the paravertebral axis from the base of the skull to the pelvis, as well as PCCs that are confined to the adrenal medulla (Else et al., 2018). Therefore, the diagnosis of a hereditary PGL/PCC syndrome should be suspected in any individual with a diagnosis of PGL or PCC, and the current recommendation is to offer germline genetic studies to all of these patients, regardless of family history or age at presentation (Plouin et al., 2016; Aygun and Uludag, 2020), especially if the presentation is multiple, multifocal, recurrent, or early-onset (<35 years) (Muth et al., 2012). About 15–20% of the hereditary cases are associated with pathogenic/likely pathogenic (P/LP) germline variants in genes that encode different subunits of the succinate dehydrogenase complex-SDH (SDHA, SDHB, SDHC, and SDHD, and the cofactor SDHAF2) (Else et al., 2018; Garcia-Carbonero et al., 2021). Also, 9% of the cases are associated with Von Hippel-Lindau syndrome - VHL (VHL gene), 5% corresponds to multiple endocrine neoplasia 2 syndrome—MEN2 (RET proto-oncogene), and 2% are associated with neurofibromatosis type 1 syndrome—NF1 (NF1 gene). Other genes associated with hereditary PGL/PCC are TMEM127, MAX, FH, MEN1, EGLN2, MDH2, SLC25A11, and DLST, with lower frequencies (<1–2%) (Dahia, 2014; Garcia-Carbonero et al., 2021).
In this study, we present for the first time the clinical characterization and spectrum of causative gene variants identified in a series of unrelated Colombian patients affected with PCC and/or PGL.
2 Materials and methods
2.1 Type of study and patients
This is a registry-based study on patients affected with PPGL tumors that were included in the Hereditary Cancer Program developed in the INC-C. This institutional Program was approved by the Scientific Committee of the INC-C in 2017 and was established to offer germline genetic testing to cancer patients at the INC-C as a care service for their comprehensive management. Patients participating in this institutional Program accepted to be donors at the tissue Biobank from the INC-C named “Banco Nacional de Tumores Terry Fox” and signed an informed consent for DNA biobanking and future research studies. The Program’s registry is being implemented for epidemiological reports. A total of 20 unrelated patients affected with PGL or PCC were identified in the registry from April 2018 to December 2021. To confirm a hereditary PGL/PCC syndrome, most of them were referred from the clinical oncology or endocrinology services at the INC-C to the Institutional´s genetic counselling service, and only five of them were managed by a medical geneticist at external institutions. All patients were offered genetic testing for diagnostic purposes, given the high frequency of germline mutations reported for these patients and international recommendations. Only 1 out of the 20 patients did not accept the genetic study. Of the 19 patients with genetic results, 14 were performed at the INC-C and 5 corresponded to studies carried out in external laboratories.
2.2 DNA extraction, library preparation, and massive sequencing (NGS)
Germline DNA was extracted from peripheral blood using the Quick-DNA TM Miniprep Plus Kit (Zymo Research, United States) following the manufacturer’s instructions. DNA was quantified with the Qubit dsDNA BR kit (Invitrogen) and the NanoDrop™ 2000 (Thermo Scientific) equipment, and DNA quality was assessed with the Bioanalyzer High Sensitivity DNA Analysis Kit (Agilent). Library preparation and sequencing was performed as recommended by the manufacturer’s protocol for the Nextera Flex for Enrichment Illumina kit and the Canadian Consortia Inherited Cancer a customized probe panel (reference # 20011891; Illumina Inc., San Diego, United States), that targets 105 genes known to be strongly associated with inherited cancers—or candidate genes—and detects single nucleotide variants (SNVs) and small insertions and deletions (INDELs) (Table 1). Additionally, the multigene panel design allows inferring possible copy number variants (CNVs) in all genes with bioinformatics methods by using sequencing data. Briefly, the DNA sample was enriched for the target regions using the protocol by Nextera Flex for Enrichment (Illumina Inc., San Diego, CA) based on hybridization, and the libraries were sequenced using paired-end technology (2 × 251 cycles) in a MiSeq instrument (Illumina Inc., San Diego, CA). Unless otherwise indicated, all target regions were sequenced to a depth greater than 100X (if an allelic depth greater than 50X was not achieved, complementary analyses of the region of interest were performed by orthogonal methods). This assay focuses on the coding sequences of the genes included. Promoter regions, non-transcribed regions, and other non-coding regions are not included. NGS analysis was previously standardized and validated in our laboratories to sequence 12 samples simultaneously and according to the Analytical Performance results we obtained a sensitivity of 99.89% and a specificity of 99.99% for the accurate detection of SNVs and small INDELs genetic variants using the commercial bioinformatics software developed by Sophia Genetics (Saint-Sulpice, Switzerland).
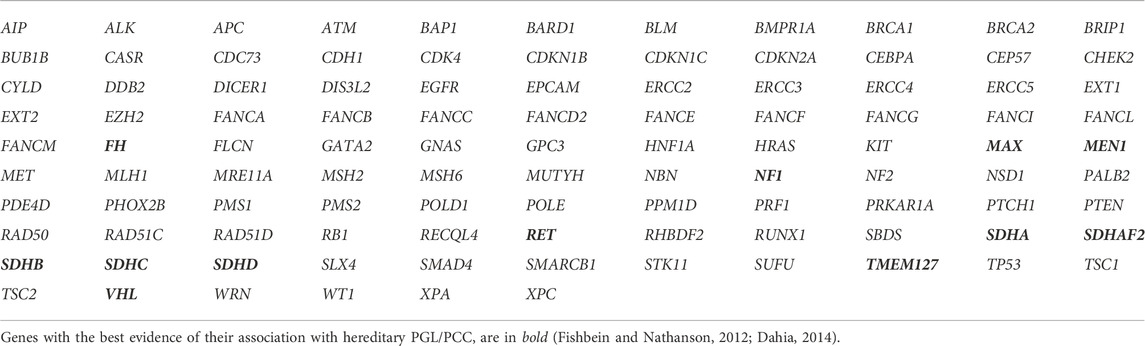
TABLE 1. Genes included in the multigene panel.
