 Hanna Moczulska1*
Hanna Moczulska1* Michal Pietrusinski1
Michal Pietrusinski1 Karolina Zezawska1
Karolina Zezawska1 Marcin Serafin1Beata Skoczylas1Tomasz Jachymski2Katarzyna Wojda2Piotr Sieroszewski2
Marcin Serafin1Beata Skoczylas1Tomasz Jachymski2Katarzyna Wojda2Piotr Sieroszewski2 Maciej Borowiec1
Maciej Borowiec1- 1Department of Clinical Genetics, Medical University of Lodz, Lodz, Poland
- 2Department of Fetal Medicine and Gynecology, Medical University of Lodz, Lodz, Poland
Objective: Tetrasomy 9p and trisomy 9p are rare chromosomal aberrations. The phenotypes of tetrasomy 9p and trisomy 9p are variable. Most cases are diagnosed in the postnatal period. The study aims to analyze the prenatal phenotype of tetrasomy 9p and trisomy 9p in terms of ultrasound and screening tests.
Methods: A set of 1573 prenatal tests performed from 2016 to 2021 was reviewed to identify all cases with trisomy 9p and tetrasomy 9p. In four cases with 9p gain, non-invasive and invasive test results were analyzed.
Results: Four cases with the 9p gain were diagnosed in the prenatal period: two cases with tetrasomy 9p and two cases with trisomy 9p. Nasal bone hypoplasia and ventriculomegaly are common features of 9p gain. In two out of four cases with the 9p gain, an increased risk of trisomy 21 was found in the combined first-trimester screening test.
Conclusion: Trisomy 9p and tetrasomy 9p are characterized by a variable phenotype in the prenatal period, manifesting in genetically abnormal fetuses. The tetrasomy 9p and trisomy 9p may suggest trisomy 21 in the first trimester.
Introduction
Tetrasomy 9p is a rare chromosomal aberration caused by the presence of four copies of the short arm of chromosome 9. The extra two copies form an additional isochromosome. Most cases of tetrasomy 9p occur de novo during maternal meiosis II nondisjunction (Dutly et al., 1998). The short arm is duplicated and the long arm is lost. About 60 cases of tetrasomy 9p have been published, including over 20 prenatal cases (Vinkšel et al., 2019). Individuals with tetrasomy 9p demonstrated brain, heart, and genitourinary system malformations. Additionally, facial dysmorphia and intellectual disability were observed in patients with tetrasomy 9p. Fewer and less severe abnormalities were observed in cases with mosaicism (Chen et al., 2014). Symptoms vary in affected individuals, depending on the type and percentage of cells containing tetrasomy 9p. A prenatal diagnosis of mosaic tetrasomy 9p is rare. A false negative result in pregnancies with fetal mosaic tetrasomy 9p may be obtained, because the mosaic level of tetrasomy 9p may decrease after long-term tissue culture (Chen et al., 2014).
The phenotype of trisomy 9p is milder. More than 150 cases of trisomy 9p have been described. Most cases with trisomy 9p demonstrate a parental reciprocal translocation between chromosome nine and another autosome. In 9p trisomy, duplication may involve part of the short arm, the whole short arm, or the short arm, and part of the long arm of chromosome 9. The symptoms of 9p trisomy may be similar in affected individuals, regardless of the size of the duplicate 9p part. Trisomy 9p is usually diagnosed in the postnatal period (Guilherme et al., 2014). Little information has been published about the fetal phenotype of trisomy 9p; however, it is likely that trisomy 9 has a milder phenotype that does not allow detection during pregnancy.
As gains in 9p can be diagnosed invasively with karyotyping or aCGH and not by routine screening tests, the aim of our study was to analyze the prenatal phenotype of cases with trisomy 9p and tetrasomy 9p in ultrasound and screening tests.
Materials and methods
A sample of 1573 prenatal tests (1409 amniocentesis and 164 chorionic villus sampling) performed in the Department of Clinical Genetics of the Medical University of Lodz from 2016 to 2021 were reviewed to identify cases with trisomy 9p and tetrasomy 9p. The search identified four cases with 9p gain; in these cases, non-invasive and invasive test results were analyzed. In addition, a review of the literature was performed.
