 Tao Xiao
Tao Xiao Liefen He1
Liefen He1 Yonghong Zhang
Yonghong Zhang Shiou Yih Lee
Shiou Yih Lee
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 14 September 2022
Sec. Evolutionary and Population Genetics
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.998575
Owing to its attractive flower shape and color, Renanthera (Orchidaceae), comprising about 19 species, has significant ornamental value as a houseplant, in floral design and in landscape gardens. Two species of Renanthera are categorized as endangered and critically endangered in China’s Red List and international trade in these orchids is currently strictly monitored by the Convention on International Trade in Endangered Species of Wild Fauna and Flora (CITES). This paper reports on the de novo assembled and annotated plastome of four species of Renanthera; R. citrina, R. coccinea, R. imschootiana, and R. philippinensis. The length of the plastome sequences ranged from 144,673 bp (R. imschootiana) to 149,007 bp (R. coccinea) with GC content of 36.6–36.7%. The plastomes showed a typical quadripartite structure, including a large single-copy (84,241–86,404 bp), a small single-copy (11,468–12,167 bp), and a pair of inverted repeats (24,482–25,715 bp) regions. Of the 120 genes detected, 74 were protein coding, 38 were tRNA, and eight were rRNA genes. The plastome of Renanthera is rather conserved, but nucleotide variations that could distinguish them apart are noticeable—the total number of tandem repeats ranged from 62 (in R. imschootiana) to 74 (in R. citrina); while the number of long repeats ranged from 21 (in R. imschootiana and R. philippinensis) to 43 (in R. citrina). Three hypervariable regions (psbI-trnS-GCU, trnG-GCC, rpl32) were identified. Phylogenetic analyses based on the CDS using maximum likelihood (ML) and Bayesian inference (BI) revealed that Renanthera is closely related to Holcoglossum, Neofinetia, Pendulorchis, and Vanda. The relationship between the four species of Renanthera was fully resolved; a monophyletic clade was formed and R. coccinea was recorded as the first to diverge from the rest. The genetic data obtained from this study could serve as a useful resource for species identification in Renanthera as well as contribute to future research on the phylogenomics of Orchidaceae.
Renanthera Lour. (Aeridinae, Vandeae, Orchidaceae) comprises about 19 species of epiphytic and/or lithophytic perennial herbs, mainly distributed in China, India, Indonesia, Malaysia, New Guinea, Philippines Vietnam, and the Solomon Islands (Chen and Jeffrey, 2009; Chase et al., 2015). Members of Renanthera produce a spectacular branched inflorescence containing numerous flowers with large, yellow, orange and red lateral sepals (Luo et al., 2012). Due to the magnificent floral features, these orchids have received great attention by orchid breeders and collectors as houseplants, floral design, and landscape gardening. However, their over-exploitation in the wild has disturbed their original habitat severely and threatened their survival in the wild. As a result, all members of Renanthera, except for R. imschootiana, which is listed in of the Convention on International Trade in Endangered Species of Wild Fauna and Flora (CITES) since 1975 (UNEP-WCMC, 2020). In China, three species are recorded, including R. citrina, R. coccinea, and R. imschootiana (Chen and Jeffrey, 2009); however, two of them, R. coccinea, and R. imschootiana are categorized as endangered and critically endangered, respectively, in the Red List of China Higher Plants (Qin et al., 2017).
In general, the morphological features, such as the number of pollinia, presence or absence of a nectar spur, shape and size of labellum, and shape of the column-foot, have been used as key characters in the taxonomic classification of Aeridinae (Pridgeon et al., 2014). However, the true phylogenetic relationships at subtribe level were unclear due to multiple evolutionary stages in the evolution of character states over a short period of time (Topik et al., 2005). Genetic studies on Renanthera are limited. Previous molecular phylogenetic studies of Aeridinae using a combined dataset of four plastid regions (atpI-atpH, matK, psbA-trnH, trnL-trnF) and the nuclear ribosomal internal transcribed spacer (ITS) region reportedly resolved the phylogenetic relationship within Renanthera (Zou et al., 2015). However, the phylogenetic tree was not supported with a strong backbone. The lack of informative sites resulted in a number of uncertainties in the molecular placements of many genera in the subtribe.
With the development of next-generation sequencing (NGS) technology, sequencing of the complete organellar genome has become simpler and affordable. The use of genome-scale data to resolve relationships in complicated plant groups appears promising. Coupled with advanced bioinformatic analytic tools, the phylogenetic relationships of many members of Orchidaceae, which has been uncertain for many years, were resolved (Li et al., 2019; Kim et al., 2020a). The complete plastid genome (plastome) sequences of 10 taxa of Vandeae, Kim et al. (2020b) revealed the molecular placement of Vandeae in the Epidendroideae. The phylogenetic relationship among members in Vandeae, with a focus on Aeridinae, was further identified in a phylogenetic reconstruction based on complete plastome sequences (Liu et al., 2020). Despite the well-supported tree reported by Liu et al. (2020), members of Renanthera were not included.
As an understudied yet threatened species in China, three species that are native to China, including R. citrina, R. coccinea, and R. imschootiana, as well as a domesticated species, R. philippinensis, were included in this study. To provide genetic data at genome level, we sequenced and characterized the complete plastomes of four species of Renanthera. At the same time, we analyzed the sequence repeats, the contraction and expansion of the inverted repeat (IR) region, identified the variable sites, and revealed the phylogenetic relationships between the four species of Renanthera to further elucidate evolutionary patterns in Renanthera at the plastome level. The findings obtained through this study yielded useful genomic information on these threatened species that may aid in conservation strategies.
Four Renanthera species, including R. citrina, R. coccinea, R. imschootiana, and R. philippinensis, were collected from natural populations, except for R. philippinensis which is a cultivated species from an orchid nursery in Guangdong. The orchids are transferred and planted in the greenhouse of Yunnan Normal University, Kunming, China (24°52'1.41“N, 102°51'19.39”E). Voucher specimens are deposited in the Herbarium of Yunnan Normal University (YNUB) (Table 1). Total genomic DNA was extracted from fresh leaves using the E.Z.N.A® HP Plant DNA Kit (Omega Bio-tek, GA, United States) based on the manufacturer’s protocol. Shotgun libraries (350 bp) were prepared using a TruSeq DNA Sample Prep Kit (Illumina, United States). Next-generation sequencing (NGS) was conducted on an Illumina Hiseq 2000 sequencing platform (Illumina, CA, United States), from which approximately 6 Gb of raw NGS data was obtained. The raw reads were filtered using the NGS QC Toolkit (Patel and Jain, 2012) to remove low quality reads and to trim off the sequence adaptors. The filtered reads were then fed into the GetOrganelle (Jin et al., 2020) pipeline for genome de novo assembly. Gene annotation was carried out using GeSeq (Tillich et al., 2017) based on default parameters, and was checked through Geneious Prime v2020.0.3 (Kearse et al., 2012). The circular map of the genome was illustrated using OGDRAW (Greiner et al., 2019).