2.3 Variant calling, interpretation of genetic data, and mutation detection
Sequence reads in FastQC files were aligned to the hg19 human reference genome with the Burrows-Wheeler Aligner (BWA) tool. Variant calling and annotation of SNVs and INDELs were carried out with the SOPHiA DDM® platform using the ILL1IC1G3_TSC algorithm (Sophia Genetics, Saint-Sulpice, Switzerland). This algorithm also allows inferring CNVs from sequence data for all genes included in the panel, and the detection of ALU elements. The genetic variants detected by the SOPHiA algorithm were reviewed by an oncogeneticist and a biologist trained in genetics for interpretation and delivery of genetic results at the post-test consultation. The Human Genome Variation Society (HGVS) nomenclature (http://www.hgvs.org/) was used in the genetic report, and the five-tier criteria of the American College of Medical Genetics and Genomics (ACMG) for variant classification were implemented for variant classification (Richards et al., 2015). The genetic variants reported in this manuscript were included in an institutional automated or manual curation exercise, to update their classification.
2.4 Large deletions/duplication analyses
The gold-standard method to identify large genomic deletions and duplications is Multiplex Ligation-dependent Probe Amplification (MLPA). Among the eight patients describe here as carriers of the exon 1 deletion of the SDHB gene, two corresponded to cases with external results that included both NGS and MLPA analysis, and these cases were included in the Hereditary Cancer Program through the tissue Biobank as positive controls for future standardization purposes. The other six, corresponded to cases in which a CNV in the SDHB was detected through NGS at our genetic laboratory. All of these cases were also analyzed with MLPA at an external genetic laboratory. Positive control samples from our tissue Biobank were provided for this step.
Briefly, the SALSA MLPA Probemix P226 SDH was used for confirmation purposes. This consists in a semi-quantitative assay for both an in vitro diagnostic and research use. Regions covered with this assay are: SDHD 11q23.1; SDHB 1p36.1; SDHC 1q23.3; SDHAF1 19q13.12; SDHAF2 11q12.2 (https://www.mrcholland.com/product/P226/2401?). Detection by fragment analysis was performed following the manufacturer’s instructions (MRC-Holland, Amsterdam, Netherlands): 1) DNA extraction and denaturation; 2) Hybridization with the specific probes; 3) Ligation and amplification by PCR reaction; 4) Capillary electrophoresis of the amplified products in the ABI 3500 Genetic Analyzer sequencer (Applied Biosystems); and 5) Analysis of results with the program bioinformatician Coffalyzer.
2.5 Statistical analysis
The diagnosis of a hereditary PGL/PCC syndrome was established with the identification of a germline P/LP variant in the affected patient, in a gene known to be associated with increased susceptibility to developing PGL or PCC tumors, such as: MAX, NF1, RET, SDHA, SDHAF2, SDHB, SDHC, SDHD, TMEM127, VHL, FH and MEN1 (Table 1). Demographic data (sex, age at diagnosis), tumor type, clinical presentation, and results of the molecular study, were analyzed. Data were summarized using descriptive statistics.
3 Results
3.1 Demographic and clinical characteristics of patients
Of the 19 patients with genetic results, the majority were women (n = 12, 63.16%) and the mean age at first presentation was 34.8 years (range 9–60 years). Three patients (15.79%) had PGL or PCC of pediatric presentation (<18 years). Overall, 68.42% (13/19) had catecholamine-secreting tumors (or their metabolites), and 47.37% (9/19) had metastatic tumors (Table 2).
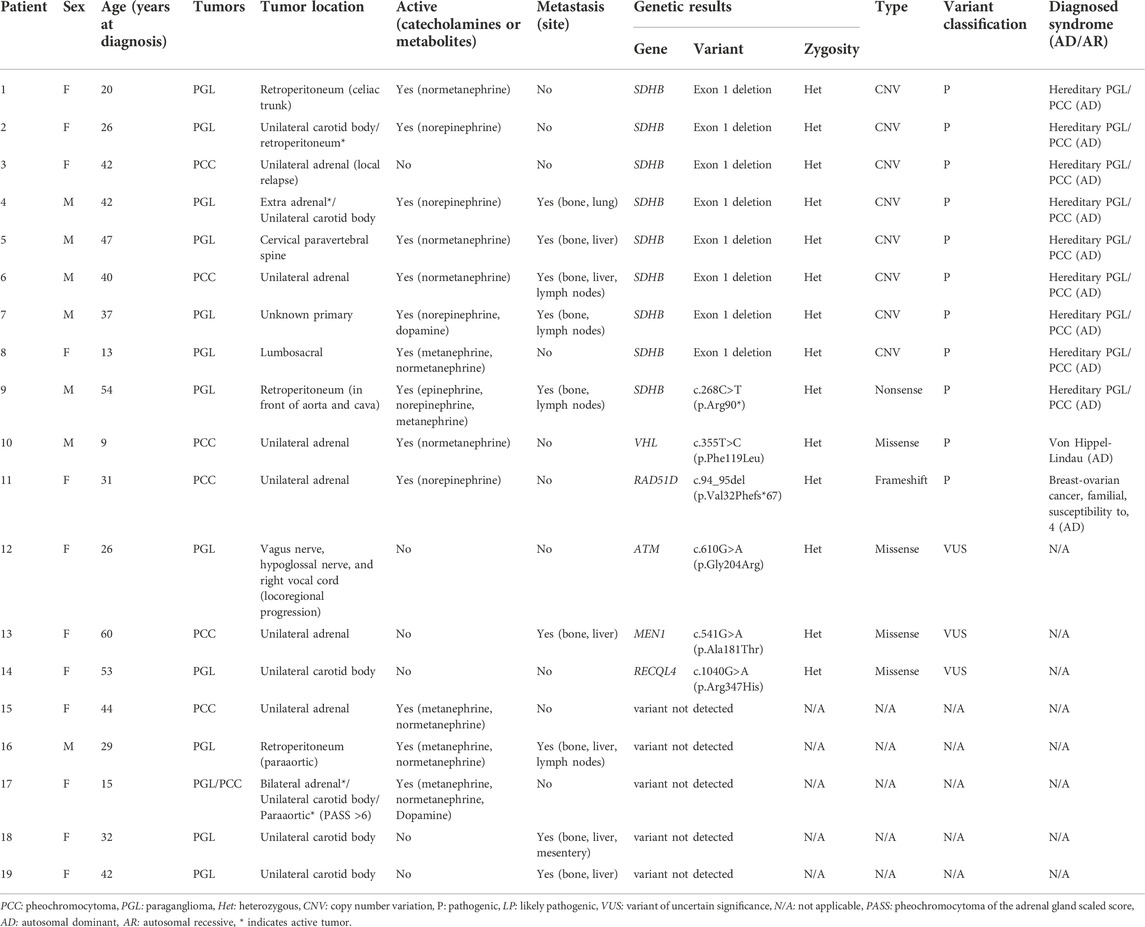
TABLE 2. Clinical characteristics and genetic test results of patients with pheochromocytoma/paraganglioma (n = 19).
