 Haiyan Luo1,2†
Haiyan Luo1,2† Ting Huang1,2†Qing Lu1,2Liuyang Zhang1,2Yonghua Xu1,2Yan Yang1,2Zhen Guo2,3Huizhen Yuan1,2Yinqin Shen1,2Shuhui Huang1,2Bicheng Yang1,2
Ting Huang1,2†Qing Lu1,2Liuyang Zhang1,2Yonghua Xu1,2Yan Yang1,2Zhen Guo2,3Huizhen Yuan1,2Yinqin Shen1,2Shuhui Huang1,2Bicheng Yang1,2 Yongyi Zou1,2*Yanqiu Liu1,2*
Yongyi Zou1,2*Yanqiu Liu1,2*- 1Department of Medical Genetics, Jiangxi Maternal and Child Health Hospital, Nanchang, Jiangxi, China
- 2Jiangxi Key Laboratory of Birth Defect Prevention and Control, Jiangxi Maternal and Child Health Hospital, Nanchang, Jiangxi, China
- 3Department of Ultrasound, Jiangxi Maternal and Child Health Hospital, Nanchang, Jiangxi, China
Background and aims: Hemoglobinopathy associated with the HBB gene, with its two general subtypes as thalassemia and abnormal hemoglobin (Hb) variants, is one of the most prevalent hereditary Hb disorders worldwide. Herein we aimed to elucidate the prevalence of ß-thalassemia and abnormal hemoglobin variants and the prenatal diagnosis of the HBB gene in Jiangxi Province, southern central China.
Methods: Hematological indices and capillary Hb electrophoresis were conducted for 136,149 subjects who were admitted to Jiangxi Maternal and Child Health Hospital and requested for hemoglobinopathy investigation. Routine α- and ß-globin genotyping were performed by gap-polymerase chain reaction (Gap-PCR) and reverse dot-blot (RDB) hybridization for the 11,549 individuals suspected to be thalassemia carriers. For participants whose genotypes could not explain their hematological indices, further Sanger sequencing and Gap-PCR were conducted for the detection of rare or novel variants in related globin genes. Prenatal diagnosis was performed for 77 pregnant couples both carrying ß-thalassemia trait at appropriate gestational ages.
Results: Among the 11,549 subjects, 2,548 individuals were identified with HBB-associated hemoglobinopathy based on molecular analysis. A total of 2,358 subjects were identified as ß-thalassemia heterozygous carriers and nine cases were diagnosed as compound heterozygous ß-thalassemia. Additionally, 125 cases were detected with composite α- and ß-thalassemia and the remaining 56 individuals with abnormal Hb variants in the HBB. A total of 35 types of variants were identified in the HBB gene, including 26 types of ß-thalassemia and nine types of abnormal Hb variants. Four novel variants were firstly reported, including one variant in HBA2 and three variants in HBB. Overall, 77 prenatal samples underwent ß-thalassemia molecular diagnosis; 20 fetuses were identified with normal ß-thalassemia genotypes, 30 fetuses as ß-thalassemia heterozygotes, 11 as homozygotes, and 16 as compound heterozygotes in HBB.
Conclusion: We have demonstrated a relatively high prevalence rate at 1.872% of ß-hemoglobinopathies including common and rare ß-thalassemia as well as abnormal Hb variants among large child-bearing population in the Jiangxi area of southern central China for the first time. Our data presents that prenatal diagnosis is an effective way to prevent and control birth defects of ß-thalassemia.
Introduction
Hemoglobinopathies are one of the most common monogenic diseases worldwide (Ghosh et al., 2020). In the human body, hemoglobin (Hb) is a tetramer composed of two α-like and two ß-like globin chains, with the main function to deliver oxygen to the tissues (Ahmed et al., 2020). ß-Associated hemoglobinopathy is a group of hematological diseases caused by mutations in ß-globin genes, including ß-thalassemia and structural Hb variants (Kohne, 2011). ß-thalassemia (β-thal) is caused by reduced output of ß-globin chains, known as β+, or absent production of ß-globin chains, known as β0 (Origa, 2017). The altered production of normal ß-globin chain results in an imbalance of alpha/nonalpha chain synthesis, which is the major determinant of the clinical and hematological severity of ß-thal (Muncie and Campbell, 2009). The most common mutation of ß-thalassemia is small nucleotide variation (SNV), while a few others appear to be large deletions (Schwartz et al., 1988). The structural hemoglobin (Hb) variant, which leads to variable symptoms from almost asymptomatic to severe hemolytic anemia, is characterized by structural abnormalities caused by alteration of the globin peptide chain conformation in the ß-globin chains, which is typically based on single amino acid substitution resulting from a point mutation (Clarke and Higgins, 2000; Taher et al., 2018). Most individuals with heterozygous Hb variants exhibit normal hematological parameters but may display distinct Hb fractions by capillary electrophoresis (Keren et al., 2008; Altinier et al., 2013).
Despite a high prevalence in tropical and subtropical regions, ß-thal and structural Hb variants in HBB are now widely distributed worldwide because of population migration, and show abundant genetic diversity (Weatherall and Clegg, 2001). So far, there have been over 947 variants in HBB reported worldwide according to the online hemoglobin database (http://globin.bx.psu.edu/cgi-bin/hbvar/query_vars3). Furthermore, increasing reports on the prevalence and mutation spectrum of ß-hemoglobinopathy among different ethnic groups and different regions have improved the prevention of this major health problem in recent decades. China, as a country located in southeastern Asia, has high incidence of ß-hemoglobinopathy, especially for ß-thal in the southern provinces (Long et al., 2014). Many large-scale studies among the Chinese southern population, such as those from Hainan, Guangdong, Guangxi, and Fujian Province, have revealed the frequency and distribution of ß-thalassemia mutations in these specific geographic areas (Zhuang et al., 2020; Huang H. et al., 2021; Zhou et al., 2021; Wang et al., 2022). Several other studies have revealed the prevalence and mutation spectrum of abnormal Hb variants in some provinces of China (Lou et al., 2014; Huang et al., 2019; Xu A. et al., 2020). However, no previous study has focused on ß-hemoglobinopathy among large child-bearing age population in the Jiangxi area, a province located in southern central China next to Fujian and Guangdong Province.
Here, we explored the prevalence and mutation spectrum of ß-associated common and rare thalassemia and structural Hb variants among large-scale population of reproductive ages in Jiangxi Province, and provided prenatal diagnosis for those at risk of giving birth to a child with severe ß-thal intermediate or ß-thal major. Our work may provide a basis for genetic counseling, public education, and as a starting point to improve the prevention of ß-thal.