All prenatal genetic tests were ordered in response to a diagnosis of high-risk pregnancy in the Department of Clinical Genetics and the Department of Fetal Medicine and Gynecology of the Medical University of Lodz. The study itself was conducted in a tertiary referral center. The study group comprised women at high risk of having a child with a genetic disease. All patients were Caucasian. The patients were tested in the National Health System’s prenatal screening program. The criteria for inclusion in the prenatal testing program comprises any of the following: the age above 35 years, the occurrence of a chromosomal aberration in the previous fetus or child, structural chromosomal aberration in the mother or father of the fetus, a significantly increased risk of having a child with a monogenic or multifactorial disease, abnormal fetal ultrasound or biochemical tests results that indicate increased the risk of chromosomal abnormalities or fetal abnormalities. Detailed ultrasound examinations were performed in all cases. In most cases, fetal echocardiography was performed, especially in cases of suspected fetal heart defects, in fetuses with non-cardiac defects, fetal chromosomal abnormalities, and in cases with a family history of heart defects. All scans were carried out by certified sonographers (certificates of the Foetal Medicine Foundation and the Polish Society of Gynaecologists and Obstetricians). Non-invasive tests consisted of the combined test (reimbursed in the National Health System’s prenatal screening program) and the non-invasive prenatal screening testing NIPT (not reimbursed in Poland). Invasive procedures were indicated by abnormal fetal ultrasound, high risk of aneuploidy in screening tests, or history indicating an increased risk of genetic disease in the fetus. Fetal material was collected by amniocentesis or chorionic villus sampling. In each case, the clinical geneticist selected the appropriate genetic test. In our department, the first-line test is aCGH (array-based comparative genomic hybridization). In selected cases, the diagnosis is completed by an assessment of karyotype or molecular tests. aCGH was performed using Agilent, GenetiSure Pre-Screen Kit 8x60K with a resolution of approximately 0.50 Mb. According to standard protocols, samples were cultured for classical cytogenetic analysis. The chromosomes obtained in the metaphase were subjected to Giemsa staining after trypsin treatment and analyzed on the Cytovision karyotyping platform (Cytovision DM2500) (Caspersson et al., 1968; Sumner, 1982). Karyotype and aCGH results are described according to the International System for Human Cytogenomic Nomenclature (ISCN 2020) (Liehr, 2021). Schematic drawings of the observed chromosomal aberrations for all 4 cases are demonstrated in Figure 1. The study was conducted according to the Declaration of Helsinki on human subject research. The participants gave their written informed consent authorizing us to perform genetic tests and use the data for research and education.
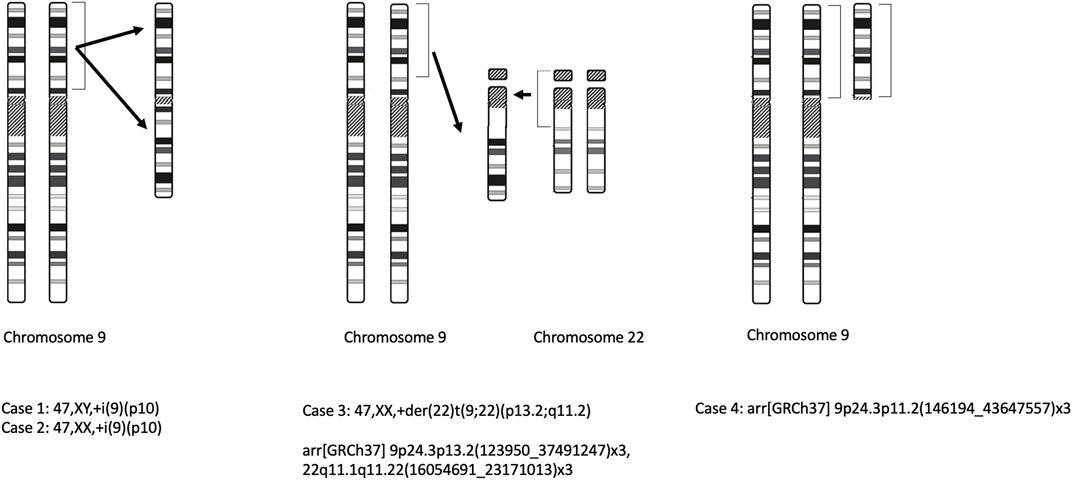
FIGURE 1. Schematic drawings of the observed chromosomal aberrations (case 1, 2, 3, 4).