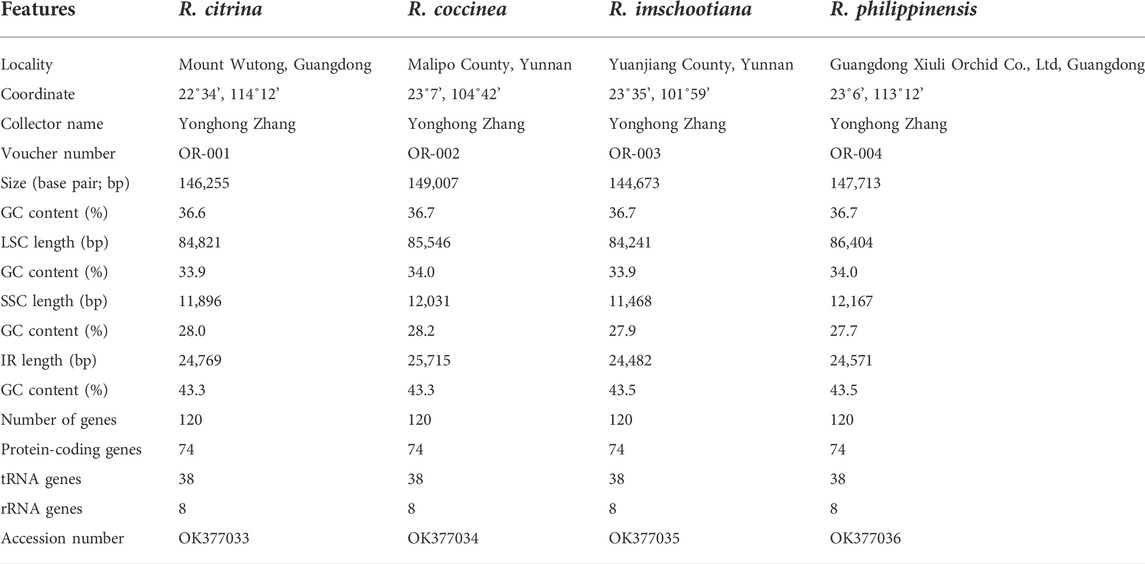
TABLE 1. Complete plastid genome features of four species of Renanthera.
Simple sequence repeats (SSRs) were calculated using MISA-web (Beier et al., 2017), in which the parameters for SSR identification were set at minimal repeat numbers of 10, 5, 4, 3, 3, and 3 for mono-, di-, tri-, tetra-, penta- and hexa- nucleotide repeats, respectively. For large repeats, REPuter was used to identify forward, palindrome, reverse, and complementary long repeats (Kurtz and Schleiermacher, 1999). The hamming distance was set as 3, while the minimal repeat size was 30 bp.
The nucleotide sequence of each protein-coding gene in the plastome was extracted using Geneious Prime v2020.0.3 (Kearse et al., 2012). The relative synonymous codon usage (RSCU) for each protein-coding gene was calculated using MEGA7 (Kumar et al., 2016).
Chloroplast genomes across the four species of Renanthera were compared on the mVISTA tool (Frazer et al., 2004) using Shuffle-LAGAN mode with the sequence of Renanthera coccinea as a reference. To detect the expansion and contraction of the inverted repeat (IR) region, the boundaries between the IR and single-copy regions (IR/SC) of the plastomes were visualized using IRscope (Amiryousefi et al., 2018). Prior to nucleotide divergence analysis, the plastome sequences were aligned using MAFFT v7 (Katoh and Standley, 2013). The nucleotide variability of the four plastomes of Renanthera were calculated using DnaSP v5.10 (Librado and Rozas, 2009), in which highly mutational hotspot regions were identified through a sliding window analysis. The length of the window frame was set at 1,000 bp and a 500 bp step size was selected. The number and location of the single nucleotide polymorphisms (SNPs) and variable sites in the plastome alignment were also determined using the same program.
Phylogenetic analysis was carried out based on CDSs of 30 species of Orchidaceae. The CDSs were extracted, ClustalW-aligned, and concatenated alphabetically using Geneious Prime v2020.0.3 (Kearse et al., 2012). Three closely related species, Tridactyle tridactylites (Angraecinae, Vandeae; MW760855), Cattleya crispata (Epidendroideae; KP168671), and Cymbidium ensifolium (Epidendroideae; KT722983) were included as outgroups. Phylogenetic trees were constructed using two methods: maximum likelihood (ML) and Bayesian inference (BI). For ML, the concatenated dataset was fed into the RAxML v8.2.11 pipeline (Stamatakis, 2014) and analysis was carried out based on general time-reversible (GTR) with the gamma distributed (+G) (=GTR + G) substitution model. Each branch node was assessed under 1,000 bootstrap replicates. For BI, the concatenated dataset was fed into the MrBayes v3.2.6 pipeline (Ronquist et al., 2012), in which a mix substitution type and a 4 by 4 nucleotide substitution model was selected for the likelihood model. Markov chain Monte Carlo simulation was performed twice independently for 2,000,000 generations. Four chains were selected and data sampling was conducted every 100 generations. The first 25% of trees was discarded as burn-in. Both pipelines are available in the CIPRES Science Gateway (Miller et al., 2011). Final tree results for both analyses were visualized using FigTree v1.4.4 (Rambaut, 2018).
The plastomes of the four species of Renanthera displayed a typical quadripartite structure that consisted of a genome size ranging from 144,673 bp (R. imschootiana) to 149,007 bp (R. coccinea) (Figure 1). The plastomes contained a pair of IRs that were separated by a large single-copy (LSC) region and a small single-copy (SSC) region. The size of each region varied across all species (Table 1). For the IR region, the size ranged from 24,482 bp (R. imschootiana) to 25,715 bp (R. coccinea); while the SSC and the LSC regions were 11,468 bp (R. imschootiana) to 12,167 bp (R. philippinensis) and 84,241 bp (R. imschootiana) to 86,404 bp (R. philippinensis), respectively. The overall GC contents for all the plastomes were 36.7%, except for R. citrina, which was 36.6%. All four plastomes were predicted with the same number of genes, which was 120. The genes include 74 CDS, 38 rRNA, and eight tRNA genes. Among these genes, 17 were duplicates in the IR regions, including five CDS (rpl2, rpl23, rps7, rps19, ycf2), eight tRNA (trnA-UGC, trnH-GUG, trnI-CAU, trnI-GAU, trnL-CAA, trnN-GUU, trnR-ACG, trnV-GAC), and four rRNA (rrn4.5, rrn5, rrn16, rrn23) genes (Table 2).