Materials and methods
Subjects
A total of 136,149 subjects of child-bearing age admitted to Jiangxi Maternal and Child Hospital for routine examination from January 2015 to December 2021 were enrolled in our study. The investigated individuals were mainly acquired from the Outpatient Department of the Assisted Reproductive Centre, Medical Genetics Centre, Obstetrics & Gynecology Department of our hospital. All of the included individuals came from rural or urban areas in Jiangxi Province and were aged from 18 to 50 years. This study was approved by the Ethics Committees of Jiangxi Maternal and Child Health Hospital, Jiangxi Province, China. All of the medical tests were performed after obtaining informed consent from the participants.
Hematological studies and hemoglobin electrophoresis analysis
Conventional peripheral complete blood count (CBC) and capillary Hb electrophoresis were primarily screened for all the participants. Two EDTA-Na tubes with 2 ml of peripheral venous blood were obtained from each subject for routine blood examination and hemoglobin electrophoresis screening. Erythrocyte correlative indices were detected using a Sysmex XE-2100 blood analyzer (Sysmex Corporation) according to the standard operating procedure. The composition and content of hemoglobin was analyzed by a Sebia capillary electrophoresis system (Sebia, Inc.) according to the manufacturer’s instructions. Participants with erythrocyte indices with mean corpuscular volume (MCV) < 82 fL and (or) corpuscular hemoglobin (MCH) values <27 pg were taken as suspected thalassemia carriers. Subjects with hemoglobin A2 (HbA2) < 2.5% and HbA2 > 3.5% were suspected as probable α-thalassemia and ß-thalassemia carriers, respectively. Individuals screened with abnormal hemoglobin fraction or HbF values >5% were suspected as abnormal Hb variant or rare ß-thalassemia carriers.
Routine thalassemia genetic analysis
A total of 11,549 subjects (female: 7,911, male: 3,638), including suspected thalassemia carriers and a few individuals who demanded genetic testing, underwent routine molecular detection for thalassemia. Firstly, 2 ml of peripheral venous blood was collected in an EDTA tube, and the genomic DNA of the peripheral venous blood was extracted using the GeneRotex Nucleic Acid Extraction System (GeneRotex, Xian, China). A NanoDrop spectrophotometer (Thermo Scientific, Wilmington, DE, United States) was used to qualify and quantify DNA. Gap-PCR and PCR-RDB (Yaneng BioSciences, Shenzhen) were used to detect three common α-thalassemia deletions (-SEA, -α3.7, and -α4.2) and three α-thalassemia point mutations (Hb Constant spring (Hb CS) (CD142, TAA > CAA), Hb Quong Sze (Hb QS) (CD125, CTG > CCG), and Hb Westmead (CD122, CAC > CAG), as well as 16 types of SNVs in HBB, including CD41-42 (-TCTT), CD43 (G > T), IVS-II-654 (C > T), CD17 (A > T), CD14-15 (+G), −28 (A > G), −29 (A > G), CD71-72 (+A), CD26 (G > A), IVS-I-1 (G > T), IVS-I-1 (G > A), CD27-28 (+C), IVS-I-5 (G > C), Cap+40–43 (-AAAC), initiation codon (ATG > AGG), and CD31 (-C).
Rare and novel thalassemia genetic analyses
Gap-PCR was performed to detect six rare structural variations, including -THAI/, -α21.9/, -α27.6/, HK αα, anti4.2, α fusion gene for α-thal, and three rare ß-thal deletions, including Taiwanese, Gγ+(Aγδβ)0, and SEA-HPFH according to the manufacturer’s protocols (Yaneng BioSciences, Shenzhen, China). Sanger sequencing was used to detect the rare or novel SNVs in HBA1, HBA2, and HBB. The primers used in our study were listed in Table 1.

TABLE 1. Primers used for direct Sanger sequencing.
Prenatal diagnosis
Chorionic villi sampling (CVS) in the first trimester, amniocentesis in the second trimester, and umbilical cord blood sampling in the third trimester were chosen based on the gestational age and willingness of the pregnant females. All of the fetal samples were collected from the pregnant mother under B-mode ultrasound-guidance at the appropriate gestational ages, with CVS from 12 to 14 weeks, amniocentesis from 18 to 26 weeks, and fetal blood sampling from 27 to 32 weeks. About 100 mg of villi tissue, 8 ml of amniotic fluid, or 2 ml of umbilical cord blood were collected. Extraction of fetal DNA was conducted using a QIAamp DNA Mini Kit (Qiagen, Germany) according to the manufacturer’s protocols. A NanoDrop spectrophotometer (Thermo Scientific, Wilmington, DE, United States) was used to qualify and quantify the DNA.
Follow-up of pregnancy outcomes
The 77 couples were followed up by phone interview 1-year after prenatal diagnosis to record the outcomes of fetuses with severe thalassemia. Additionally, the phenotypes and development of fetuses with non-severe thalassemia were followed up 1 year after birth to validate the accuracy of the prenatal diagnosis. In our study, the mutant homozygote and double heterozygote were defined as severe forms of ß-thal.
Statistical analysis
Statistical analysis was performed using SPSS statistical software version 20.0 (International Business Machines Corporation). Descriptive analysis was used to show the genotype and allele frequencies in different populations. The ratio of ß-thalassemia alleles, αβ-thalassemia, and abnormal Hb variants was calculated.
Results
Routine thalassemia genetic analysis and rare or novel thalassemia genetic analysis
Among the 11,549 subjects, 2,457 cases were detected with mutations by routine thalassemia testing, whereas the remaining 91 cases showed continuous hypochromic microcytic anemia, an increased HbA2 or HbF value, or an abnormal hemoglobin fraction by hematological screening and underwent further rare or novel thalassemia genetic analysis. Thirty individuals displaying an increased HbA2 value were identified with rare mutations in HBB, including CD54-58 (-TATGGGCAACCCT), -72 (T > A), codon 30 (AGG > AGC), -28(A > C), -31(A > G), SEA-HPFH, Taiwanese, IVS-I-6 (T > G), IVS-I-110 (G > A), and initiation codon ATG > GTG, with ten genotypes in all. Fifty-six individuals demonstrating abnormal hemoglobin fractions were identified as abnormal Hb variant carriers, including those with Hb J-Bangkok, Hb New York, Hb G-Taipei, Hb G-Coushatta, Hb D-Los Angeles, Hb Yaizu, Hb Deer Lodge, Hb E-Saskatoon, and Hb G-Siriraj, with a total of nine types. In addition, three individuals, all of whom showed normal HbA2 values and hematological parameters, were identified with three novel variants IVS-II-47 (G > C), IVS-II-300–308 (-A), IVS-II-806 (G > C) in HBB, separately. One individual were identified with the polymorphism IVS-II-81 C>T. Furthermore, one participant was identified with novel variant c.301-41_301-39delGGC in HBA2 combined with the common ß-thal mutation IVS-II-654 (C > T).