Results
Four cases of 9p gain were diagnosed in the prenatal period: two cases with tetrasomy 9p and two cases with trisomy 9p (one case had an additional 22q11 duplication). Table 1 contains the prenatal sonographic features for the four described cases and other cases from the literature.
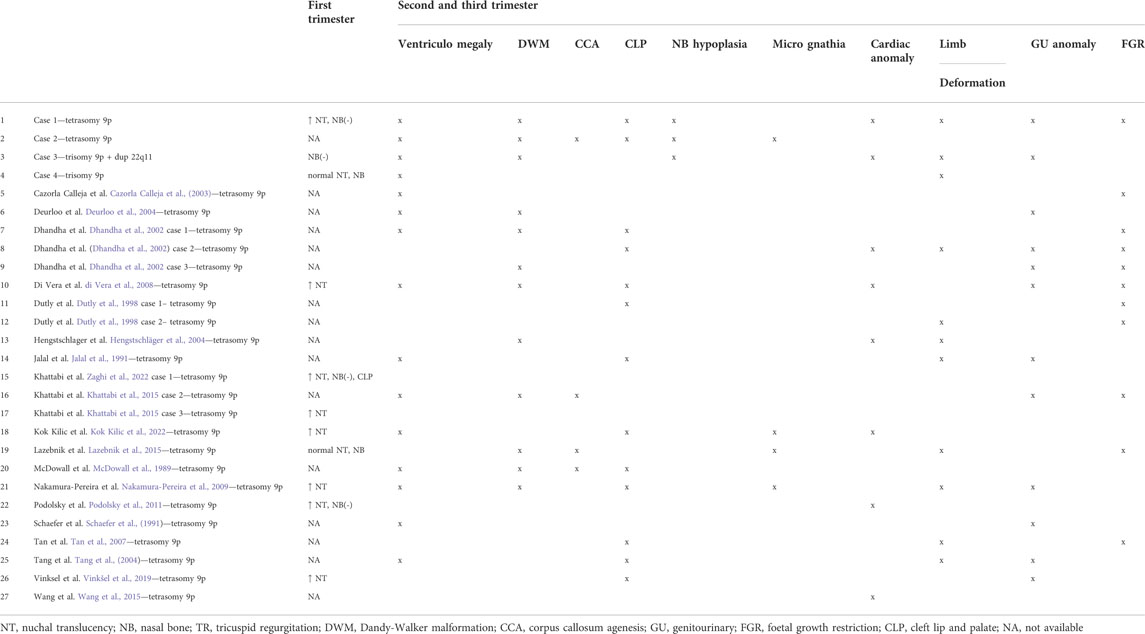
TABLE 1. Prenatal sonographic features of four described cases and cases from the literature.
Case 1
A 32-year-old gravida, G3P1A1 (gravida, para, abortus), underwent amniocentesis at 16 weeks of gestation because of a high risk of fetal trisomy 21. There was no family history of congenital malformations. The nuchal translucency (NT) measured 3.0mm, and the nasal bone was absent. The combined first-trimester screening revealed a high risk for Down syndrome 1:35 [PAPP-A (pregnancy-associated plasma protein-A) 0.57 MoM (multiple of the median); BhCG (the β-subunit of human chorionic gonadotropin) 0.46 MoM]. NIPT (non-invasive prenatal testing) indicated a low risk of trisomy 21, 18, and 13. Amniocentesis was performed at 16 weeks of gestation. Fetal aCGH demonstrated 9p24.3p13.1 gain: arr [GRCh37] 9p24.3p11.2(146194_43647557)x3. Fetal karyotype identified a complete tetrasomy of the short arm of chromosome 9 47,XY,+i(9)(p10) (Liehr, 2021). Second-trimester sonography (19 weeks of gestation) showed nasal bone hypoplasia (4.4mm; <1st percentile), cleft lip and palate, abnormal posterior fossa (Dandy-Walker malformation), ventriculomegaly (posterior ventricle Vp 10.3 mm), ventricular septal defect, and humerus and femur shortening (humerus length HL 11th percentile; femur length FL 9th percentile). The pregnancy was terminated. No postmortem examination was performed.
Case 2
33-year-old gravida, G1P0A0, was consulted in the course of the first gestation. She was exposed to harmful medications for atopic dermatitis in early pregnancy—tetracycline and retinoid derivatives. The family and medical histories were unremarkable.