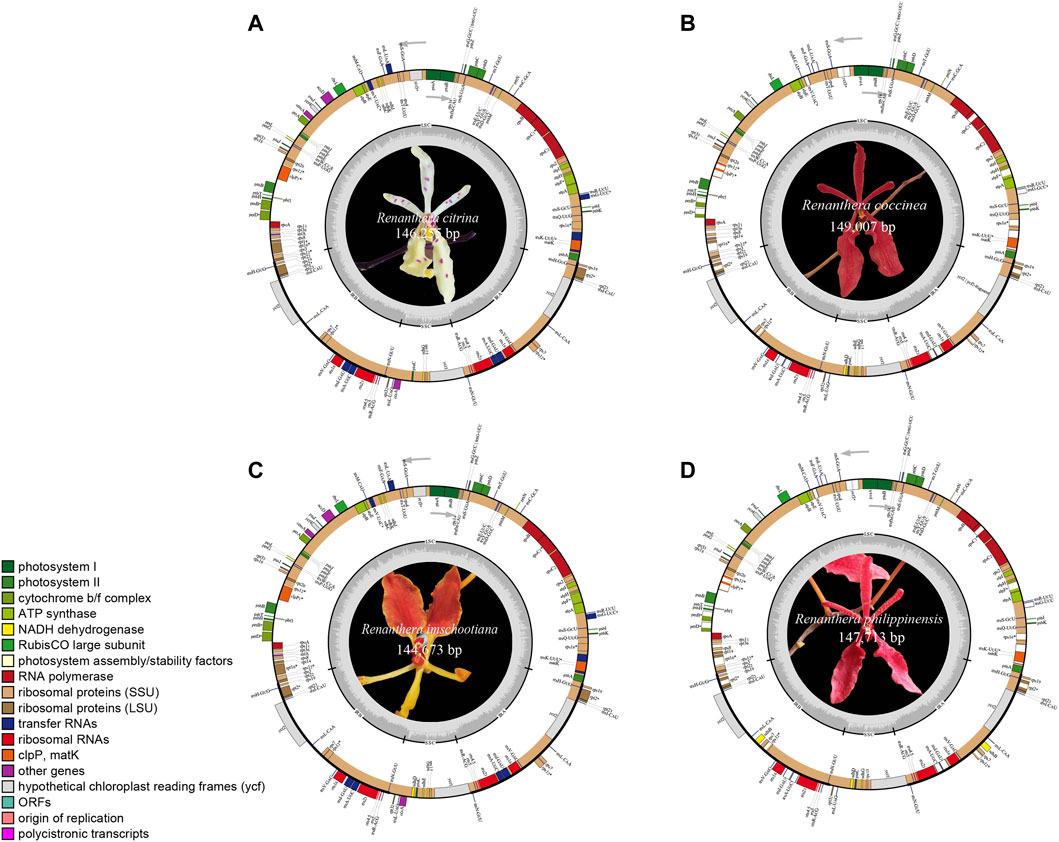
FIGURE 1. Gene map of plastid genomes of four species of Renanthera used in this study, including (A) R. citrina; (B) R. coccinea; (C) R. imschootiana; (D) R. philippinensis. Genes on inside of map are transcribed in clockwise direction; genes on outside of map are transcribed in counterclockwise direction. Different functional groups of genes are shown in different colors. Inverted repeat (IR), small single-copy (SSC), and large single-copy (LSC) regions are indicated.

TABLE 2. Genes present in the plastid genomes of four species of Renanthera used in this study.
SSRs analysis detected 74, 68, 62, and 68 SSRs in R. citrina, R. coccinea, R. imschootiana and R. philippinensis, respectively (Figure 2). Among the SSRs, the mononucleotide repeats were the most abundant; at least 41–50 mononucleotide repeats were in the four species. This was followed by the dinucleotides (9–10 repeats), trinucleotides (4—8 repeats), and tetranucleotides (3—6 repeats). For pentanucleotide repeats, all the species was recorded with one occurrence, except R. citrina, which had four. One hexanucleotide repeat was detected in R. philippinensis, but none in the others. Most of the SSRs were located in the LSC region, in which 51 (75%), 53 (71.62%), 48 (70.59%), and 47 (75.81%) were recorded in R. citrina, R. coccinea, R. imschootiana and R. philippinensis, respectively.

FIGURE 2. Distribution of sequence repeats in the plastid genomes of four species of Renanthera. (A) Number of different short sequence repeat (SSR) types detected in four plastid genomes of Renanthera; (B) Frequencies of identified SSRs in large single-copy (LSC), small single-copy (SSC) and inverted repeat (IR) regions; (C) Number of long repeats based on types; (D) Number of long repeats by sequence length.
The number of long repeats detected in the four plastomes ranged from 21 (R. imschootiana and R. philippinensis) to 43 (R. citrina). All the plastomes contained three types of long repeats that were forward, reverse, and palindromic, except for R. citrina. For R. citrina, aside from the three long repeat types, it also contained a complement type. All species, except for R. citrina, had long repeats mostly in the 30–40 bp range; the long repeat of R. citrina was 41–50 bp. For long repeats above 50 bp, R. citrina and R. imschootiana had eight and one, respectively; none was in R. coccinea and R. philippinensis.
A total of 184,687 codons was identified in the CDSs predicted across the four plastomes of Renanthera (Table 3). Among these codons, leucine (10.7%) and isoleucine (9.1%) were the most abundant amino acids, whereas cysteine (2.4%) had the lowest frequency. All four plastomes exhibited similar codon preferences, in which 32 codons were more preferred (RSCU >1), and 30 codons were least preferred (RSCU <1). No preference (RSCU = 1) was found for tryptophan (UGG) or methionine (AUG). For start codons, methionine (AUG) was more preferred than tryptophan (UGG); while for stop codons, UAA was more preferred than UAG and UGA, in the four different plastomes.