Overall population prevalence of ß-hemoglobinopathies in the jiangxi region
In our study, 11,549 individuals were suspected cases of ß-hemoglobinopathies by hematological indices or capillary analysis, and all of them underwent further genetic analysis. A total of 2,548 cases were diagnosed with ß-hemoglobinopathy, including 2,367 cases with ß-thal, 125 cases with composite α-and ß-thal, and 56 cases with abnormal Hb variants. The overall prevalence rate of ß-hemoglobinopathies in Jiangxi Province was 1.872% (2,548/13,6149), and the prevalence rates of ß-thal, coinheritance of αβ-thal and Hb variants were 1.739% (2,367/13,6149), 0.092% (125/13,6149), and 0.041% (56/13,6149), respectively. Carriers of composite α- and ß-thal and Hb variants were detected for the first time in a large population of child-bearing adults in Jiangxi and the detailed information was listed as in Table 2.
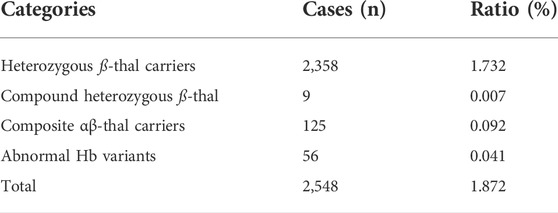
TABLE 2. Detailed constitution of ß-hemoglobinopathy among 13,6149 participants in the Jiangxi region.
Genotypes and mutation spectrum of ß-thal and structural Hb variants
We identified 83 genotypes in our cohort, with 34 of ß-thal, 39 of composite αβ-thal, and ten of abnormal Hb variants. Among the 2,367 subjects detected with ß-thal, we identified 26 kinds of ß-thal variants in all (Table.3). The most frequent genotype was βIVS−II−654 (C>T)/βN, and βCD41/42(−TTCT)/βN, with a remarkable proportion of 45.42% and 22.64%, respectively. The other common genotypes were βCD17(A>T)/βN, ß−28(A>G)/βN, and βCD27/28(+C)/βN, with the corresponding proportions of 10.69%, 7.52%, and 4.86%. As for the allele frequency, the four most frequent mutations were IVS-II-654 (C>T), CD41/42 (-TTCT), CD17 (A>T), and −28 (A>G), with corresponding proportions of 41.9%, 27.5%, 12.6%, and 9.1%, which accounted for 91.1% of all ß-thal mutant alleles. A total of 125 subjects were detected as carriers of both α- and ß-globin variants (Table.4). Among them, 89.6% of genotypes consisted of common SNVs of the ß-globin gene combined with large deletions of the α-globin gene (–SEA/αα, -α3.7/αα, -α4.2/αα). Composite–SEA/αα and βIVS−II−654 (C>T)/βN was the most common genotype. In our study, 56 subjects were identified with silent structural Hb variants in HBB, with ten genotypes in total. All of these individuals displayed normal hematological indices but abnormal hemoglobin fraction by capillary electrophoresis (Table.5). Hb J-Bangkok was the most prevalent Hb variant, accounting for 42.86%, followed by Hb New York and Hb Taipei. We also identified four rare Hb variants, including Hb Yaizu, Hb Deer Lodge, Hb E-Saskatoon, and Hb G-Siriraj. Furthermore, four novel variants were identified, including one variant c.301-41_301-39delGGC in HBA2 and three variants (c.315 + 47G>C, c.315 + 300-308delA, and c.315-45G>C) in HBB. The hematological characteristics of the three subjects with novel variants were shown in Table 6.
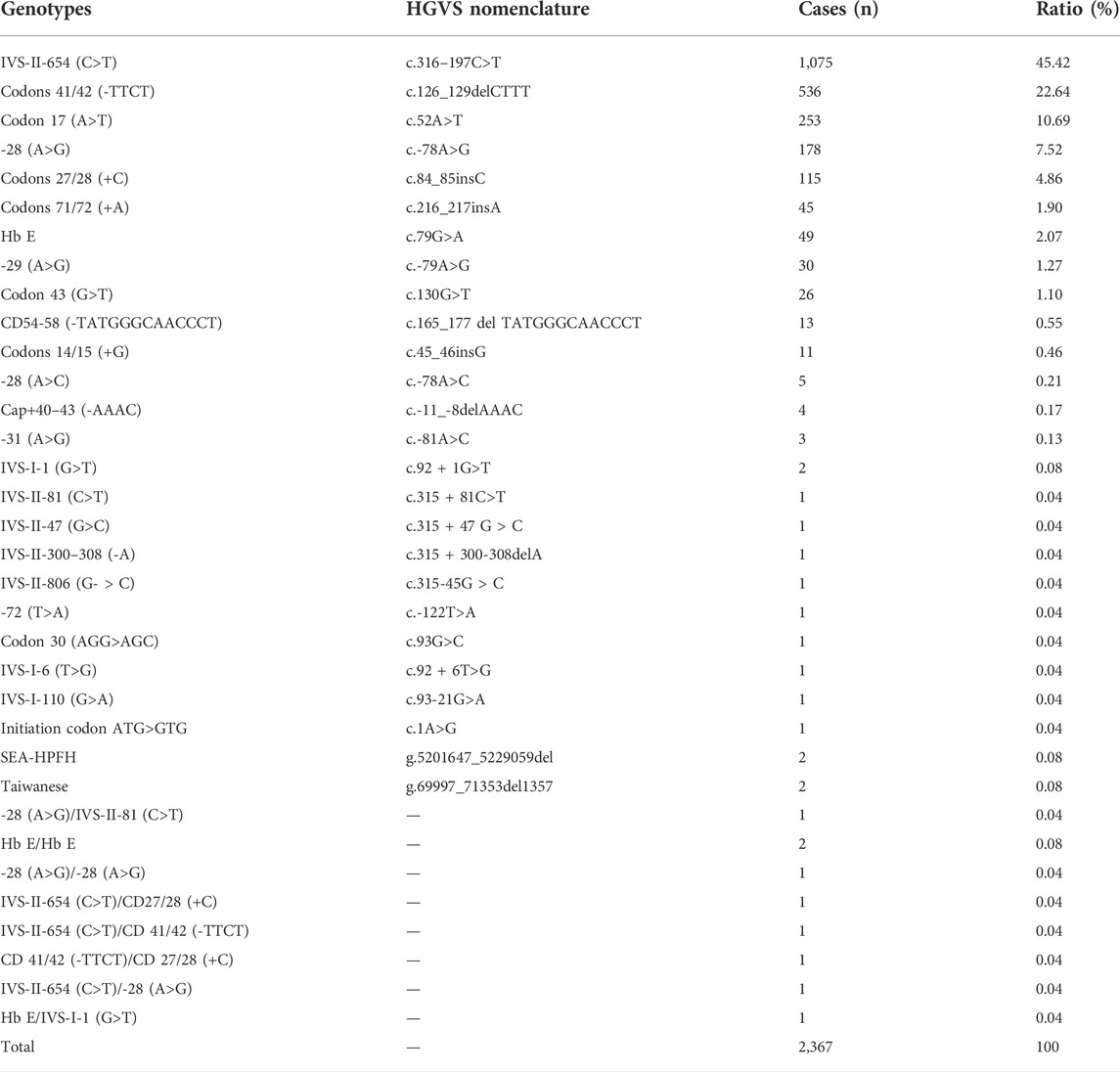
TABLE 3. Genotype constitution of ß-thalassemia in Jiangxi Province.