Regarding tumor location and multifocality or multiple presentation, five patients (26.32%) presented PGL of the head and neck, three patients (38.5%) developed extra-adrenal abdominal PGL with involvement of the retroperitoneal region, and one patient (5.2%) had a PGL located at the lumbosacral level. Multiple PCC/PGL tumors were found in 15.79% (3/19) of the patients; two of them with multiple primary PGLs located in the head and neck and abdomen (retroperitoneum), and the third one developed bilateral PCC with various PGLs tumors located in the head and neck and abdomen. Finally, six patients (31.5%) had unilateral PCC, while another patient (5.2%) presented with metastatic PGL of unknown primary origin (Table 2).
About 26.32% (5/19) of the patients also developed other primary tumors different than PPGL (Table 3). Family history of PGL or PCC tumors were reported by 15.79% (3/19) of the cases (Table 3), while another 11 patients (57.89%) reported relatives affected with different tumors (data not shown).
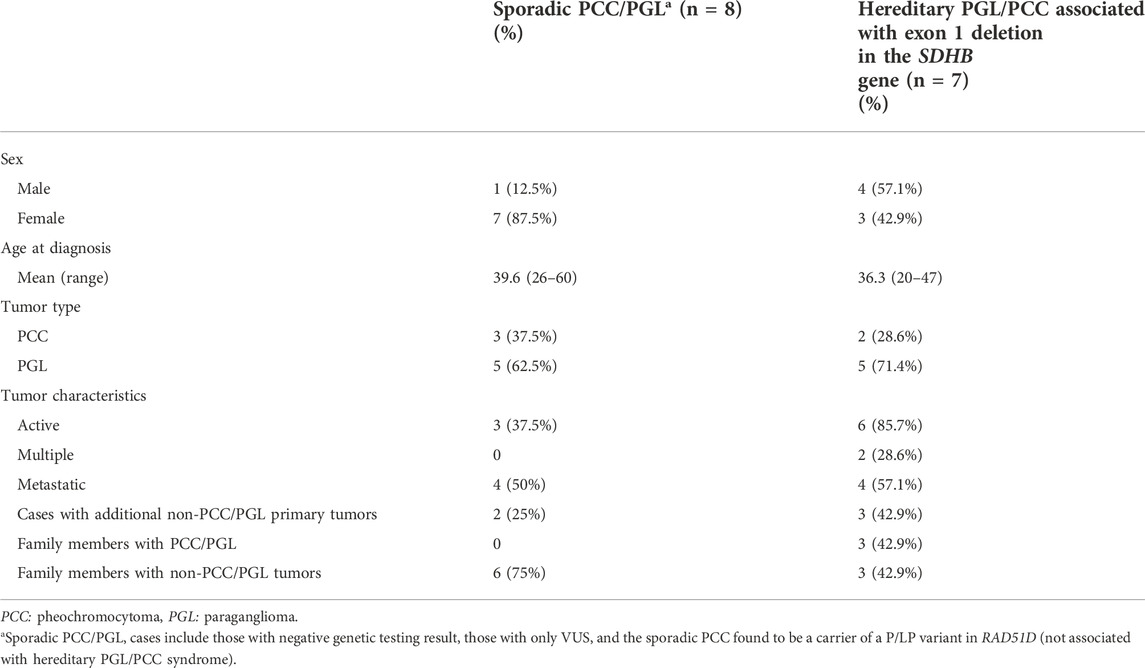
TABLE 3. Characteristics of adult patients with sporadic PCC/PGL and hereditary PGL/PCC associated with exon 1 deletion in the SDHB gene.
3.2 Germline genetic results and observed phenotypes
A heterozygous germline P/LP variant in a known PPGL susceptibility gene was detected in 52.63% (10/19) of the Colombian patients. In nine cases the affected gene was SDHB and were diagnosed with hereditary PGL/PCC syndrome (MedGen UID: 313270). Only one case was found to be a carrier of a disease-causing variant in the VHL gene and was diagnosed with VHL syndrome (MedGen UID: 42458) (Table 2). It is noteworthy that among the nine cases with a SDHB mutation, only one corresponded to a nonsense variant SDHB: c.268C>T (p.Arg90*), while the remaining eight are carriers of a recurrent CNV consisting of a large deletion of one copy of exon 1, explaining 80% (8/10) of the hereditary PGL/PCC cases and 42% (8/19) of all the cases affected with PGL or PCC tumors (Table 2; Figure 1). The patient with the nonsense variant in the SDHB gene was diagnosed at age 54 years with a metastatic secreting-PGL tumor, with neither a personal history of second primary tumors nor a family history of PGL/PCC tumors (Table 2). On the other hand, the patient with the missense mutation in the VHL gene: c.355T>C (p.Phe119Leu) corresponds to a child diagnosed at 9 years with non-metastatic secreting-PCC tumor, not associated with second primaries or positive family history (Table 2).
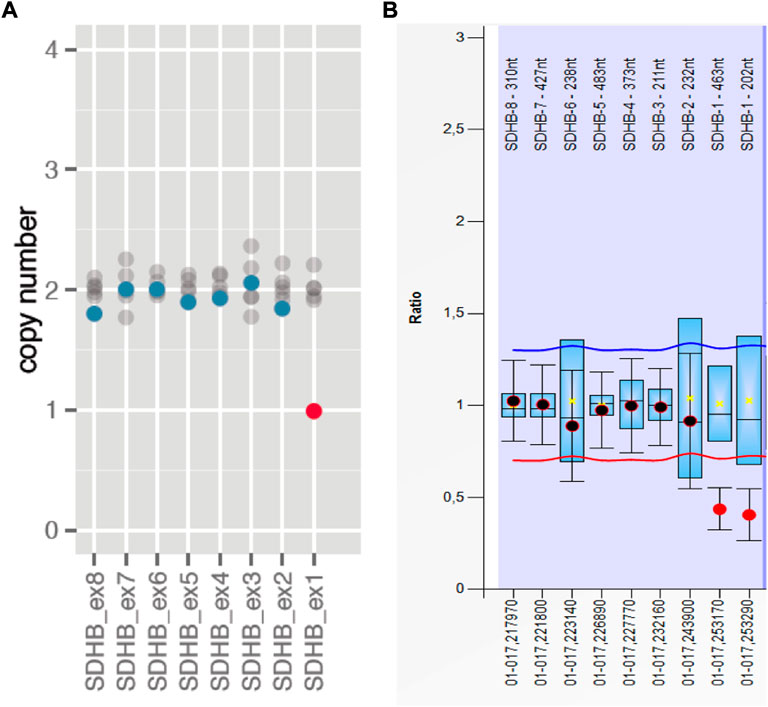
FIGURE 1. Recurrent CNV in the SDHB gene in Colombians consisting in a large deletion of one copy of exon 1. (A) The graphic shows the copy number of the carrier (dots in color blue/red) and if it is similar (blue dots) or different (red dots) compared to the median of the copy numbers of each exon (1–8) among 8 different samples (gray dots) included in the same run. Graphic generated and extracted from SOPHIA Genetics. (B) Corresponding graphic obtained with MLPA confirmatory assay.