Medical data from the first trimester were unavailable, the patient reported that the ultrasound result was normal, and the combined test was not performed. Second-trimester sonography (18 weeks of gestation) demonstrated multiple abnormalities: agenesis of the corpus callosum, ventriculomegaly (Vp 11 mm), abnormal posterior fossa (Dandy-Walker malformation), bilateral cleft lip and palate, absent nasal bones, and retrognathia. In addition, pericardial effusion, increased renal echogenicity and pyelectasis, and clenched hands were found. Diagnostic amniocentesis was performed. Fetal karyotype showed following results—47,XX,+i(9)(p10). The pregnancy was terminated. No postmortem examination was performed.
Case 3
A 32-year-old gravida, G2P2A1, underwent amniocentesis at 19 weeks of gestation because of the high risk of fetal trisomy 21 and multiple malformations. There was no family history of congenital disabilities. At 13 weeks of pregnancy, an ultrasound examination revealed absent nasal bone, tricuspid regurgitation, and bradycardia (fetal heart rate 134/min.). The combined first-trimester screening revealed a high risk for Down syndrome 1:47 (PAPP-A 0.88 MoM; BhCG 4.36 MoM). NIPT indicated a low risk of trisomy 21, 18, and 13. The scope of NIPT included risk assessment for trisomy 21, trisomy 18, trisomy 13 and sex chromosomal abnormalities. At that time, the patient decided against an invasive procedure. Second-trimester sonography demonstrated ventriculomegaly (Vp 10 mm), nasal bone hypoplasia, and ARSA (aberrant right subclavian artery). There was also suspicion of corpus callosum partial agenesis and Dandy-Walker malformation. At 19 weeks the patient underwent amniocentesis. After sampling the amniotic fluid, aCGH was performed. The following aberrations were revealed: arr[GRCh37] 9p24.3p13.2(123950_37491247)x3,22q11.1q11.22(16054691_23171013)x3. Fetal karyotype was not assessed due to cell culture failure. Sonography at 22 weeks showed Dandy-Walker malformation, mild ventriculomegaly (Vp 10 mm), hypoplasia of corpus callosum, nasal bones hypoplasia (3.9mm; <1st percentile), and shortening of the humerus and femur (FL 35mm; 3rd percentile, HL 31mm; 2nd percentile). Moreover, tricuspid regurgitation and pulmonary stenosis (Vmax = 95 cm/s) were detected.
At 28 and 32 weeks, subsequent examination showed mild pulmonary stenosis (Vmax = 105 cm/s), previously reported abnormalities in the brain, mild renal pelvis dilatation (5.5 mm), nasal bone hypoplasia (5.3mm; <1st percentile), long bones shortening (FL 35mm; 3rd percentile, HL 44mm; 1st percentile) and polyhydramnios (amniotic fluid index - AFI 30 cm). The baby was born in the 39th week of pregnancy, Apgar 9/9, weight 3000g and length 51 cm. The newborn karyotype revealed an additional derivative chromosome: 47,XX,+der (22)t (9;22) (p13.2;q11.2)dmat. The mother of the child was a carrier of a balanced translocation: 46,XX,t(9;22)(p13.2;q11.2). The father’s karyotype was normal: 46,XY.
Case 4
A 28-year-old, G3P1A1, was referred to the clinic for suspected congenital fetal defects in second-trimester sonography. First-trimester sonography was normal. The combined first-trimester screening revealed a low risk for trisomy 21, 18, and 13 (PAPP-A 0.43 MoM; BhCG 1.19 MoM); risk for trisomy 21 1:1519, risk for trisomy 18 < 1:20000 and risk for trisomy 13 1:2275. At 21 weeks of gestation, bilateral ventriculomegaly (Vp 11.8 mm), club foot, and fetal growth restriction (10th percentile) were detected. Amniocentesis was performed at 24 weeks of gestation. aCGH study demonstrated trisomy: arr[GRCh37] 9p24.3p11.2 (146194_43647557)x3 (43.5 Mbp). Negative test results for cytomegalovirus (CMV) and Toxoplasma gondii were obtained. Parents reported that the delivery was at 39 weeks, and genetic testing in the newborn confirmed prenatal diagnosis.