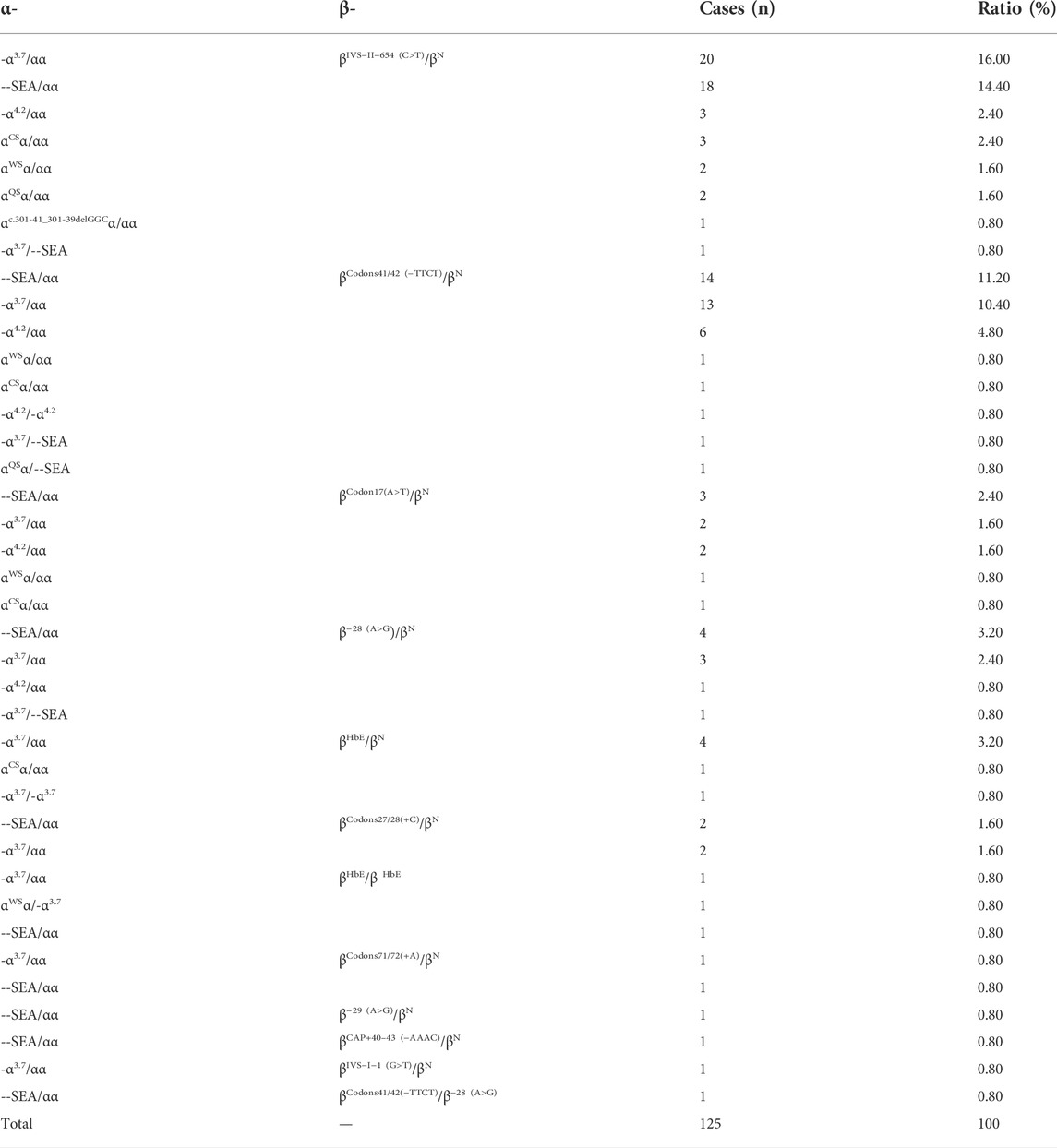
TABLE 4. Genotypes of composite α- and ß-thalassemia in Jiangxi Region.
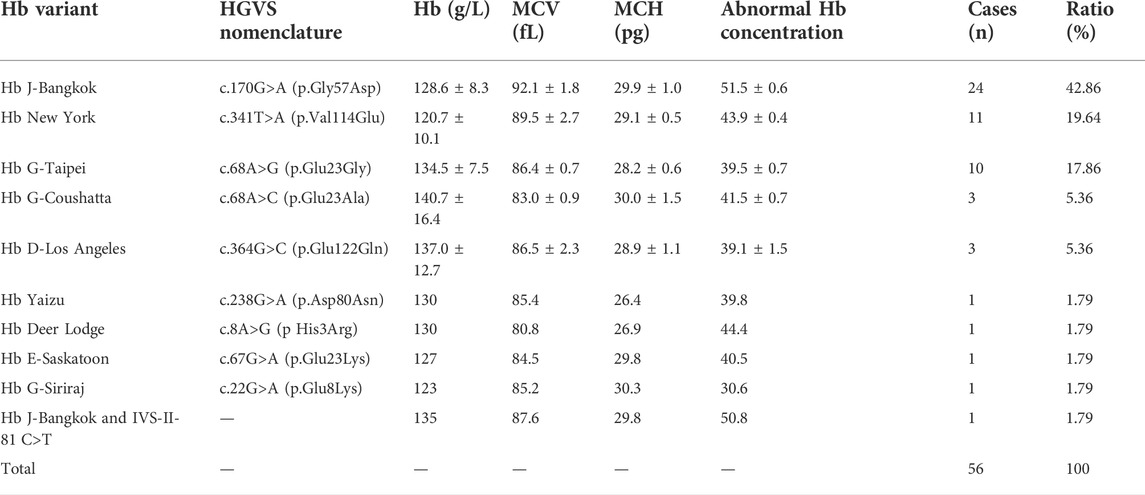
TABLE 5. Abnormal hemoglobin variants in HBB and the hematological characteristics in this cohort in Jiangxi Province.
