
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Genet., 16 August 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.983283
This article is part of the Research TopicRare DyslipidemiasView all 17 articles
 Ursa Sustar1,2
Ursa Sustar1,2 Urh Groselj1,2,3*
Urh Groselj1,2,3* Sabeen Abid Khan4
Sabeen Abid Khan4 Saeed Shafi5
Saeed Shafi5 Iqbal Khan6,7
Iqbal Khan6,7 Jernej Kovac2,8
Jernej Kovac2,8 Barbara Jenko Bizjan8
Barbara Jenko Bizjan8 Tadej Battelino1,2
Tadej Battelino1,2 Fouzia Sadiq9*
Fouzia Sadiq9*Background: Due to nonspecific symptoms, rare dyslipidaemias are frequently misdiagnosed, overlooked, and undertreated, leading to increased risk for severe cardiovascular disease, pancreatitis and/or multiple organ failures before diagnosis. Better guidelines for the recognition and early diagnosis of rare dyslipidaemias are urgently required.
Methods: Genomic DNA was isolated from blood samples of a Pakistani paediatric patient with hypertriglyceridemia, and from his parents and siblings. Next-generation sequencing (NGS) was performed, and an expanded dyslipidaemia panel was employed for genetic analysis.
Results: The NGS revealed the presence of a homozygous missense pathogenic variant c.230G>A (NM_178172.6) in exon 3 of the GPIHBP1 (glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1) gene resulting in amino acid change p.Cys77Tyr (NP_835466.2). The patient was 5.5 years old at the time of genetic diagnosis. The maximal total cholesterol and triglyceride levels were measured at the age of 10 months (850.7 mg/dl, 22.0 mmol/L and 5,137 mg/dl, 58.0 mmol/L, respectively). The patient had cholesterol deposits at the hard palate, eruptive xanthomas, lethargy, poor appetite, and mild splenomegaly. Both parents and sister were heterozygous for the familial variant in the GPIHBP1 gene. Moreover, in the systematic review, we present 62 patients with pathogenic variants in the GPIHBP1 gene and clinical findings, associated with hyperlipoproteinemia.
Conclusion: In a child with severe hypertriglyceridemia, we identified a pathogenic variant in the GPIHBP1 gene causing hyperlipoproteinemia (type 1D). In cases of severe elevations of plasma cholesterol and/or triglycerides genetic testing for rare dyslipidaemias should be performed as soon as possible for optimal therapy and patient management.
Affecting 15–20% of the population (Parhofer and Laufs, 2019; Basit et al., 2020), hypertriglyceridemia has been associated with an increased risk for pancreatitis (Carrasquilla et al.; Simha, 2020; Álvarez-López et al., 2021). In adults with severe hypertriglyceridemia only up to 2% of the cases could be explained by a monogenic variant in genes involved in triglyceride (TG) metabolism (Hegele et al., 2020). Polygenic variants of smaller effects combined with environmental factors are considered a primary cause of hypertriglyceridemia. Secondary causes include diabetes mellitus, metabolic syndrome, alcohol and commonly used drugs (Simha, 2020).
Hypertriglyceridemia is defined as fasting triglyceride (TG) levels over 2 mmol/L (180 mg/dl), whereas in severe hypertriglyceridemia fasting TG levels exceed 10 mmol/L (885 mg/dl) (Hegele et al., 2020). Patients with the most severe phenotypes start expressing clinical symptoms at a younger age, usually have serum TG levels above 11.3 mmol/L (1000 mg/dl), and in some cases also have abdominal pain related to acute pancreatitis, hepatosplenomegaly, lipemia retinalis, and eruptive xanthomata already in childhood (Gonzaga-Jauregui et al., 2014; Brown et al., 2020; Hegele et al., 2020).