Discussion
Our paper presents two prenatal cases of tetrasomy 9p and two cases of trisomy 9p. Tetrasomy 9p is characterized by a variable phenotype in the prenatal period (Dhandha et al., 2002; Vinkšel et al., 2019). The fetal phenotype of trisomy 9p is poorly described in the literature. Leichtman et al. described phenotypic overlap between tetrasomy 9p and trisomy 9p, and noted that tetrasomy 9p presents a more severe phenotype. The spectrum of symptoms may be continuous between tetrasomy 9p, mosaic tetrasomy 9p, and trisomy 9p. The phenotype may depend on the gene dosage effect (Leichtman et al., 1996). Our study indicates a similar relationship, with cases with tetrasomy 9p having a more severe phenotype (cases 1 and 2).
The clinical data of all the prenatal cases with tetrasomy 9p, plus our two cases with tetrasomy 9p and our two cases with 9p trisomy, are given in Table 1. Comparing cases from the literature is challenging, as they have been published in the last 10–20 years, and the quality of ultrasound examinations has increased significantly during this time. Nevertheless, the table shows that the most common defects in fetuses with tetrasomy 9p are ventriculomegaly, Dandy-Walker malformation, and cleft lip/palate (Table 1). These defects are quite unspecific and can be found in many genetically abnormal fetuses.
Brain defects are often reported in fetuses with tetrasomy 9p, with the most common abnormalities being ventriculomegaly and Dandy-Walker malformation. Deurloo et al. suggested that Dandy-Walker malformation may be a marker of tetrasomy 9p (Deurloo et al., 2004). In our study, all cases had ventriculomegaly, and three out of four had Dandy-Walker malformation. Agenesis of the corpus callosum is also observed in fetuses with tetrasomy 9p (McDowall et al., 1989; Lazebnik and Cohen, 2015). Two cases in our study demonstrated corpus callosum hypoplasia/agenesis (cases 2 and 3).
In cases of tetrasomy 9p, fetal facial dysmorphia consists of cleft lip and palate, micrognathia, and abnormal facial profile. Hypertelorism is often found in postnatal diagnosis (Jalal et al., 1991). Until now, binocular distance (BOD) has not been studied in prenatal cases with tetrasomy 9p, nor were the nasal bones. Podolsky et al. presented one case with the absence of nasal bone as a marker of tetrasomy 9p (Podolsky et al., 2011). Zaghi et al. described one case with tetrasomy 9p and absent nasal bone (Zaghi et al., 2022). Khattabi et al. presented a case with tetrasomy 9p and absent nasal bone diagnosed in the first trimester (Khattabi et al., 2015). In our study, three of the four fetuses had nasal bone hypoplasia. In two cases, nasal bone hypoplasia was diagnosed in the first trimester (cases 1 and 3).
Congenital heart defects are observed in 30% of fetuses with tetrasomy 9p, with the most common being defects in the ventricular septal and common atrioventricular canal, and complex heart defects (Dhandha et al., 2002; Kok Kilic et al., 2022). Interestingly, a persistent left superior vena cava is often seen, which may be the only symptom of tetrasomy 9p. Wang et al. presented a prenatal diagnosis of mosaic tetrasomy 9p in a fetus with isolated persistent left superior vena cava (Wang et al., 2015).
Defects of the genitourinary system are observed in 43% of cases with tetrasomy 9p. The severity of the lesions varies significantly from mild pyelectasia to bilateral multicystic dysplastic kidney. The amount of amniotic fluid is often abnormal and polyhydramnios is mainly observed (Tan et al., 2007). Oligohydramnios is found in fetuses with tetrasomy 9p and severe urinary tract defects (Schaefer et al., 1991).