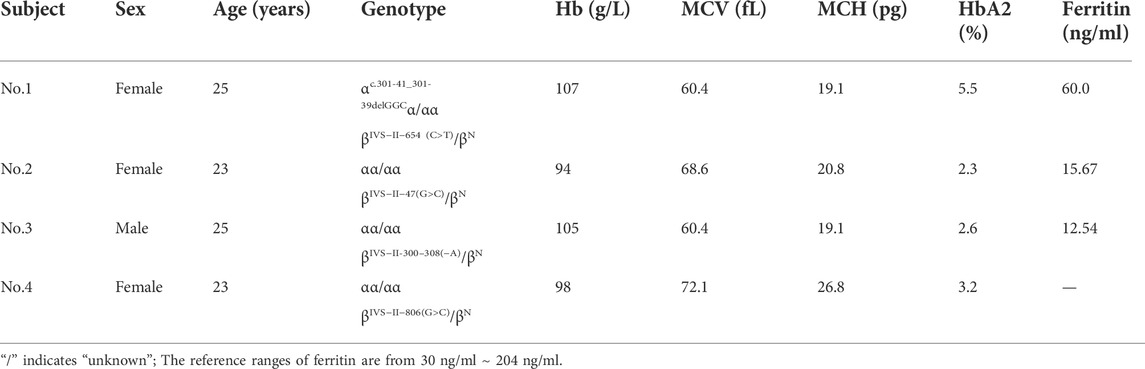
TABLE 6. Hematological characteristics of the four subjects with novel variants.
Prenatal diagnosis and postnatal follow-up outcomes
The screening strategy for pregnant or pre-pregnant couple in our study was recommended as follows: The female was genetically tested first, and if the female was determined to be a ß-thal carrier, her husband underwent further thalassemia genetic analysis. Prenatal diagnosis was recommended if both individuals in the couple were confirmed to be ß-thal trait carriers. In our study, 77 couples were identified as both being ß-thal carriers, and were considered to be at risk of having clinically affected offsprings. Prenatal diagnosis revealed 20 fetuses with normal genotypes in HBB, 26 fetuses as simple ß-thal carriers, four fetuses as composite α- and ß-thal carriers, 16 cases as compound heterozygous ß-thal patients, and 11 fetuses as homozygous ß-thal patients. The information on prenatal diagnosis was listed in detail in Supplementary Table S1.
No post-operative complications were found in 77 pregnant women with thalassemia, and 24 of the 77 pregnancies were terminated due to predicted severe ß-thal intermediate or major, including 10 with ß-thal homozygote and 14 with ß-thal double heterozygote. Following induced abortion, the fetal umbilical cord blood was sampled for genetic confirmation of ß-thal, which showed consistent results with prenatal diagnosis. All normal fetuses and those with ß-thal minor continued with the pregnancy. All of the pregnancy outcomes of the 77 families were listed in Supplementary Table S1. Telephone follow-up after the prenatal diagnosis revealed no thalassemia phenotypes in normal babies 6 months after birth, the babies with ß-thal minor had no or mild anemia symptoms and normal development milestones, while three babies with ß-thal double heterozygote demonstrated moderate anemia symptoms.
Discussion
Here, we presented a large-scale genetic analysis of HBB-associated hemoglobinopathy among adults of child-bearing age in Jiangxi Province. The results showed a high prevalence of 1.872% for hemoglobinopathy of ß-globin in China, with the respective carrier rate at 1.739% for ß-thal, 0.092% for the composite αβ-thal, and 0.041% for the abnormal Hb variant. Based on previous studies, prevalence rate of ß-hemoglobinopathies among child-bearing population in typical geographic areas such as Guangxi (Xiong et al., 2010), Guangdong (Xu et al., 2004), Hubei, (Cheng et al., 2022), and Hunan Province (He et al., 2017) were listed in Table.7. Frequencies of ß-hemoglobinopathies in Jiangxi Province showed difference from other reports in China probably mainly due to geographic and ethnic background. The carrier rate of ß-thal and αβ-thalassemia in our study showed a slightly higher prevalence rate than the previously reported 1.82% in Jiangxi Province (Lin et al., 2014) mainly because of the wider geographic areas contained and more comprehensive of rare ß-thal in our study. Furthermore, the prevalence of abnormal Hb variants in HBB (0.041%) was determined among child-bearing adults in Jiangxi Province for the first time in our study, which was significantly lower than the reported 0.222% in Southeastern China (Huang et al., 2019; Xu L. et al., 2020) and the reported 0.185% for Hb variants in HBB in Dongguan Region of Guangdong Province (Lou et al., 2014). Notably, since Hb E is a special Hb variant with ß-thal hematological trait, we classified Hb E as ß-thal instead of abnormal Hb variant in our study, otherwise the carrier rate of abnormal Hb variant would have increased to 0.086% (116/136,149). We also observed some differences in the ß-thal mutation spectrum in Jiangxi Province compared to those of other provinces in China. For example, in contrast to Guangxi, Guangdong, and Hainan (Xu et al., 2004; Xiong et al., 2010; Wang et al., 2022), CD41/42 (-TCTT) is not the most prevalent mutation in Jiangxi Province, while compared with its surrounding provinces, such as Hunan and Hubei (Liu et al., 2019; Cheng et al., 2022), Jiangxi Province shares a more similar mutation spectrum, with IVS-II-654 (C>T), CD41/42 (-TCTT), and CD17 (A>T) as the three most frequent genotypes in these areas, accounting for almost 80% of all genotypes. Likewise, the spectrum of structural Hb variants in HBB in Jiangxi Province is similar to those reported in previous studies, with the most three common variants as Hb New York, Hb J-Bangkok, and Hb G-Taipei, while Hb YaiZu, Hb Deer Lodge, and Hb E-Saskatoon had been seldom reported previously in China.