An additional patient with an incidental finding of a unilateral, non-metastatic, secreting PCC tumor at age 31 years, carries a pathogenic variant in RAD51D gene that confers an autosomal dominant predisposition to breast and ovarian cancer (MedGen UID: 481975), but is not considered to be associated with a hereditary PGL/PCC syndrome (Table 2). In accordance with the above, 57.89% (11/19) of the cases are carriers of a P/LP germline variant associated with any hereditary cancer syndrome, 15.79% (n = 3) of the patients had a variant of uncertain significance (VUS), and 26.32% (n = 5) of the cases had negative results.
Table 3 describes some characteristics among patients >18 years with sporadic PGL or PCC (n = 8; these are cases with negative results or a VUS and also includes the carrier of the pathogenic variant in RAD51D since the PPGL tumor is considered unrelated) and hereditary cases with the recurrent CNV in the SDHB gene (n = 7). The mean age at diagnosis of the sporadic cases was 39.62 years; excluding the sporadic case with RAD51D mutation, the mean age at diagnosis would be 40.85 years in the sporadic group. On the other hand, the mean age at diagnosis in the hereditary PGL/PCC group with the recurrent CNV in SDHB was 36.29 years. In both groups, a higher proportion of PGL tumors was observed (62.5% and 71.4%, respectively), with lower frequency of PCC tumors (37.5% and 28.6%, respectively).
Other characteristics to mention in the group of hereditary cases is that 85.7% had secreting-tumors, 28.6% had multiple tumors, and 57.1% were metastatic at diagnosis. In patients with sporadic PGL or PCC, the proportion of secreting-tumors was only 37.5%, none of them had multifocal disease or multiple tumors, and 50% had metastatic disease (Table 3). Also, in the group of hereditary PGL/PCC more cases developed other primary tumors different from PPGL compared to the sporadic group (42.9% vs. 25%, respectively). Finally, none of the sporadic cases reported a family history of PGL/PCC tumors, although 75% of them had a positive family history for other tumors types; while in cases with the CNV in SDHB, 42.9% reported a positive family history of PPGL tumors and another 42.9% had a positive family history for other tumor types (Table 3). As for the age at diagnosis of metastatic disease, the mean in both groups was 44.25 years (data not shown).
Regarding the three patients with pediatric presentation (not included in Table 3), in two of them with NGS and MLPA studies, a P/LP variant associated with hereditary PGL/PCC was detected (Table 2). These are Patient 8 diagnosed at age 13 years with non-metastatic secreting-PGL tumor of lumbosacral location in whom the recurrent CNV in SDHB was identified, and Patient 10 diagnosed at age 9 years with non-metastatic secreting-PCC tumor, who carries the pathogenic variant in the VHL gene. The third pediatric patient corresponds to a case with a 14-gene NGS study reported as negative (without MLPA studies according to the EMR), but with a high suspicion of a hereditary PGL/PCC syndrome since the age at diagnosis was 15 years with multiple secreting-PGL/PCC tumors (including bilateral PCC with a Pheochromocytoma of the Adrenal gland Scaled Score—PASS >6 and a second primary). This case was only evaluated by a geneticist from another institution, and in the absence of MLPA studies, it should be considered as inconclusive (Table 2).
4 Discussion
PGL/PCC-predisposing germline variants were identified in 10 (52.63%) of 19 Colombian affected cases in our study, which is higher than expected compared to what has been reported in the literature (30–40%) (Welander et al., 2011; Fishbein, 2016; Aygun and Mehmet, 2020). Of the 12 known PPGL susceptibility genes included in the multigene cancer panel used in the Hereditary Cancer Program, only SDHB and VHL were mutated. SDHB is considered one of the main susceptibility genes associated with PPGL tumors (Cascón et al., 2009; Aim et al., 2021), and in this study we identified that 90% of the hereditary cases were explained by a pathogenic variant in the SDHB gene. Moreover, mutations in SDHB gene were found in the 47.37% of all the affected patients analyzed in this study. This result is similar to reports from other studies conducted in Saudi Arabia and Chinese populations in whom SDHB mutations were the most frequently found (Saudi Arabia: 20.8% of 101 affected cases and 57.75% of 37 inherited cases; China: 29.6% of 314 affected cases and 49.46% of 93 inherited cases) (Albattal et al., 2019); but different than other reports from European populations affected with PPGL tumors, in which VHL was reportedly the most frequently mutated gene (Gimenez-Roqueplo et al., 2006). Scientific evidence supports that both the prevalence and the genetic profile of hereditary PGL/PCC varies among different ethnic populations.
SDHB acts as a tumor suppressor gene associated as a cause of hereditary PPGL (Astuti et al., 2001). The SDHB protein (OMIM185470) corresponds to the iron-sulfur catalytic subunit of the heterotetrameric succinate dehydrogenase (SDH) complex, a component of the tricarboxylic acid cycle and the mitochondrial respiratory chain (complex II) (Aim et al., 2021). As of February 2022, 388 variants have been reported in the database of SDH mutations in the LOVD system (Bayley et al., 2005). Recent reports from an international initiative for a curated SDHB variant database, composed of 223 variants from 737 patients worldwide, highlights that 44% of these variants were missense, 17% were frameshift indels, 10% affects splice site, 9% were nonsense, 6% corresponded to large rearrangements and 14% to other variant types (Aim et al., 2021). This type of variant distribution persisted within the group of curated P/LP genetic variants in SDHB, with 34%, 26%, 15%, 14%, 9% and 3%, respectively (Aim et al., 2021). In contrast, in our study of unrelated cases, 89% (8/9) of the P/LP genetic variants in the SDHB gene corresponded to a large deletion of one copy of exon 1 of the gene, while the remaining 11% corresponded to a nonsense variant.