The abnormalities caused by tetrasomy 9p were usually found in the second or third trimester. Single reports of tetrasomy 9p have been reported after first-trimester diagnosis (Nakamura-Pereira et al., 2009; Kok Kilic et al., 2022). Fortunately, modern ultrasound technology allows faster and more accurate early diagnosis of developmental abnormalities. In our group, nuchal translucency was increased in one case with 9p gain, and nasal bone hypoplasia was observed in three cases: two cases were diagnosed in the first trimester. A combined first-trimester screening test indicated a high risk of trisomy 21 in two fetuses (case 1, case 3). Our findings suggest that the clinical picture of the 9p gain in the first trimester may suggest the most common trisomies. Therefore invasive testing should be recommended in cases with a high risk for trisomy 21 (>1:100) and normal NIPT, including aCGH. Common features include increased NT, absence of nasal bones, tricuspid regurgitation, and abnormal first-trimester maternal serum screening test. Our cases demonstrated decreased levels of PAPP-A (0.57 MoM, 0,88 MoM and 0.43 MoM) and variable free B-hCG levels (0.46 MoM, 4.3 MoM and 1.19 MoM). Khattabi et al. described a case with tetrasomy 9p in which maternal serum screening indicated a high risk of trisomy 21 (Khattabi et al., 2015); however, Lazebnik et al. presented a prenatal case with tetrasomy 9p with a low risk for trisomy 21, 18, and 13 (serum analytes: 1.11 MoM PAPP-A and 1.75 MoM BhCG) (Lazebnik and Cohen, 2015). The relationship between first-trimester maternal screening and the fetal 9p gain has not been investigated; as such, more cases are needed to thoroughly analyze the relationship between PAPP-A and BhCG levels and the fetal 9p gain.
Non-invasive prenatal testing (NIPT) is not routinely used in the prenatal screening of trisomy 9p and tetrasomy 9p, and hence few publications discuss this possibility. Wang et al. presented a case with mosaic tetrasomy 9p in the fetus, in which NIPT revealed “extra” genetic material derived from chromosome 9 (Wang et al., 2015). The first case of maternal mosaic tetrasomy 9p being incidentally detected on NIPT was reported by Shu et al.; in this case, NIPT was performed twice and both results revealed multiple chromosomal aberrations including elevation in DNA from chromosome 9p. The scope of NIPT included risk assessment for trisomy 21, trisomy 18, trisomy 13, sex chromosomal abnormalities, and genome-wide chromosomal aberrations at a resolution of 3 Mb or above. Amniocentesis was performed and a normal fetal aCGH result was obtained. Maternal blood karyotype revealed mos 47,XX,+dic(9;9)(q21.1;q21.1)[24]/46,XX[9], maternal fibroblasts were not examined. Subtelomeric multiplex ligation-dependent probe amplification (MLPA) was performed on uncultured maternal blood and on the maternal buccal swab. The blood test confirms mosaic 9p duplication, and the buccal swab result was normal. As the amniocentesis was normal, no clinical description or genetic testing was performed on the child after delivery (Shu et al., 2021). This case confirms that some cases with mosaic tetrasomy 9p may go undiagnosed in the general population.
In our study, the karyotype of case 3 shows the presence of derivative chromosome 22: 47,XX,+der(22)t(9;22)(p13.2;q11.2)dmat. 22q11.2 duplication syndrome is a condition characterized by variable clinical phenotype that includes heart defects, urogenital abnormalities, velopharyngeal insufficiency with or without cleft palate, mild learning difficulties with some individuals being essentially normal. The duplicated region contains 30 to 40 genes, however for many of these genes, little is known about their function. Genes that may influence the phenotype may be similar to genes involved in the same deleted region in DiGeorge syndrome (DGS; OMIM 188400) and velocardiofacial syndrome (VCFS; OMIM 192430) (mainly TBX1, HIRA, CRKL).
Trisomy 9p and tetrasomy 9p can be diagnosed using classical cytogenetic analysis and aCGH. The karyotype identifies the structure of a chromosomal aberration. aCGH offers higher test resolution than traditional G-band karyotyping and allows for more precise localization of the chromosomal breakpoints. Nowadays whole exome sequencing (WES) is sometimes performed as a first-line genetic test. WES can also diagnose chromosomal aberrations; however, it is not a good tool to diagnose them. The biggest advantage of microarrays over WES methods is the quality of the data post-processing in detecting specific copy number variations (CNVs). This is because WES methods mimic a traditional microarray method when so-called “pseudo probes” are created from the next-generation sequencing (NGS) reads to establish a log2 ratio value, which can then be used to estimate the actual copy number. In contrast, microarrays have “real probes” and during experiments, the signal intensity of these probes is compared directly to reference probes. Thus microarrays offer better overall genomic coverage and can detect intronic and intergenic alterations.
Conclusion
Trisomy 9p and tetrasomy 9p are characterized by a variable phenotype in the prenatal period which can be found in many genetically abnormal fetuses. The clinical picture of the 9p gain in the first trimester may suggest trisomy 21.
Data availability statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Ethics statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. The patients/participants provided their written informed consent to participate in this study.