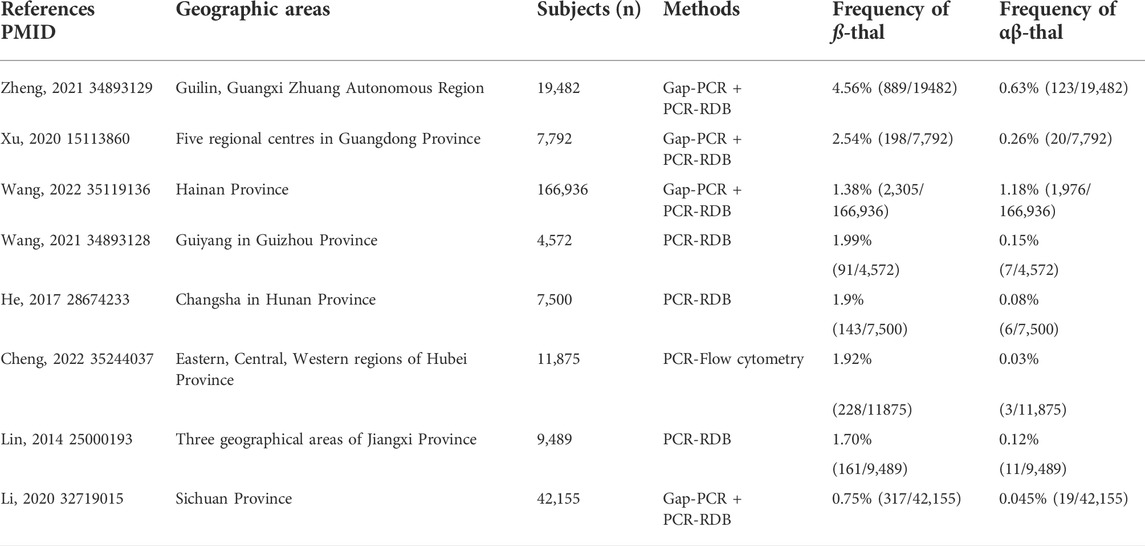
TABLE 7. Comparison of ß-thal frequency among reproductive-age adults in different geographic areas in China.
β-thalassemia is clinically classified into four types, including ß-thal silent, ß-thal trait, ß-thal intermediate, and ß-thal major. ß-thalassemia intermediate, with the genotype of β+/β+ or β+/β0, has a more complex molecular basis than the other three types (Thein, 2013). Patients affected with ß-thal intermediate usually present with diverse phenotypes from mild-to-moderate anemia that does not require lifelong dependence on blood transfusion. In contrast, individuals with ß-thal major, with the genotype of β+/β0 or β0/β0 and severe anemia (Hb persistently <70 g/L), require regular blood transfusions and standardized iron-free therapy to survive (Writing Group For Practice Guidelines For Diagnosis And Treatment Of Genetic Diseases Medical Genetics Branch Of Chinese Medical Association et al., 2020). Cure of this disease depends on successful bone-marrow transplantation, which is expensive and has treatment-associated complications (Ali et al., 2021). Therefore, severe ß-thal intermediate or ß-thal major presents a major public health problem and a social burden in highly prevalent areas. For the prevention of this disease, prenatal diagnosis is the most useful method for a continuous decline in birth rates of babies with ß-thal major (Huang L. F. et al., 2021). Genotyping is strongly recommended for inclusion in the prenatal diagnosis of ß-thal, since precise prenatal diagnosis is crucial to prevent the development of thalassemia and for genetic counseling. Apart from prenatal diagnosis, pre-implantation genetic testing (PGT) for thalassemia has emerged as a new choice for some families, which, although expensive, causes less harm to the pregnant females.
Accurate genetic testing is the basis of effective birth defect prevention of thalassemia in pre-pregnancy or prenatal genetic counseling. Although traditional genetic testing is a low-cost and time-saving strategy, it only focuses on the most common regional mutations, which may lead to missed diagnosis of rare or novel variants. Therefore, further supplementary molecular testing is required for individuals whose genotypes cannot explain the hematological indices in clinical practice. In our study, 30 participants with increased HbA2 values and microcytic hypochromic hematological profiles were identified with rare ß-thal mutations. Overall, 10 types of rare ß-thal mutations that had not been previously reported in Jiangxi Province were identified, representing 1.18% (30/2,548) of all the mutations, with IVS-I-6 (T>G) identified for the first time worldwide. Among the 30 cases with rare ß-thal mutations, the most common genotype was CD54-58 (-TATGGGCAACCCT), followed by -28 (A>C), suggesting that these two mutations show a more frequent carrier rate than some of the 17 point mutations tested by traditional PCR-RDB method. In addition, we also identified four novel variants, including HBA2:c.301-41_301-39delGGC, HBB:IVS-II-47 (G>C), IVS-II-300–308 (-A), and IVS-II-806 (G>C), even though their pathogenesis still requires further study. Notably, we found that participants with the variant β+ Cap+40–43 (-AAAC) identified by traditional thalassemia genetic testing, did not demonstrate the hematological characteristics of increased HbA2 values or microcytic hypochromic when iron-deficiency is excluded. As reported by Li, the pathogenicity of this variant remains uncertain and may be regarded as a silent ß-thal allele (Zhao et al., 2020). Moreover, based on our findings, we propose that the two frequent mutations CD54-58 (-TATGGGCAACCCT) and -28 (A>C) should be included into traditional genetic testing based on specific geographic background in Jiangxi Province. Secondly, hematological indices of CBC test and hemoglobin electrophoresis are both indispensable for the correct recognition of ß-hemoglobinopathies. Finally, more comprehensive and efficient molecular testing approaches may be considered for identification of both common and rare variants of thalassemia simultaneously.