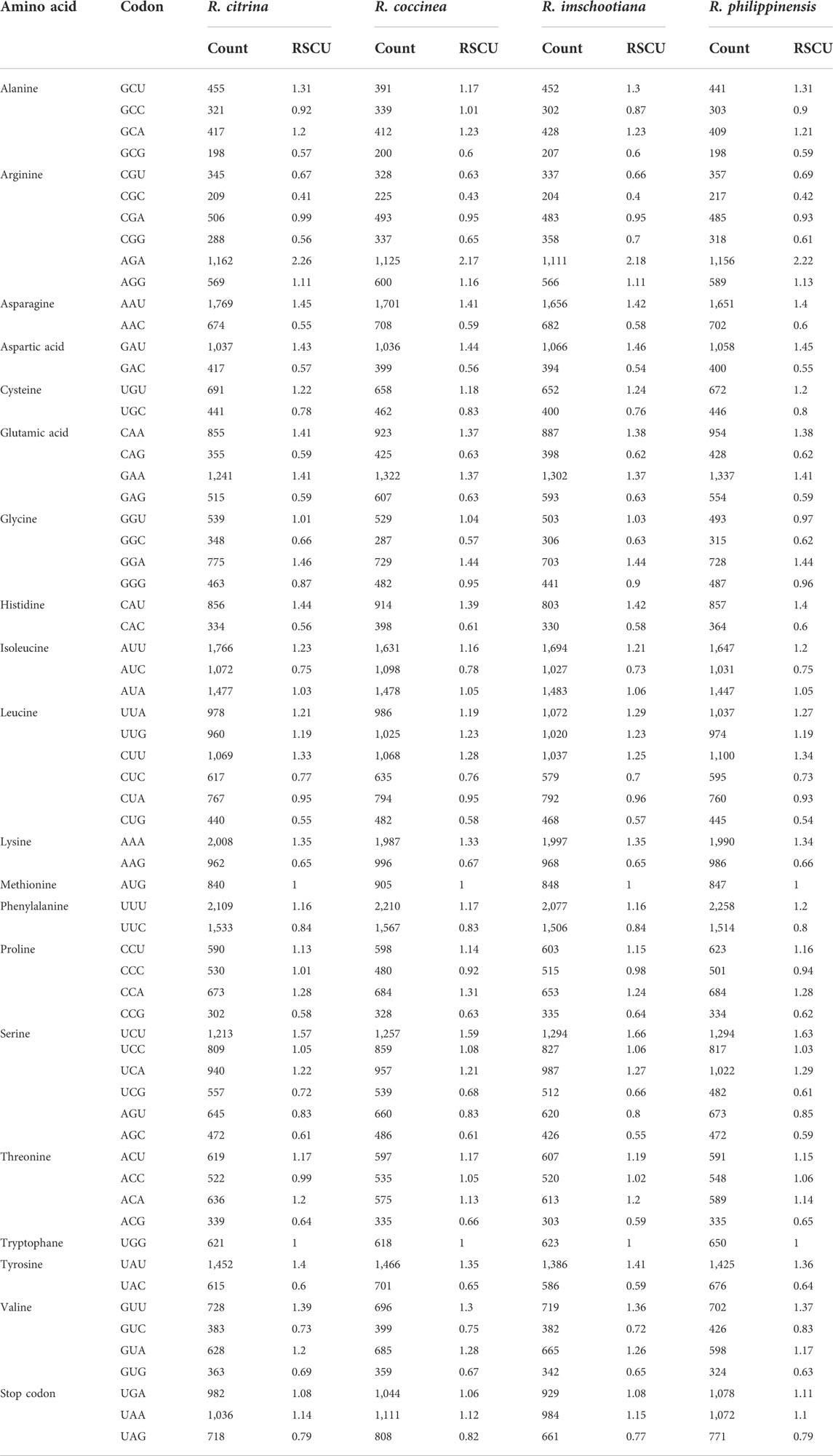
TABLE 3. Relative synonymous codon usage values (RSCU) in plastid genome of four species of Renanthera.
The positions of the IR junctions were well-conserved across the four species of Renanthera (Figure 3). At the junction between the LSC and IRb (JLB), genes rpl22 and rps19 were adjacent to the junction; the rpl22 gene of LSC crossed over into IRb. For the junction between the LSC and IRa (JLA), genes rps19 and psbA were adjacent to the junction. The rpl32 and trnN genes were adjacent to the junction between SSC and IRb (JSB); while the trnN and ycf1 genes were adjacent to the junction between SSC and IRa (JSA). The ycf1 gene of R. citrina crossed over JSA, into the IRa region; while the ycf1 genes for the other three species were still intact within the SSC region.
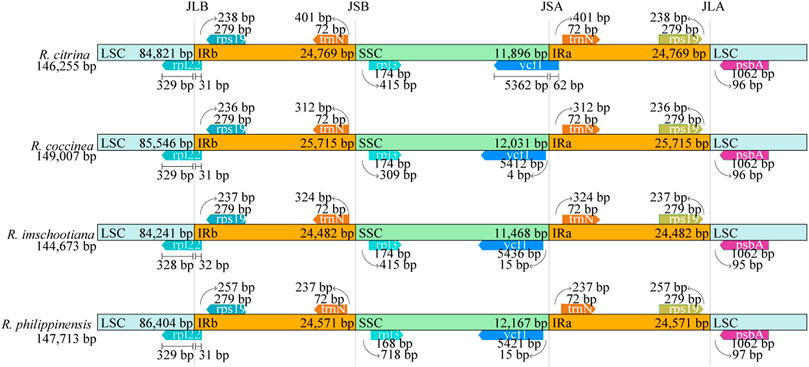
FIGURE 3. Comparison of the junctions of LSC, SSC, and IR regions among four plastid genomes of Renanthera.
Comparative analysis of the complete chloroplast genome can reveal differences between different species (Figure 4). Altogether, the genome sequence alignment of the four species of Renanthera exhibit a high degree of similarity. Three genes, ndhJ, ndhK, and ndhC, were obviously lost in R. citrina and R. imschootiana. Additionally, a distinct large gap was observed from the rps7 gene to trnL-CAA in the IRa and IRb of R. citrina, R. imschootiana and R. philippinensis. The nucleotide diversity (Pi) value for the four plastomes ranged from 0 to 0.04533, with an average of 0.0086. At a cut-off point set at Pi ≥ 0.035, three hypervariable regions, including psbI-trnS-GCU, trnG-GCC, rpl32, were identified (Figure 5). Among the three hypervariable regions, two were in the LSC region, while one was in the SSC region. None was detected in the IR region.

FIGURE 4. Complete plastid genome comparison of four species of Renanthera. The plastid genome of R. coccinea was selected as reference.