Author contributions
HM, MP, KZ, PS and MB contributed to conception and design of the study. HM, MS, TJ, KW, BS, PS, MB organized the database. HM wrote the first draft of the manuscript. MP, AZ wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Caspersson, T., Farber, S., Foley, G. E., Kudynowski, J., Modest, E. J., Simonsson, E., et al. (1968). Chemical differentiation along metaphase chromosomes. Exp. Cell Res. 49, 219–222. doi:10.1016/0014-4827(68)90538-7
Cazorla Calleja, M. R., Verdú, A., and Félix, W. (2003). Dandy-Walker malformation in an infant with tetrasomy 9p. Brain Dev. 25, 220–223. doi:10.1016/s0387-7604(02)00211-5
Chen, C. P., Wang, L. K., Chern, S. R., Wu, P. S., Chen, Y. T., Kuo, Y. L., et al. (2014). Mosaic tetrasomy 9p at amniocentesis: Prenatal diagnosis, molecular cytogenetic characterization, and literature review. Taiwan. J. Obstet. Gynecol. 53, 79–85. doi:10.1016/j.tjog.2013.12.002
Deurloo, K. L., Cobben, J. M., Heins, Y. M., de Ru, M., Wijnaendts, L. C. D., and van Vugt, J. M. G. (2004). Prenatal diagnosis of tetrasomy 9p in a 19-week-old fetus with dandy-walker malformation: A case report. Prenat. Diagn. 24, 796–798. doi:10.1002/pd.850
Dhandha, S., Hogge, W. A., Surti, U., and McPherson, E. (2002). Three cases of tetrasomy 9p. Am. J. Med. Genet. 113, 375–380. doi:10.1002/ajmg.b.10826
di Vera, E., Liberati, M., Celentano, C., Calabrese, G., Guanciali-Franchi, P. E., Morizio, E., et al. (2008). Rhombencephalosynapsis in a severely polymalformed fetus with non-mosaic tetrasomy 9p, in intracytoplasmic-sperm-injection pregnancy. J. Assist. Reprod. Genet. 25, 577–580. doi:10.1007/s10815-008-9257-7
Dutly, F., Balmer, D., Baumer, A., Binkert, F., and Schinzel, A. (1998). Isochromosomes 12p and 9p: Parental origin and possible mechanisms of formation. Eur. J. Hum. Genet. 6, 140–144. doi:10.1038/sj.ejhg.5200168
Guilherme, R. S., Meloni, V. A., Perez, A. B. A., Pilla, A. L., de Ramos, M. A. P., Dantas, A. G., et al. (2014). Duplication 9p and their implication to phenotype. BMC Med. Genet. 15, 142. doi:10.1186/s12881-014-0142-1
Hengstschläger, M., Bettelheim, D., Drahonsky, R., Repa, C., Deutinger, J., and Bernaschek, G. (2004). Prenatal diagnosis of tetrasomy 9p with Dandy-Walker malformation. Prenat. Diagn. 24, 623–626. doi:10.1002/pd.933
Jalal, S. M., Kukolich, M. K., Garcia, M., Benjamin, T. R., and Day, D. W. (1991). Tetrasomy 9p: An emerging syndrome. Clin. Genet. 39, 60–64. doi:10.1111/j.1399-0004.1991.tb02986.x
Khattabi, E. L., Jaillard, L., Andrieux, S., Pasquier, J., Perrin, L., Capri, L., et al. (2015). Clinical and molecular delineation of Tetrasomy 9p syndrome: Report of 12 new cases and literature review. Am. J. Med. Genet. A 167, 1252–1261. doi:10.1002/ajmg.a.36932
Kok Kilic, G., Pariltay, E., Karaca, E., Durmaz, B., Ekici, H., Imamoglu, M., et al. (2022). Prenatal diagnosis of a case with tetrasomy 9p confirmed by cytogenetics, FISH, microarray analysis and review. Taiwan. J. Obstet. Gynecol. 61, 122–126. doi:10.1016/j.tjog.2021.10.003
Lazebnik, N., and Cohen, L. (2015). Prenatal diagnosis and findings of tetrasomy 9p. J. Obstet. Gynaecol. Res. 41, 997–1002. doi:10.1111/jog.12706