Despite these strengths, certain limitations of our study should also be acknowledged. As mutations in HBD can cover the typical increased HbA2 value for individuals with ß-thal (Chen et al., 2020), missed diagnosis may occur based on the current hematological screening strategy as the hematological parameters might be normal or borderline indices for some rare thalassemia mutation types or Hb variants. Thus, it is possible that silent mutations in α-globin genes, α triplications, or Hb variants without abnormal Hb composition were missed in this study. In recent years, the combined Gap-PCR and next-generation sequencing (NGS) has emerged and used as an alternative more complete and versatile molecular approach for thalassemia genetic testing (Shang et al., 2017), even though it is considered scalable, cost-effective, and comprehensive. Besides, another alternative efficient, accurate, and cost-effective single platform technology generating longer PCR fragments including intergenic and intragenic regions to identify other rare variants undetectable by the typical panel test have been studied and validated (Xu L. et al., 2020). Studies revealed that this new emerging methods with universal approach to comprehensive analysis of variants in homologous genes HBA1/HBA2, cis or trans configuration, and complex structural rearrangements are hardly associated with false-negative and false-positive results (Liang et al., 2021).
Conclusion
In summary, we have provided the most comprehensive mutation spectrum of thalassemia and abnormal Hb variants mutation spectrum in the HBB gene among adults of reproductive-age in Jiangxi Province to date. We also provided intensive genetic counseling and prenatal diagnosis for couples with indications to avoid the birth of fetuses with ß-thal intermediate or major. Our work may provide as a baseline for genetic counseling, public education, and as a reference to improve the prevention of ß-thal.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by The studies involving human participants were reviewed and approved by Ethics Committee of Jiangxi Maternal and Child Health Hospital (No. EC-KT-202216). The patients/participants provided their written informed consent to participate in this study.
Author contributions
HL and TH conceived and designed the study. QL and YX performed the hematological screening tests, LZ, YY, and YS conducted some of the genetic detection and ZG conducted the B-mode ultrasound-guidance during the fetal sampling. HY performed the CVS, amniocentesis or umbilical cord blood sampling. SH and BY contributed to track the objects of research. YZ and YL provided writing guidance and correction. HL and TH wrote the manuscript together. All the authors have read and approved the manuscript.
Funding
This study was supported by Jiangxi Provincial Key Laboratory of Birth Defect for Prevention and Control (No. 20202BCD42017 to YL). National Natural Science Foundation of China (Grant No. 82160318 to YZ), Key Research and Development Program of Jiangxi Province (Grant No. 20192BBGL70006 to ZG), Provincial Health Commission Program of Jiangxi (Grant No.SKJP220211066 to TH, No. SKJP220211247 to HL and 202130792 to YZ).
Acknowledgments
We are grateful to all subjects participating in this study and thanks are also expressed to Dr. Chen for his kind assistance with the manuscript preparation.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.992073/full#supplementary-material
References
Ahmed, M. H., Ghatge, M. S., and Safo, M. K. (2020). Hemoglobin: Structure, function and Allostery. Subcell. Biochem. 94, 94345–94382. doi:10.1007/978-3-030-41769-7_14
Ali, S., Mumtaz, S., Shakir, H. A., Khan, M., Tahir, H. M., Mumtaz, S., et al. (2021). Current status of beta-thalassemia and its treatment strategies. Mol. Genet. Genomic Med. 9 (12), e1788. doi:10.1002/mgg3.1788
Altinier, S., Varagnolo, M., Zaninotto, M., and Plebani, M. (2013). Identification and quantification of hemoglobins in whole blood: The analytical and organizational aspects of capillarys 2 Flex Piercing compared with agarose electrophoresis and HPLC methods. Clin. Chem. Lab. Med. 51 (4), 791–797. doi:10.1515/cclm-2012-0061
Chen, M., Huang, H., Chen, L., Lin, N., Zhang, M., Lin, Y., et al. (2020). First report of the spectrum of δ-globin gene mutations among women of reproductive age in Fujian area-Discrimination of δ-thalassemia, α-thalassemia, and Iron Deficiency Anemia. J. Clin. Lab. Anal. 34 (11), e23479. doi:10.1002/jcla.23479
Cheng, Y., Chen, M., Ye, J., Yang, Q., Wang, R., Liu, S., et al. (2022). The prevalence and outcomes of α- and β-thalassemia among pregnant women in Hubei Province, Central China: An observational study. Med. (Abingdon) 101 (9), e28790. doi:10.1097/MD.0000000000028790
Clarke, G. M., and Higgins, T. N. (2000). Laboratory investigation of hemoglobinopathies and thalassemias: Review and update. Clin. Chem. 46 (8), 1284–1290. doi:10.1093/clinchem/46.8.1284
Ghosh, K., Ghosh, K., Agrawal, R., and Nadkarni, A. H. (2020). Recent advances in screening and diagnosis of hemoglobinopathy. Expert Rev. Hematol. 13 (1), 13–21. doi:10.1080/17474086.2019.1656525
He, J., Zeng, H., Zhu, L., Li, H., Shi, L., and Hu, L. (2017). Prevalence and spectrum of thalassaemia in Changsha, Hunan province, China: Discussion of an innovative screening strategy. J. Genet. 96 (2), 327–332. doi:10.1007/s12041-017-0779-6
Huang, H., Chen, M., Chen, L., Zhang, M., Wang, Y., Lin, N., et al. (2021). Prenatal diagnosis of thalassemia in 695 pedigrees from southeastern China: A 10-year follow-up study. J. Clin. Lab. Anal. 35 (10), e23982. doi:10.1002/jcla.23982
Huang, H., Xu, L., Chen, M., Lin, N., Xue, H., Chen, L., et al. (2019). Molecular characterization of thalassemia and hemoglobinopathy in Southeastern China. Sci. Rep. 9 (1), 3493. doi:10.1038/s41598-019-40089-5
Huang, L. F., Yu, L. L., Nikuze, L., Singh, S., Jiang, J. X., Jiang, J. L., et al. (2021). Spectrum of β-thalassemia mutations in some areas of Guangxi Zhuang Autonomous region of southern China: A study on a Pediatric population aged 0-15 Years. Hemoglobin 45 (5), 318–321. doi:10.1080/03630269.2022.2041435
Keren, D. F., Hedstrom, D., Gulbranson, R., Ou, C. N., and Bak, R. (2008). Comparison of Sebia Capillarys capillary electrophoresis with the Primus high-pressure liquid chromatography in the evaluation of hemoglobinopathies. Am. J. Clin. Pathol. 130 (5), 824–831. doi:10.1309/AJCPQY80HZWHHGZF
Kohne, E. (2011). Hemoglobinopathies: Clinical manifestations, diagnosis, and treatment. Dtsch. Arztebl. Int. 108 (31-32), 532–540. doi:10.3238/arztebl.2011.0532
Liang, Q., Gu, W., Chen, P., Li, Y., Liu, Y., Tian, M., et al. (2021). A more universal approach to comprehensive analysis of thalassemia alleles (CATSA). J. Mol. Diagn. 23 (9), 1195–1204. doi:10.1016/j.jmoldx.2021.06.008
Lin, M., Zhong, T. Y., Chen, Y. G., Wang, J. Z., Wu, J. R., Lin, F., et al. (2014). Molecular epidemiological characterization and health burden of thalassemia in Jiangxi Province, P. R. China. PLoS ONE 9 (7), e101505. doi:10.1371/journal.pone.0101505
Liu, Q., Jia, Z. J., Xi, H., Liu, J., Peng, Y., and Wang, H. (2019). Analysis on the genotype of 5018 cases of thalassemia in Hunan area. Zhongguo Shi Yan Xue Ye Xue Za Zhi 27 (6), 1938–1942. doi:10.19746/j.cnki.issn.1009-2137.2019.06.037
Long, J., Huang, G., Liang, W., Liang, B., Chen, Q., Xie, J., et al. (2014). The prevalence of schizophrenia in mainland China: Evidence from epidemiological surveys. Acta Psychiatr. Scand. 130 (4), 244–256. doi:10.1111/acps.12296
Lou, J. W., Wang, T., Liu, Y. H., He, Y., Zhong, B. M., Liu, J. X., et al. (2014). Prevalence and molecular characterization of structural hemoglobin variants in the Dongguan region of Guangdong province, southern China. Hemoglobin 38 (4), 282–286. doi:10.3109/03630269.2014.928779
Muncie, H. L., and Campbell, J. (2009). Alpha and beta thalassemia. Am. Fam. Physician 80 (4), 339–344.