FIGURE 5. Nucleotide diversity (Pi) in the complete plastid genomes of four species of Renanthera. Window length: 1,000 bp, step size: 500 bp.
Plastome alignment of the four species of Renanthera resulted in a 152,882 bp long sequence alignment. Across the alignment, 433 parsimony informative sites and 1,838 singletons were identified. A total of 2,271 SNPs was identified. SNPs in the LSC region were the most abundant (n = 1,708), followed by 360 and 203 of them in the SSC and IR regions, respectively.
Phylogenetic analyses using both ML and BI methods based on the CDS sequences of 30 species of Orchidaceae revealed similar topological structure in the phylogenetic trees; hence, only the ML tree is presented, with the values of the bootstrap support (BS) as well as the posterior probability (PP) of the ML and BI trees indicated on each branch node (Figure 6). In general, the phylogenetic relationship within Renanthera was well-resolved (BS ≥ 75%, PP ≥ 0.90); R. coccinea clustered with R. philippinensis and R. citrina clustered with R. imschootiana. Renanthera is closely-related to the Holcoglossum + Neofinetia + Pendulorchis + Vanda clade. In general, all the branch nodes in the phylogenetic tree were strongly supported in the BI analysis; while seven branch nodes were indicated with moderate bootstrap support values in the ML analysis.
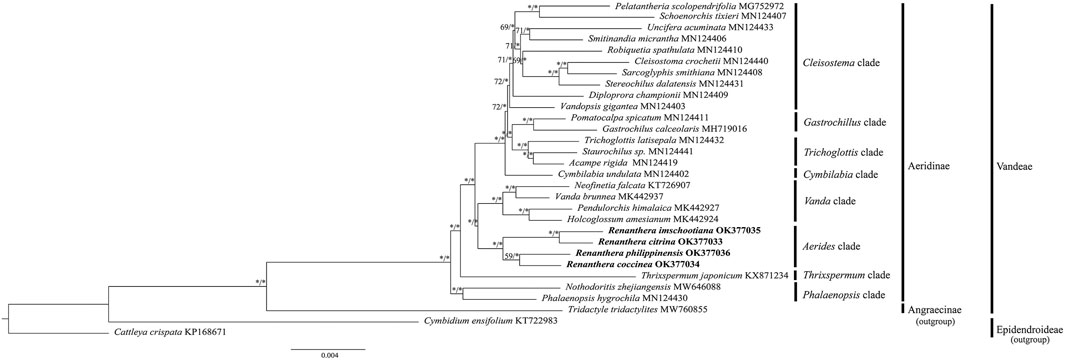
FIGURE 6. Phylogenetic trees based on CDS sequences of 30 species of Orchidaceae. Three species, Tridactyle tridactylites (Angraecinae, Vandeae; MW760855), Cattleya crispata (Epidendroideae; KP168671), and Cymbidium ensifolium (Epidendroideae; KT722983) were included as outgroups. Numbers on each node represent bootstrap support (BS) and posterior probability (PP) values. Branches indicated with BS ≥ 75 and PP ≥ 0.90 are considered as well-supported, which are represented with an asterisk (*).
In this study, we obtained novel plastome sequences of four species of Renanthera using next-generation sequencing techniques. Although there is a published report on the plastome sequence of R. imschootiana (Xin et al., 2020), information on its plastome sequence was not publicly available. Thus, it was not incorporated into this study. In this study, it was observed that the structure, gene content, and gene order in the four species of Renanthera were highly conserved. The plastome is generally maternal inherited in most angiosperms, including orchids, and usually comes with little recombination and a highly conserved structure across closely-related species (Wicke et al., 2011). Based on the study of Liu et al. (2020), the plastomes for members of Aeridinae were reported to range from 146,000 to 150,000 bp, except in two species, Schoenorchis seidenfadenii (142,859 bp) and Uncifera acuminata (145,191 bp). The species of Renanthera with the smallest plastome size, R. imschootiana, had a relatively smaller plastome size when compared to most of the members of Aeridinae; the three other species of Renanthera had a plastome size similar to most of the members of Aeridinae. The highly conserved number of genes present in the plastome of Renanthera suggested that the difference in plastome size could be due to the presence of indels in the intergenic spacer regions. However, similar to the rest of the members of Aeridinae, the overall GC content of Renanthera was not more than 37%.
The plastid ndh genes experienced independent loss in species of Renanthera (Table 2). Absence or pseudogenization of ndh genes in the plastomes of members of Aeridinae or Vandeae is considered a common phenomenon (Kim et al., 2020b; Jiang et al., 2022). The literature (Kim et al., 2015; Kim et al., 2020a) has reported that epiphytic orchids lose or pseudogenize their ndh genes when compared to terrestrial orchids, which is consistent with our findings for the four species of Renanthera in this study, which are epiphytes. Members of Renanthera rarely grows on the ground, they are usually epiphytic on tree trunks and/or lithophytic on rocks along valleys; thus, it is speculated that other species of Renanthera would undergo ndh gene loss, similar to those reported in this study. While it was proposed that the loss of ndh genes could be random in photosynthetic lineages, ndh gene loss is not only confined to Orchidaceae, but also reported in other autotrophic plants (Sabater, 2021). In general, ndh genes mediate cyclic electron transfer, which can protect plants from photoinhibition caused by various environmental stresses, and play an important role in maintaining efficient photosynthesis (Mi, 2016). The loss of ndh genes in the orchid plastome may be due to gene substitution or gene translocation from the plastome to the mitochondrial or nuclear genome (Lin et al., 2015). Usually, the loss of ndh genes in non-angiosperms, as in Pinus and Welwitschia (McCoy et al., 2008; Zeb et al., 2020), is not believed to be life-threatening. However, it could provide inverse effect to angiosperms as it will prevent plants from evolving and diversifying (Sabater, 2021), and will reduce the ecological adaptations of the species (Sun et al., 2020).
SSR are widely found in various species and are useful for studies of molecular evolution and genetic diversity (Doorduin et al., 2011). Of the 62–74 SSRs detected in the four species of Renanthera, the majority of them were identified from the LSC (73.2%) region, follow by SSC (19.4%) and IR (7.4%) region. Therefore, the distribution of SSRs in the chloroplast genome of Renanthera is uneven. The SSRs numbers in the LSC region of plastome of R. coccinea and R. philippinensis are 53 and 47, respectively. The distribution of SSR in the four species of Renanthera was consistent with that of most angiosperms (Xu et al., 2021; Sun et al., 2022; Xia et al., 2022).