Leichtman, L. G., Zackowski, J. L., Storto, P. D., and Newlin, A. (1996). Non-mosaic tetrasomy 9p in a liveborn infant with multiple congenital anomalies: Case report and comparison with trisomy 9p. Am. J. Med. Genet. 63, 434–437. doi:10.1002/(SICI)1096-8628(19960614)63:3<434::AID-AJMG4>3.0.CO;2-R
Liehr, T. (2021). International system for human cytogenetic or cytogenomic nomenclature (ISCN): Some thoughts. Cytogenet. Genome Res. 161, 223–224. doi:10.1159/000516654
McDowall, A. A., Blunt, S., Berry, A. C., and Fensom, A. H. (1989). Prenatal diagnosis of a case of tetrasomy 9p. Prenat. Diagn. 9, 809–811. doi:10.1002/pd.1970091110
Nakamura-Pereira, M., do Cima, L. C., Llerena, J. C., Guerra, F. A., and Peixoto-Filho, F. M. (2009). Sonographic findings in a case of tetrasomy 9p associated with increased nuchal translucency and Dandy-Walker malformation. J. Clin. Ultrasound 37, 471–474. doi:10.1002/jcu.20612
Podolsky, R., Saltzman, D., Auerbach, M., and Roman, A. S. (2011). Absent nasal bone as a marker of tetrasomy 9p. Prenat. Diagn. 31, 1313. doi:10.1002/pd.2877
Schaefer, G. B., Domek, D. B., Morgan, M. A., Muneer, R. S., and Johnson, S. F. (1991). Tetrasomy of the short arm of chromosome 9: Prenatal diagnosis and further delineation of the phenotype. Am. J. Med. Genet. 38, 612–615. doi:10.1002/ajmg.1320380422
Shu, W., Cheng, S. S. W., Xue, S., Chan, L. W., Soong, S. I., Kan, A. S. Y., et al. (2021). First case report of maternal mosaic tetrasomy 9p incidentally detected on non-invasive prenatal testing. Genes 12, 370. doi:10.3390/genes12030370
Sumner, A. T. (1982). The nature and mechanisms of chromosome banding. Cancer Genet. Cytogenet. 6, 59–87. doi:10.1016/0165-4608(82)90022-X
Tan, Y. Q., Chen, X. M., Hu, L., Guan, X. Y., and Lu, G. X. (2007). Prenatal diagnosis of nonmosaic tetrasomy 9p by microdissection and FISH: Case report. Chin. Med. J. 120, 1281–1283. doi:10.1097/00029330-200707020-00016
Tang, W., Boyd, B. K., Hummel, M., and Wenger, S. L. (2004). Prenatal diagnosis of tetrasomy 9p. Am. J. Med. Genet. A 126A, 328. doi:10.1002/ajmg.a.20511
Vinkšel, M., Volk, M., Peterlin, B., and Lovrecic, L. (2019). A systematic clinical review of prenatally diagnosed tetrasomy 9p. Balk. J. Med. Genet. 22, 11–20. doi:10.2478/bjmg-2019-0012
Wang, H., Xie, L. S., Wang, Y., and Mei, J. (2015). Prenatal diagnosis of mosaic tetrasomy 9p in a fetus with isolated persistent left superior vena cava. Taiwan. J. Obstet. Gynecol. 54, 204–205. doi:10.1016/j.tjog.2014.12.005
Keywords: tetrasomy 9p, trisomy 9p, prenatal diagnosis, amniocentesis, microarrary
Citation: Moczulska H, Pietrusinski M, Zezawska K, Serafin M, Skoczylas B, Jachymski T, Wojda K, Sieroszewski P and Borowiec M (2022) Cases of tetrasomy 9p and trisomy 9p in prenatal diagnosis—Analysis of noninvasive and invasive test results. Front. Genet. 13:994455. doi: 10.3389/fgene.2022.994455
Received: 14 July 2022; Accepted: 01 September 2022;
Published: 26 September 2022.
Edited by:
Aleksandra Jezela-Stanek, National Institute of Tuberculosis and Lung Diseases, PolandReviewed by:
Brigitte Strizek, University Hospital Bonn, GermanyIzabela Laczmanska, Wroclaw Medical University, Poland
Copyright © 2022 Moczulska, Pietrusinski, Zezawska, Serafin, Skoczylas, Jachymski, Wojda, Sieroszewski and Borowiec. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hanna Moczulska, aGFubmEubW9jenVsc2thQHVtZWQubG9kei5wbA==