Schwartz, E., Cohen, A., and Surrey, S. (1988). Overview of the beta thalassemias: Genetic and clinical aspects. Hemoglobin 12 (5-6), 551–564. doi:10.3109/03630268808991644
Shang, X., Peng, Z., Ye, Y., AsanZhang, X., Chen, Y., et al. (2017). Rapid Targeted next-generation sequencing platform for molecular screening and clinical genotyping in subjects with hemoglobinopathies. EBioMedicine 23, 23150–23159. doi:10.1016/j.ebiom.2017.08.015
Taher, A. T., Weatherall, D. J., and Cappellini, M. D. (2018). Thalassaemia. Lancet 391 (10116), 155–167. doi:10.1016/S0140-6736(17)31822-6
Thein, S. L. (2013). The molecular basis of β-thalassemia. Cold Spring Harb. Perspect. Med. 3 (5), a011700. doi:10.1101/cshperspect.a011700
Wang, M., Zhang, X., Zhao, Y., Lu, Z., and Xiao, M. (2022). Prevalence and genetic analysis of thalassemia in childbearing age population of Hainan, the Free Trade Island in Southern China. J. Clin. Lab. Anal. 36 (3), e24260. doi:10.1002/jcla.24260
Weatherall, D. J., and Clegg, J. B. (2001). Inherited haemoglobin disorders: An increasing global health problem. Bull. World Health Organ. 79 (8), 704–712.
Writing Group For Practice Guidelines For Diagnosis And Treatment Of Genetic Diseases Medical Genetics Branch Of Chinese Medical Association Shang, X., Wu, X., Zhang, X., Feng, X., and Xu, X. (2020). Clinical practice guidelines for beta-thalassemia. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 37 (3), 243–251. doi:10.3760/cma.j.issn.1003-9406.2020.03.004
Xiong, F., Sun, M., Zhang, X., Cai, R., Zhou, Y., Lou, J., et al. (2010). Molecular epidemiological survey of haemoglobinopathies in the Guangxi Zhuang Autonomous Region of southern China. Clin. Genet. 78 (2), 139–148. doi:10.1111/j.1399-0004.2010.01430.x
Xu, A., Chen, W., Xie, W., Wang, Y., and Ji, L. (2020). Hemoglobin variants in southern China: Results obtained during the measurement of glycated hemoglobin in a large population. Clin. Chem. Lab. Med. 59 (1), 227–232. doi:10.1515/cclm-2020-0767
Xu, L., Mao, A., Liu, H., Gui, B., Choy, K. W., Huang, H., et al. (2020). Long-molecule sequencing: A new approach for identification of clinically significant DNA variants in α-thalassemia and β-thalassemia carriers. J. Mol. Diagn. 22 (8), 1087–1095. doi:10.1016/j.jmoldx.2020.05.004
Xu, X. M., Zhou, Y. Q., Luo, G. X., Liao, C., Zhou, M., Chen, P. Y., et al. (2004). The prevalence and spectrum of alpha and beta thalassaemia in Guangdong province: Implications for the future health burden and population screening. J. Clin. Pathol. 57 (5), 517–522. doi:10.1136/jcp.2003.014456
Zhao, Y., Jiang, F., and Li, D. Z. (2020). Hematological characteristics of β-globin gene mutation -50 (G>A) (HBB: c.-100G>A) carriers in mainland China. Hemoglobin 44 (4), 240–243. doi:10.1080/03630269.2020.1793774
Zhou, B. Y., Wang, Y. X., Xu, S. S., Gu, H., and Li, M. Z. (2021). Molecular spectrum of α- and β-thalassemia among Young individuals of Marriageable age in Guangdong province, China. Biomed. Environ. Sci. 34 (10), 824–829. doi:10.3967/bes2021.112
Keywords: HBB, hemoglobinopathy, mutation spectrum, prenatal diagnosis, central China-southern central China
Citation: Luo H, Huang T, Lu Q, Zhang L, Xu Y, Yang Y, Guo Z, Yuan H, Shen Y, Huang S, Yang B, Zou Y and Liu Y (2022) Molecular prevalence of HBB-associated hemoglobinopathy among reproductive-age adults and the prenatal diagnosis in Jiangxi Province, southern central China. Front. Genet. 13:992073. doi: 10.3389/fgene.2022.992073
Received: 12 July 2022; Accepted: 08 September 2022;
Published: 28 September 2022.
Edited by:
Aleksandra Jezela-Stanek, National Institute of Tuberculosis and Lung Diseases, PolandReviewed by:
Efthymia Vlachaki, Aristotle University of Thessaloniki, GreeceLing Qiu, Peking Union Medical College Hospital (CAMS), China
Copyright © 2022 Luo, Huang, Lu, Zhang, Xu, Yang, Guo, Yuan, Shen, Huang, Yang, Zou and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongyi Zou, em91eW9uZ3lpQGdtYWlsLmNvbQ==; Yanqiu Liu, bHlxMDkxNEAxMjYuY29t
†These authors have contributed equally to this work