The codon usage among the four plastomes of Renanthera was consistent across the four species; the CDSs of the four species of Renanthera had the same preferences in codon usage for all 20 amino acids, indicating that the nucleotide sequences in the CDS of the four species were identical, which is deemed accurate. This is because plastome sequences of closely related species have greater conservatism as they were inherited from their most recent common ancestor (Khan et al., 2021). Among these amino acids, leucine (n = 19,700) is the most frequently used, cysteine (n = 4,422) is the least universal amino acid in the cp genomes of these species. Alternatively, the high leucine frequency in the four plastomes can be attributed to the fact that leucine is largely needed in the chloroplasts due to its important function in photosynthesis-related metabolism (Knill et al., 2009). Cysteine, which was the least abundant amino acid among all the CDS analyzed, is a reactive element that can be considered toxic when it is accumulated above a certain level allowed by the host (Hildebrandt et al., 2015). This is consistent with the codon preference of other genera in the Orchidaceae, such as Thuniopsis and Polystachya. (Jiang et al., 2022; Li et al., 2022).
Contraction and expansion of the IR region is usually observed in most angiosperms (Li et al., 2019), including Renanthera. This was true when the 3’ end of the rpl22 gene of all four species of Renanthera that should be intact in the LSC region extended into the IRb region for 31–32 bp. The ycf1 gene in the SSC region of R. citrina was observed crossing over JSA, extending into the IRa region; while ycf1 of the remaining three species of Renanthera were intact in the SSC region. Generally, angiosperms retain the structure and size of the cp genome (Ebert and Peakall, 2010). However, due to evolutionary events such as contraction and expansion in the genome, variation can occur in the boundaries of inverted repeats (IRs) and single copy regions (LSC and SSC) (Raubeson et al., 2007) allowing certain genes to enter the IR or SC regions. Successive IR expansions have shown the importance of the JLA and JLB junctions for the analysis of evolutionary processes, providing clues about the origin and evolution of species (Zhang et al., 2013). Yet, in the case of Renanthera, although with the limited sampling size reported in this study, the finding suggested that the IR boundary shifts in Renanthera can be considered to be relatively minor, as this can be related to the small difference observed in the genome size across different species.
Sequence divergence analysis conducted on the four plastomes revealed that the IR region was highly conserved when compared to the single-copy regions. To our knowledge, there are no published reports on the molecular identification of the species of Renanthera. We speculated that this is either due to difficulty in species sampling, or the genus has not received attention. As molecular genetic tools could aid in the identification of the species of orchids and forensic detection at border controls (De and Pathak, 2018), the identification of useful gene regions in Renanthera will definitely contribute to safeguarding these orchids by security agencies. Molecular genetic tools such as DNA barcoding are among the promising techniques in the identification of species of land plants (Hollingsworth, 2011). The gene loci used in DNA barcoding usually contains sufficient informative sites that can delimit closely related species efficiently. The application of highly variable regions derived from the plastome sequence as potential DNA barcoding loci has been proven to be promising, as demonstrated in several findings (Vaughn et al., 2014; Lee et al., 2021). Three highly variable regions were identified in this study to facilitate marker development of Renanthera. It is noteworthy that an increased sampling size is needed to obtain reliable results (Li et al., 2015). The findings from this study may contribute to the development of DNA barcoding markers from Renanthera.
Despite being a valuable and threatened orchid, there are no reports on the phylogenetic relationships in Renanthera. According to the phylogenetic analysis using short gene sequences, Renanthera is monophyletic (Zou et al., 2015). However, due to small sampling size, we were not able to provide supporting evidence on this matter based on our phylogenetic analysis. Interestingly, the Aerides clade, where Renanthera is placed, was sister to Vanda clade when using the complete plastome sequence dataset. This finding was incongruent with the results by Zou et al. (2015) that used short gene sequences. Given that there are no known records on natural hybridization among species of Vandeae, the phylogenetic tree based on the plastome dataset is considered reliable (Liston et al., 2021). However, increased taxon samplings could reduce computational errors in phylogenetic tree reconstructions (Zwickl and Hillis, 2002); Zou et al. (2015) included 74 genera of Aeridinae in their analysis. In this study, only 24 genera of Aeridinae were included and the Aerides clade was represented by only one genus, Renanthera. Despite the fact that the phylogenetic analysis was well-resolved based on the plastome dataset, the finding by Zou et al. (2015) on the molecular placement of the Aerides clade in Aeridinae could be more reliable due to greater taxon sampling size. Thus, to ascertain the molecular placement of the Aerides clade in Aeridinae at the plastome level, it is suggested to include plastome data for several taxa of the Aerides clade such as species of Aerides, Arachnis, and Rhynchostylis.
Our study revealed that the plastome of Renanthera is well-conserved across four species, R. citrina, R. coccinea, R. imschootiana, and R. philippinensis. Phylogenetic analysis based on the CDS sequences revealed that Renanthera is closely related to Holcoglossum, Neofinetia, Pendulorchis, and Vanda and the relationship of the four species of Renanthera was monophyletic. The genomic data obtained from this study will provide a useful resource for conservation, as well as to contributing in studies on the phylogeny and evolution of Renanthera and Orchidaceae.
The data presented in the study are deposited in the GenBank of NCBI repository, accession numbers OK377033—OK377036..
LH and YZ collected the plant materials and performed the experiments. TX and LH assembled the sequences and analyzed the data. TX and LH wrote the manuscript. LY, YZ, and SL conceived the research and revised the manuscript. All authors read and approved the final manuscript.
This study was supported by the National Natural Science Foundation of China (31760048).
We thank all the peer reviewers and editors for their opinions and suggestions.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Amiryousefi, A., Hyvönen, J., and Poczai, P. (2018). IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. doi:10.1093/bioinformatics/bty220
Beier, S., Thiel, T., Münch, T., Scholz, U., and Mascher, M. (2017). MISA-Web: A web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi:10.1093/bioinformatics/btx198
Chase, M. W., Cameron, K. M., Freudenstein, J. V., Pridgeon, A. M., Schuiteman, A., van den Berg, C., et al. (2015). An updated classification of Orchidaceae. Bot. J. Linn. Soc. 177, 151–174. doi:10.1111/boj.12234
Chen, X. Q., and Jeffrey, J. (2009). “Renanthera Flora of China,”. Editors Z. Y. Wu, and H. R. Peter (Beijing: Science Press), 451–452.
De, L. C., and Pathak, P. (2018). Conservation, management and utilization of orchid genetic resources. J. Orchid. Soc. India 32, 81–91.