Exon 1 deletion in the SDHB gene has been previously reported in the literature in unrelated patients and families (McWhinney et al., 2004; Cascón et al., 2006; Cascon et al., 2007), leading to the hypothesis that this deletion may be related to a hotspot or a founder effect (Cascón et al., 2006). The molecular characterization of the deletion revealed the same breakpoints in the unrelated Spanish families studied, all originating from a small, restricted area in the northeast of the Iberian Peninsula, and that these differed from the breakpoints found in a French patient (Cascon et al., 2007). Reviewing the city of origin of the eight carriers of the recurrent CNV in the SDHB gene reported in this study, all but one were born in Bogotá and one patient was born in another city located 378 km southwest of Bogotá (called Neiva); nevertheless, both cities are located in the Andean region of the country. It would be interesting to test whether a founder effect similar to that observed in Spanish families is responsible for this apparent high prevalence in our Colombian patients, or if this high frequency could be explained by the presence of mutational hot spots in the SDHB gene. Germline haplotype information could help to better unravel the mechanisms associated with the reported recurrent mutation.
It has been reported that in carriers of P/LP variants in the SDHB gene, the average age at diagnosis of PPGL tumors is 31.7 years, with a range of 3–75 years; thus, the follow-up recommended in these carriers is in the childhood stage (Muth et al., 2018). In our study of Colombian cases with SDHB exon 1 deletion, the mean age at diagnosis of PPGL tumors (including adults and children) was 33.38 years (13–47 years), which is comparable to what has been reported in the literature for carriers of this large deletion (8–30 years) (McWhinney et al., 2004). In general, SDHB mutations have been associated primarily with PCC-type tumors (Ricketts et al., 2010); however, PGLs have previously been described as the primary tumor type diagnosed in cases with exon 1 deletion in the SDHB gene (Astuti et al., 2001; Cascón et al., 2006; Cascon et al., 2007). This is consistent with our study, since in 62% (6/8) of the patients with this large deletion in the SDHB gene, the initial finding was a PGL tumor. Moreover, variable expressivity was observed in relation to diverse primary PGL tumor locations and metastatic behavior, which is consistent with what has been previously reported (Ricketts et al., 2010; Else et al., 2018).
In our study, we did not observed differences regarding the age at diagnosis of metastatic disease between the group of patients with SDHB exon 1 deletion and the group of patients with sporadic PGL or PCC tumors, while a trend towards an earlier age at diagnosis of metastatic disease has been reported in patients carrying mutation in SDHx genes (especially SDHB) (Fishbein et al., 2017). Nevertheless, there is a discussion in the literature about a possible detection bias because in these patients a greater alert for follow-up can be generated due to the malignant potential attributed to the alteration in SDH activity (Fishbein et al., 2017; Tabebi et al., 2022). For example, it has been described that inactivating mutations in SDH complex suppress its activity, resulting in the accumulation of succinate, an oncometabolite that generates a pseudohypoxic phenotype and contributes to cancer, and that the biological effect of knocking down the SDHB gene is associated with a positive effect on cell proliferation, which is related to increased chromaffin cell metabolism and their adaptation to glutamine use as an alternative source of energy, thus promoting the activity of the oxidative phosphorylation system (Tabebi et al., 2022).
In the European-American-Pheochromocytoma-Paraganglioma-Registry (EAPPR), an 80% of 177 unrelated pediatric PCC/PGL patients had a germline mutation in a susceptibility gene; 38% of the patients had a second primary PGGL tumor and this trend increased with age, essentially in cases with hereditary disease, until reaching a frequency of 50% at 30 years after the initial diagnosis. Of the hereditary cases, 9% had malignant tumors and these were commonly associated with mutated SDHB (Bausch et al., 2014). Among the pediatric cases included in our study, Patient 17 had an initial diagnosis of left PCC at age 15 years, and second PPGL primary tumors at age 22 and 30 years managed by the oncology and endocrinology services at the INC-C, but with negative germline genetic result performed in an external laboratory. This case was evaluated by an extra-institutional medical geneticist and according to the EMR was concluded as not associated with a hereditary PGL/PCC syndrome; however, considering the high suspicion of a hereditary PGL/PPC and the elevated prevalence of exon 1 deletion in the SDHB gene documented in our study, it is necessary to complement the genetic studies in this patient with MLPA.
Another well-known syndrome associated with inherited PGL/PCC tumors is VHL disease, which is also associated with a variety of benign and malignant tumors, mainly retinal and cerebellar tumors, and spinal hemangioblastoma and renal cell carcinoma (van Leeuwaarde et al., 2018). This syndrome is caused by mutations in the VHL gene and has been reported to account for 49% of pediatric patients with PCC/PGL (Bausch et al., 2014). In our study, we only identified a VHL germline mutation in 1 out of 3 (33%) affected pediatric patients, corresponding to a 10% (1/10) of all of the hereditary cases.
Given the high frequency of PPGL tumors with germline mutations found in our study, we support the recommendation that genetic testing should be considered in all patients with PPGL (Plouin et al., 2016; Muth et al., 2018; Aygun and Mehmet, 2020). Equally important is to assess how complete or informative the genetic results are, by recording the type of genetic testing, the covered genes, the molecular techniques implemented, and the overall quality of the report. In our study, we used a large multigene panel that allows the analysis of 105 known cancer susceptibility genes (and candidate genes), including MAX, NF1, RET, SDHA, SDHAF2, SDHB, SDHC, SDHD, TMEM127, VHL, FH and MEN1 associated with PGL/PCC predisposition. Other genes less frequently associated with inherited PGL/PCC cases (<1–2%) were not analyzed. These genes are EGLN1, EGLN2, MDH2, SLC25A11, DLST and KIF1B (Fishbein and Nathanson, 2012; Dahia, 2014; Garcia-Carbonero et al., 2021). We recognized that this could be a limitation of the study, specially for the interpretation of negative cases; nevertheless, we consider that our results are comparable with other publications since these less characterized genes are not usually included in commercial multigene PGL/PCC panels.