Doorduin, L., Gravendeel, B., Lammers, Y., Ariyurek, Y., Chin-A-Woeng, , and Vrieling, K. (2011). The complete chloroplast genome of 17 individuals of pest species Jacobaea vulgaris: SNPs microsatellites and barcoding markers for population and phylogenetic studies. DNA Res. 18, 93–105. doi:10.1093/dnares/dsr002
Ebert, D., and Peakall, R. (2010). Chloroplast simple sequence repeats (cpSSRs): Technical resources and recommendations for expanding cpSSR discovery and applications to a wide array of plant species. Mol. Ecol. Resour. 9, 673–690. doi:10.1111/j.1755-0998.2008.02319.x
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., and Dubchak, I. (2004). Vista: Computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. doi:10.1093/nar/gkh458
Greiner, S., Lehwark, P., and Bock, R. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64. doi:10.1093/nar/gkz238
Hildebrandt, T. M., Nesi, A. N., Araújo, W. L., and Braun, H. P. (2015). Amino acid catabolism in plants. Mol. Plant 8, 1563–1579. doi:10.1016/j.molp.2015.09.005
Hollingsworth, P. M. (2011). Refining the DNA barcode for land plants. Proc. Natl. Acad. Sci. U. S. A. 108, 19451–19452. doi:10.1073/pnas.1116812108
Jiang, H., Tian, J., Yang, J. X., Dong, X., Zhong, Z. X., Mwachala, G., et al. (2022). Comparative and phylogenetic analyses of six Kenya Polystachya (Orchidaceae) species based on the complete chloroplast genome sequences. BMC Plant Biol. 22, 177. doi:10.1186/s12870-022-03529-5
Jin, J. J., Yu, W. B., Yang, J. B., Song, Y., dePamphilis, C. W., Yi, T. S., et al. (2020). GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 241. doi:10.1186/s13059-020-02154-5
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi:10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi:10.1093/bioinformatics/bts199
Khan, A. L., Asaf, S., LubnaAl-Rawahi, A., and Al-Harrasi, A. (2021). Decoding first complete chloroplast genome of toothbrush tree (Salvadora persica L.): Insight into genome evolution, sequence divergence and phylogenetic relationship within brassicales. BMC Genomics 22, 312. doi:10.1186/s12864-021-07626-x
Kim, H. T., Kim, J. S., Moore, M. J., Neubig, K. M., Williams, N. H., Whitten, W. M., et al. (2015). Seven new complete plastome sequences reveal rampant independent loss of the ndh gene family across Orchids and associated instability of the inverted repeat/small single-copy region boundaries. PLoS One 10, e0142215. doi:10.1371/journal.pone.0142215
Kim, Y. K., Jo, S. J., Cheon, S. H., Joo, M. J., Hong, J. R., Kwak, M., et al. (2020a). Plastome evolution and phylogeny of Orchidaceae, with 24 new sequences. Front. Plant Sci. 11, 22. doi:10.3389/fpls.2020.00022
Kim, Y. K., Jo, S. J., Cheon, S. H., Kwak, M., Kim, Y. D., and Kim, K. J. (2020b). Plastome evolution and phylogeny of subtribe Aeridinae (Vandeae, Orchidaceae). Mol. Phylogenet. Evol. 144, 106721. doi:10.1016/j.ympev.2019.106721
Knill, T., Reichelt, M., Paetz, C., Gershenzon, J., and Binder, S. (2009). Arabidopsis thaliana encodes a bacterial-type heterodimeric isopropylmalate isomerase involved in both Leu biosynthesis and the Met chain elongation pathway of glucosinolate formation. Plant Mol. Biol. 71, 227–239. doi:10.1007/s11103-009-9519-5
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi:10.1093/molbev/msw054
Kurtz, S., and Schleiermacher, C. (1999). REPuter: Fast computation of maximal repeats in complete genomes. Bioinformatics 15, 426–427. doi:10.1093/bioinformatics/15.5.426
Lee, S. Y., Chen, Z., Chen, J., Fan, Q., Liu, J., and Liao, W. (2021). Authentication of medicinal herb Wikstroemia indica using novel DNA markers derived from the chloroplast genome sequences. J. Appl. Res. Med. Aromat. Plants 24, 100315. doi:10.1016/j.jarmap.2021.100315
Li, L., Wu, Q. P., Fang, L., Wu, K. L., Li, M. Z., and Zeng, S. J. (2022). Comparative chloroplast genomics and phylogenetic analysis of Thuniopsis and closely related genera within Coelogyninae (Orchidaceae). Front. Genet. 13, 850201. doi:10.3389/fgene.2022.850201
Li, X. W., Yang, Y., Henry, R. J., Rossetto, M., Wang, Y. T., and Chen, S. L. (2015). Plant DNA barcoding: From gene to genome. Biol. Rev. Camb. Philos. Soc. 90, 157–166. doi:10.1111/brv.12104
Li, Y. X., Li, Z. H., Schuiteman, A., Chase, M. W., Li, J. W., Huang, W. C., et al. (2019). Phylogenomics of Orchidaceae based on plastid and mitochondrial genomes. Mol. Phylogenet. Evol. 139, 106540. doi:10.1016/j.ympev.2019.106540
Librado, P., and Rozas, J. (2009). DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi:10.1093/bioinformatics/btp187
Lin, C. S., Chen, J. J., Huang, Y. T., Chan, M. T., Daniell, H., Chang, W. J., et al. (2015). The location and translocation of ndh genes of chloroplast origin in the Orchidaceae family. Sci. Rep. 5, 9040. doi:10.1038/srep09040
Liston, A., Weitemier, K. A., Letelier, L., Podani, J., Zong, Y., Liu, L., et al. (2021). Phylogeny of crataegus (rosaceae) based on 257 nuclear loci and chloroplast genomes: Evaluating the impact of hybridization. PeerJ 9, e12418. doi:10.7717/peerj.12418
Liu, D. K., Tu, X. D., Zhao, Z., Zeng, M. Y., Zhang, S., Ma, L., et al. (2020). Plastid phylogenomic data yield new and robust insights into the phylogeny of Cleisostoma-Gastrochilus clades (Orchidaceae, Aeridinae). Mol. Phylogenet. Evol. 145, 106729. doi:10.1016/j.ympev.2019.106729
Luo, J. P., Wu, K. L., and Weng, S. F. (2012). Research progress on Renanthera. Guangdong Agric. Sci. 39, 69–72. doi:10.16768/j.issn.1004-874x.2012.10.060
McCoy, S. R., Kuehl, J. V., Boore, J. L., and Raubeson, L. A. (2008). The complete plastid genome sequence of Welwitschia mirabilis: An unusually compact plastome with accelerated divergence rates. BMC Evol. Biol. 8, 130. doi:10.1186/1471-2148-8-130
Mi, H. L. (2016). The regulation of NAD(P)H dehydrogenase complexes bound in thylakoid membranes in photosynthesis. J. Plant Physiol. 52, 1457–1465. doi:10.13592/j.cnki.ppj.2016.1025
Miller, M. A., Pfeiffer, W., and Schwartz, T. (2011). The CIPRES science gateway: A community resource for phylogenetic analyses. Proc. TeraGrid 2011 Conf. Extrem. Digit. Discov. TG’11 41, 1–8. doi:10.1145/2016741.2016785
Patel, R. K., and Jain, M. (2012). NGS QC toolkit: A toolkit for quality control of next generation sequencing data. PLoS One 7, e30619. doi:10.1371/journal.pone.0030619
Pridgeon, A. M., Cribb, P. J., Chase, M. W., and Rasmussen, F. N. (2014). “Epidendroideae (Part 3),” in Genera orchidacearum volume 6. Editors A. M. Pridgeon, P. J. Cribb, M. W. Chase, and F. N. Rasmussen (UK: Oxford University Press), 554.