The penetrance for SDHB-associated PPGL tumors has been estimated as 21% at age 70 years and 30.6% at age 80 years (Hes et al., 2010; Rijken et al., 2016; Andrews et al., 2018). In our study, a family history of PPGL was reported in 37.5% (3/8) of the patients with exon 1 deletion in the SDHB gene. Medical reports of genetic testing and biochemical- or imaging-surveillance screening for SDHB-related tumors detection among the carriers’ relatives, were not available since they were not being followed in the INC-C. We deem this to be a limitation of the study, since we could not determined the penetrance for PPGL or other related tumors.
Future studies for calculating variant penetrance using large family pedigrees and/or population data from greater cohorts are possible with the increasing accessibility of high-throughput technologies. The study design should include follow-up data obtained from cascade testing and surveillance screening. For individuals with a genetic diagnosis of hereditary PGL/PCC, cascade testing should be offered to first-degree relatives. Genetic testing for under-age relatives should be individualized according to the gene involved (Greer et al., 2017; Muth et al., 2018). Screening recommendations for presymptomatic SDHB-carriers include annual biochemical plasma or urine testing (comprising normetanephrine and metanephrine), accompanied by whole-body magnetic resonance imaging (RWB-MRI) every 2 years, starting 15 years. In younger children, it is recommended to design an individual surveillance program together with a pediatrician and based on family history; the recommendation to initiate surveillance 5 years before the age of the youngest affected individual in the family is generally accepted (Muth et al., 2018). Clinical examinations should be performed at least twice a year (Muth et al., 2018). Regarding follow-up after treatment in PPGL patients with a high-risk genetic disease, the European Society of Endocrinology recommends testing plasma or urinary metanephrines every year for local or metastatic recurrences or new tumors for life (Plouin et al., 2016).
It is beyond the scope of this article to comprehensively cover and discuss the complex management options for PPGL patients, which require the involvement of an expert multidisciplinary team to evaluate different factors of the individual (e.g., age, genetic profile) and the tumor (e.g., primary tumor site, hormone secretion profile, molecular profile). For interested readers, recent scientific literature about this topic can be reviewed in the following cited references (Roman-Gonzalez et al., 2018; Pryma et al., 2019; Tanabe and Naruse, 2020; Garcia-Carbonero et al., 2021).
Our findings allow us to conclude that germline mutations in SDHB play an important role in hereditary PGL/PCC syndromes in Colombians. Even though most of the variants reported in the SDHx genes (including SDHB) are missense mutations, the large deletion of exon 1 in SDHB is a recurrent mutation among our patients, explaining 42% (8/19) of the PCC and/or PGL cases, in which a variable expressivity of the phenotype associated with this large deletion was observed. The evaluation of large deletions in the SDHB gene should be performed in patients with PCC and/or PGL in our country.
Data availability statement
The data presented in the study are deposited in the NCBI SRA Bioproject repository (https://www.ncbi.nlm.nih.gov/sra), under the accession number PRJNA907395.
Author contributions
MM and MS (conceptualization; data curation; formal analysis; funding acquisition; investigation; project administration; supervision; validation; preparation; writing review and editing); MM and AR (data curation); AR, ST, JR and VM (investigation); MS, AH, AR, JR and JM (project administration; resources); GG, AG, AGC, MT, AC, LF, and PA (writing review and editing).
Funding
MS (C19990300218—Program for the Creation of a National Network of Hereditary Cancer in Colombia, INC-C); MS (C19990300210—Institutional Program for the Identification and Management of Families with Suspected Hereditary Cancer, INC-C).
Acknowledgments
The authors gratefully acknowledge the INC-C and its staff for their contribution with data from the Hereditary Cancer Program Registry.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ACMG, American College of Medical Genetics and Genomics; CNV, Copy Number Variation; HGVS, Human Genome Variation Society; INDEL, Insertion-Deletion Mutation; INC-C, Instituto Nacional de Cancerología, Colombia; 131I MIBG, 131I-meta-iodobenzylguanidine; MEN2, Multiple Endocrine Neoplasia Type 2; MLPA, Multiplex Ligation-Dependent Probe Amplification; N/A, Not Applicable; NGS, Next Generation Sequencing; P/LP, Pathogenic or Likely Pathogenic; PCC, Pheochromocytoma; PGL, Paraganglioma; PPGL, Pheochromocytomas and Paragangliomas in combination; PRRT, Peptide Receptor Radionuclide Therapy; RWB-MRI, Whole-Body Magnetic Resonance Imaging; SDHB, Succinate Dehydrogenase Subunit B; SDHx, Genes encoding subunits of Succinate Dehydrogenase Enzyme; SNV, Single Nucleotide Variant; VHL, Von Hippel-Lindau; VUS, Variant of Uncertain Significance.
References
Aim, L. B., Maher, E. R., Cascon, A., Barlier, A., Giraud, S., Ercolino, T., et al. (2021). International initiative for a curated SDHB variant database improving the diagnosis of hereditary paraganglioma and pheochromocytoma. J. Med. Genet. 0, 1–8. doi:10.1136/jmedgenet-2020-107652
Albattal, S., Alswailem, M., Moria, Y., Al-Hindi, H., Dasouki, M., Abouelhoda, M., et al. (2019). Mutational profile and genotype/phenotype correlation of non-familial pheochromocytoma and paraganglioma. Oncotarget 10, 5919–5931. doi:10.18632/oncotarget.27194
Andrews, K. A., Ascher, D. B., Pires, D. E. V., Barnes, D. R., Vialard, L., Casey, R. T., et al. (2018). Tumour risks and genotype–phenotype correlations associated with germline variants in succinate dehydrogenase subunit genes SDHB, SDHC and SDHD. J. Med. Genet. 55, 384–394. doi:10.1136/jmedgenet-2017-105127
Astuti, D., Latif, F., Dallol, A., Dahia, P. L. M., Douglas, F., George, E., et al. (2001). Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am. J. Hum. Genet. 69, 49–54. doi:10.1086/321282
Aygun, N., and Mehmet, U. (2020). Pheochromocytoma and paraganglioma: From clinical findings to diagnosis. Sisli Etfal Hastan. Tip. Bul. 8, 271–280. doi:10.14744/SEMB.2020.14826
Aygun, N., and Uludag, M. (2020). Pheochromocytoma and paraganglioma: From epidemiology to clinical findings. Sisli Etfal Hastan. Tip. Bul. 54, 159–168. doi:10.14744/SEMB.2020.18794
Bausch, B., Wellner, U., Bausch, D., Schiavi, F., Barontini, M., Sanso, G., et al. (2014). Long-term prognosis of patients with pediatric pheochromocytoma. Endocr. Relat. Cancer 21, 17–25. doi:10.1530/ERC-13-0415
Bayley, J.-P., Devilee, P., and Taschner, P. E. (2005). The SDH mutation database: An online resource for succinate dehydrogenase sequence variants involved in pheochromocytoma, paraganglioma and mitochondrial complex II deficiency. J. Med. Genet. 6, 785–792. doi:10.1186/1471-2350-6-39
Benn, D. E., Robinson, B. G., and Clifton-Bligh, R. J. (2015). 15 years of paraganglioma: Clinical manifestations of paraganglioma syndromes types 1–5. Endocr. Relat. Cancer 22, T91–T103. doi:10.1530/ERC-15-0268
Berends, A. M. A., Buitenwerf, E., de Krijger, R. R., Veeger, N. J. G. M., van der Horst-Schrivers, A. N. A., Links, T. P., et al. (2018). Incidence of pheochromocytoma and sympathetic paraganglioma in The Netherlands: A nationwide study and systematic review. Eur. J. Intern. Med. 51, 68–73. doi:10.1016/j.ejim.2018.01.015
Boyd, J., Leung, A. A., Sadrzadeh, H. S., Pamporaki, C., Pacak, K., Deutschbein, T., et al. (2019). A high rate of modestly elevated plasma normetanephrine in a population referred for suspected PPGL when measured in a seated position. Eur. J. Endocrinol. 181, 301–309. doi:10.1530/EJE-19-0176
Cascon, A., Landa, I., Lopez-Jimenez, E., Diez-Hernandez, A., Buchta, M., Montero-Conde, C., et al. (2007). Molecular characterisation of a common SDHB deletion in paraganglioma patients. J. Med. Genet. 45, 233–238. doi:10.1136/jmg.2007.054965
Cascón, A., Montero-Conde, C., Ruiz-Llorente, S., Mercadillo, F., Letón, R., Rodríguez-Antona, C., et al. (2006). GrossSDHB deletions in patients with paraganglioma detected by multiplex PCR: A possible hot spot? Genes Chromosom. Cancer 45, 213–219. doi:10.1002/gcc.20283
Cascón, A., Pita, G., Burnichon, N., Landa, I., López-Jiménez, E., Montero-Conde, C., et al. (2009). Genetics of pheochromocytoma and paraganglioma in Spanish patients. J. Clin. Endocrinol. Metab. 94, 1701–1705. doi:10.1210/jc.2008-2756
Dahia, P. L. M. (2014). Pheochromocytoma and paraganglioma pathogenesis: Learning from genetic heterogeneity. Nat. Rev. Cancer 14, 108–119. doi:10.1038/nrc3648
Else, T., Greenberg, S., and Fishbein, L. (2018). Hereditary paraganglioma-pheochromocytoma syndromes. Editors M. P. Adam, D. B. Everman, and G. M. Mirzaa (Seattle, WA: University of Washington, Seattle). GeneReviews® [Internet]. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1548/.
Farrugia, F., Martikos, G., Tzanetis, P., Charalampopoulos, A., Misiakos, E., Zavras, N., et al. (2017). Pheochromocytoma, diagnosis and treatment: Review of the literature. Endocr. Regul. 51, 168–181. doi:10.1515/enr-2017-0018
Favier, J., Amar, L., and Gimenez-Roqueplo, A.-P. (2015). Paraganglioma and phaeochromocytoma: From genetics to personalized medicine. Nat. Rev. Endocrinol. 11, 101–111. doi:10.1038/nrendo.2014.188
Fishbein, L., Ben-Maimon, S., Keefe, S., Cengel, K., Pryma, D. A., Loaiza-Bonilla, A., et al. (2017). SDHB mutation carriers with malignant pheochromocytoma respond better to CVD. Endocr. Relat. Cancer 24, L51–L55. doi:10.1530/ERC-17-0086
Fishbein, L., and Nathanson, K. L. (2012). Pheochromocytoma and paraganglioma: Understanding the complexities of the genetic background. Cancer Genet. 205, 1–11. doi:10.1016/j.cancergen.2012.01.009
Fishbein, L. (2016). Pheochromocytoma and paraganglioma: Genetics, diagnosis, and treatment. Hematol. Oncol. Clin. North Am. 30, 135–150. doi:10.1016/j.hoc.2015.09.006
Furlan, A., Dyachuk, V., Kastriti, M. E., Calvo-Enrique, L., Abdo, H., Hadjab, S., et al. (2017). Multipotent peripheral glial cells generate neuroendocrine cells of the adrenal medulla. Science 357, eaal3753. doi:10.1126/science.aal3753
Garcia-Carbonero, R., Matute Teresa, F., Mercader-Cidoncha, E., Mitjavila-Casanovas, M., Robledo, M., Tena, I., et al. (2021). Multidisciplinary practice guidelines for the diagnosis, genetic counseling and treatment of pheochromocytomas and paragangliomas. Clin. Transl. Oncol. 23, 1995–2019. doi:10.1007/s12094-021-02622-9
Gimenez-Roqueplo, A.-P., Dahia, P., and Robledo, M. (2012). An update on the genetics of paraganglioma, pheochromocytoma, and associated hereditary syndromes. Hormone Metabolic Res. 44, 328–333. doi:10.1055/s-0031-1301302
Gimenez-Roqueplo, A.-P., Lehnert, H., Mannelli, M., Neumann, H., Opocher, G., Maher, E. R., et al. (2006). Phaeochromocytoma, new genes and screening strategies. Clin. Endocrinol. 65, 699–705. doi:10.1111/j.1365-2265.2006.02714.x