Qin, H. N., Yang, Y., Dong, S. Y., He, Q., Jia, L. N., Yu, S. X., et al. (2017). Threatened species list of China’s higher plants. Biodiv Sci. 25, 696–744. doi:10.17520/biods.2017144
Rambaut, A. (2018). FigTree v1.4.4. Available at: http://tree.bio.ed.ac.uk/software/figtree/.
Raubeson, L. A., Peery, R., Chumley, T. W., Dziubek, C., Fourcade, H. M., Boore, J. L., et al. (2007). Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genomics 8, 174. doi:10.1186/1471-2164-8-174
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi:10.1093/sysbio/sys029
Sabater, B. (2021). On the edge of dispensability, the chloroplast ndh genes. Int. J. Mol. Sci. 22, 12505. doi:10.3390/ijms222212505
Stamatakis, A. (2014). RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi:10.1093/bioinformatics/btu033
Sun, Y. X., Deng, T., Zhang, A. D., Moore, M. J., Landis, J. B., Lin, N., et al. (2020). Genome sequencing of the endangered Kingdonia uniflora (Circaeasteraceae, Ranunculales) reveals potential mechanisms of evolutionary specialization. iScience 23, 101124. doi:10.1016/j.isci.2020.101124
Sun, Y., Zou, P. S., Jiang, N. N., Fang, Y. F., and Liu, G. F. (2022). Comparative analysis of the complete chloroplast genomes of nine Paphiopedilum species. Front. Genet. 12, 772415. doi:10.3389/fgene.2021.772415
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq-versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. doi:10.1093/nar/gkx391
Topik, H., Yukawa, T., and Ito, M. (2005). Molecular phylogenetics of subtribe Aeridinae (Orchidaceae): Insights from plastid matK and nuclear ribosomal ITS sequences. J. Plant Res. 118, 271–284. doi:10.1007/s10265-005-0217-3
UNEP-WCMC (Comps) (2020). Checklist of CITES species. Cambridge, United Kingdom: CITES Secretariat. AvaliableAt: https://checklist.cites.org/(Accessed June 27, 2022).
Vaughn, J. N., Chaluvadi, S. R., TusharRangan, L., and Bennetzen, J. L. (2014). Whole plastome sequences from five ginger species facilitate marker development and define limits to barcode methodology. PLoS One 9, e108581. doi:10.1371/journal.pone.0108581
Wicke, S., Schneeweiss, G. M., dePamphilis, C. W., Müller, K. F., and Quandt, D. (2011). The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi:10.1007/s11103-011-9762-4
Xia, C. L., Wang, M. J., Guan, Y. H., and Li, J. (2022). Comparative analysis of the chloroplast genome for aconitum species: Genome structure and phylogenetic relationships. Front. Genet. 13, 878182. doi:10.3389/fgene.2022.878182
Xin, J., Xin, Y. X., Yao, G. Q., Yang, L. Y., Li, F., and Tang, J. R. (2020). Complete chloroplast genome sequence of tropical orchid Renanthera imschootiana. Mitochondrial DNA Part B 5, 2785–2786. doi:10.1080/23802359.2020.1778571
Xu, J., Liu, C., Song, Y., and Li, M. F. (2021). Comparative analysis of the chloroplast genome for four pennisetum species: Molecular structure and phylogenetic relationships. Front. Genet. 12, 687844. doi:10.3389/fgene.2021.687844
Zeb, U., Dong, W. L., Zhang, T. T., Wang, R. N., Shahzad, K., Ma, X. F., et al. (2020). Comparative plastid genomics of Pinus species: Insights into sequence variations and phylogenetic relationships. J. Syst. Evol. 58, 118–132. doi:10.1111/jse.12492
Zhang, H. Y., Li, C., Miao, H. M., and Xiong, S. J. (2013). Insights from the complete chloroplast genome into the evolution of Sesamum indicum L. PLoS One 8, e80508. doi:10.1371/journal.pone.0080508
Zou, L. H., Huang, J. X., Zhang, G. Q., Liu, Z. J., and Zhuang, X. Y. (2015). A molecular phylogeny of Aeridinae (Orchidaceae: Epidendroideae) inferred from multiple nuclear and chloroplast regions. Mol. Phylogenet. Evol. 85, 247–254. doi:10.1016/j.ympev.2015.02.014
Keywords: renanthera, chloroplast genome, comparative analysis, phylogenomics, vandaeae
Citation: Xiao T, He L, Yue L, Zhang Y and Lee SY (2022) Comparative phylogenetic analysis of complete plastid genomes of Renanthera (Orchidaceae). Front. Genet. 13:998575. doi: 10.3389/fgene.2022.998575
Received: 20 July 2022; Accepted: 22 August 2022;
Published: 14 September 2022.
Edited by:
Serena Aceto, University of Naples Federico II, ItalyReviewed by:
Yongqi Zheng, Chinese Academy of Forestry, ChinaCopyright © 2022 Xiao, He, Yue, Zhang and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yonghong Zhang, ZGFwaG5lY25AYWxpeXVuLmNvbQ==; Shiou Yih Lee, c2hpb3V5aWgubGVlQG5ld2ludGkuZWR1Lm15
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.