Greer, M.-L. C., Voss, S. D., and States, L. J. (2017). Pediatric cancer predisposition imaging: Focus on whole-body MRI. Clin. Cancer Res. 23, e6–e13. doi:10.1158/1078-0432.CCR-17-0515
Hes, F. J., Weiss, M. M., Woortman, S. A., de Miranda, N. F., van Bunderen, P. A., Bonsing, B. A., et al. (2010). Low penetrance of a SDHB mutation in a large Dutch paraganglioma family. BMC Med. Genet. 11, 92. doi:10.1186/1471-2350-11-92
Jain, A., Baracco, R., and Kapur, G. (2020). Pheochromocytoma and paraganglioma—An update on diagnosis, evaluation, and management. Pediatr. Nephrol. 35, 581–594. doi:10.1007/s00467-018-4181-2
Kastriti, M. E., Kameneva, P., Kamenev, D., Dyachuk, V., Furlan, A., Hampl, M., et al. (2019). Schwann cell precursors generate the majority of chromaffin cells in zuckerkandl organ and some sympathetic neurons in paraganglia. Front. Mol. Neurosci. 12, 6. doi:10.3389/fnmol.2019.00006
Lenders, J. W. M., Pacak, K., Walther, M. M., Linehan, W. M., Mannelli, M., Friberg, P., et al. (2002). Biochemical diagnosis of pheochromocytoma. JAMA 287, 1427–1434. doi:10.1001/jama.287.11.1427
McWhinney, S. R., Pilarski, R. T., Forrester, S. R., Schneider, M. C., Sarquis, M. M., Dias, E. P., et al. (2004). Large germline deletions of mitochondrial complex II subunits SDHB and SDHD in hereditary paraganglioma. J. Clin. Endocrinol. Metab. 89, 5694–5699. doi:10.1210/jc.2004-0769
Muth, A., Abel, F., Jansson, S., Nilsson, O., Ahlman, H., and Wängberg, B. (2012). Prevalence of germline mutations in patients with pheochromocytoma or abdominal paraganglioma and sporadic presentation: A population-based study in western Sweden. World J. Surg. 36, 1389–1394. doi:10.1007/s00268-012-1430-6
Muth, A., Crona, J., Gimm, O., Elmgren, A., Filipsson, K., Stenmark Askmalm, M., et al. (2018). Genetic testing and surveillance guidelines in hereditary pheochromocytoma and paraganglioma. J. Intern. Med. 285, 187–204. doi:10.1111/joim.12869
Neumann, H. P. H., Bausch, B., McWhinney, S. R., Bender, B. U., Gimm, O., Franke, G., et al. (2002). Germ-line mutations in nonsyndromic pheochromocytoma. N. Engl. J. Med. 346, 1459–1466. doi:10.1056/NEJMoa020152
Oleaga, A., and Goñi, F. (2008). Pheochromocytoma: Diagnostic and therapeutic update. Endocrinol. Nutr. 55, 202–216. doi:10.1016/S1575-0922(08)70669-7
Plouin, P. F., Amar, L., Dekkers, O. M., Fassnacht, M., Gimenez-Roqueplo, A. P., Lenders, J. W. M., et al. (2016). European Society of Endocrinology Clinical Practice Guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. Eur. J. Endocrinol. 174, G1–G10. doi:10.1530/EJE-16-0033
Pryma, D. A., Chin, B. B., Noto, R. B., Dillon, J. S., Perkins, S., Solnes, L., et al. (2019). Efficacy and safety of high-specific-activity 131 I-MIBG Therapy in patients with advanced pheochromocytoma or paraganglioma. J. Nucl. Med. 60, 623–630. doi:10.2967/jnumed.118.217463
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Ricketts, C. J., Forman, J. R., Rattenberry, E., Bradshaw, N., Lalloo, F., Izatt, L., et al. (2010). Tumor risks and genotype–phenotype–proteotype Analysis in 358 patients with germline mutations in SDHB and SDHD. Hum. Mutat. 31, 41–51. doi:10.1002/humu.21136
Rijken, J. A., Niemeijer, N. D., Corssmit, E. P. M., Jonker, M. A., Leemans, C. R., Menko, F. H., et al. (2016). Low penetrance of paraganglioma and pheochromocytoma in an extended kindred with a germline SDHB exon 3 deletion. Clin. Genet. 89, 128–132. doi:10.1111/cge.12591
Roman-Gonzalez, A., Zhou, S., Ayala-Ramirez, M., Shen, C., Waguespack, S. G., Habra, M. A., et al. (2018). Impact of surgical resection of the primary tumor on overall survival in patients with metastatic pheochromocytoma or sympathetic paraganglioma. Ann. Surg. 268, 172–178. doi:10.1097/SLA.0000000000002195
Tabebi, M., Kumar Dutta, R., Skoglund, C., Söderkvist, P., and Gimm, O. (2022). Loss of SDHB induces a metabolic switch in the hPheo1 cell line toward enhanced OXPHOS. Int. J. Mol. Sci. 23, 560. doi:10.3390/ijms23010560
Tanabe, A., and Naruse, M. (2020). Recent advances in the management of pheochromocytoma and paraganglioma. Hypertens. Res. 43, 1141–1151. doi:10.1038/s41440-020-0531-0
van Leeuwaarde, R. S., Ahmad, S., Links, T. P., and Giles, R. H. (2018). “Von hippel-lindau syndrome,” in GeneReviews® [internet]. Editors M. P. Adam, H. H. Ardinger, and R. A. Pagon. Seattle. (WA).
Welander, J., Söderkvist, P., and Gimm, O. (2011). Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomas. Endocr. Relat. Cancer 18, R253–R276. doi:10.1530/ERC-11-0170
Keywords: pheochromocytomas, paragangliomas, germline mutation, succinate dehydrogenase subunit B, human, neoplastic syndromes, hereditary
Citation: Manotas MC, Rivera AL, Gómez AM, Abisambra P, Guevara G, Medina V, Tapiero S, Huertas A, Riaño-Moreno J, Mejía JC, Gonzalez-Clavijo AM, Tapiero-García M, Cuéllar-Cuéllar AA, Fierro-Maya LF and Sanabria-Salas MC (2023) SDHB exon 1 deletion: A recurrent germline mutation in Colombian patients with pheochromocytomas and paragangliomas. Front. Genet. 13:999329. doi: 10.3389/fgene.2022.999329
Received: 20 July 2022; Accepted: 08 November 2022;
Published: 04 January 2023.
Edited by:
Madson Almeida, University of São Paulo, BrazilReviewed by:
Sergei Tevosian, University of Florida, United StatesLuis Jaime Castro-Vega, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Copyright © 2023 Manotas, Rivera, Gómez, Abisambra, Guevara, Medina, Tapiero, Huertas, Riaño-Moreno, Mejía, Gonzalez-Clavijo, Tapiero-García, Cuéllar-Cuéllar, Fierro-Maya and Sanabria-Salas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: María Carolina Sanabria-Salas, bWFjc2FuYWJyaWFzYUB1bmFsLmVkdS5jbw==">csanabria@cancer.gov.cobWFjc2FuYWJyaWFzYUB1bmFsLmVkdS5jbw==
†These authors have contributed equally to this work and share first authorship