Lipoprotein lipase (LPL) mediates the hydrolysis of triglycerides packed in lipoproteins such as chylomicrons and very-low-density lipoprotein (VLDL) (Wu et al., 2021). Many factors interact with LPL affecting TG metabolism. Dysfunction of LPL and other factors interacting with LPL may lead to hypertriglyceridemia (Liu et al., 2018). Besides LPL there are other genes involved in the LPL-mediated lipolysis of chylomicrons and VLDL: ANGPTL4 (angiopoietin-like 4), APOC2 (apoprotein C-II), APOA5 (apolipoprotein A-V), LMF1 (lipase maturation factor 1), and GPIHBP1 (glycosylphosphatidylinositol-anchored high-density lipoprotein binding protein 1) (Hegele et al., 2020; Kersten, 2021).
GPIHBP1 binds and transports LPL to the capillary lumen from interstitial space, where it hydrolyses TG and triglyceride-rich lipoproteins (TRLs) (Supplementary Figure S1). In patients with GPIHBP1 deficiency, LPL is mislocalized and intravascular hydrolysis of triglycerides is impaired (Beigneux et al., 2017; Hegele et al., 2020; Wu et al., 2021). The consequence is low plasma levels of LPL, and severe hypertriglyceridemia (Beigneux et al., 2017). The prevalence of type I hyperlipoproteinemia because of a pathogenic variant in the GPIHBP1 is estimated between 1:500,000 to 1:1,000,000 (Gonzaga-Jauregui et al., 2014).
The National Medical Ethics Committee approved the study in Slovenia (0120-14/2017/5, and 0120-273/2019/9), and the Ethics Committee approved the study in Pakistan (033-523-2019). The principles of the Declaration of Helsinki were followed. Written consent of the patient’s parents was obtained before inclusion.
Serum samples were analyzed for lipids including TC, low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C) and TG. TC, TG and LDL-C were measured by a homogenous enzymatic method (Cobas 8000 c502 module, Roche, United States).
Lipid levels were considered as normal if: TC < 4.4 mmoL/L (170 mg/dl), LDL-C <2.8 mmoL/L, (110 mg/dl) HDL-C >1.2 mmoL/L (45 mg/dl), TG < 0.8 mmoL/L (75 mg/dl) for children <9 years of age and <1 mmoL/L (90 mg/dl) if > 9 years of age. Lipid levels were considered as elevated/lowered if: TC > 5.2 mmoL/L (200 mg/dl), LDL-C >3.4 mmoL/L (130 mg/dl), HDL-C <1 mmoL/L (40 mg/dl), and TG > 1.1 mmol/L (100 mg/dl) for children <9 years of age and >1.5 mmoL/L (130 mg/dl) if > 9 years of age. The lipid levels in-between the cut-offs were considered borderline (De Jesus, 2011).
All genetic analyses were performed at the University Children’s Hospital Ljubljana in Slovenia in the same way as Slovenian national genetic testing for the universal familial hypercholesterolemia (FH) screening program in preschool children (Groselj et al., 2018, 2022; Sustar et al., 2022). Genomic DNA was isolated from the patient’s and his family members (mother, father and sister) peripheral blood using a Flexigene kit (Qiagen). xGen® Lockdown® NGS Probes (IDT, United States) for detection of coding and promoter regions for the genes associated with dyslipidemia were used (ABCA1, ABCG5, ABCG8, ALMS1, APOA1, APOA5, APOB, APOC2, APOC3, APOE, CREB3L3, GPIHBP1, LDLR, LDLRAP1, LIPA, LMF1, LPL, and PCSK9). Samples were sequenced on MiSeq sequencer with MiSeq Reagent Kit (Illumina, United States) following the manufacturer’s protocol including recommendations for quality control parameters. For variant annotation and filtration, the VafAFT tool was applied (Desvignes et al., 2018). The detected variants were classified by the American College of Medical Genetics and Genetics and the Association for Molecular Pathology (ACMG-AMP) (Richards et al., 2015) classification criteria as (likely) benign, variants of uncertain significance (VUS) and (likely) pathogenic. The pathogenic variant in the GPIHBP1 gene was reconfirmed by targeted Sanger DNA sequencing.
We gathered all accessible scientific case report publications for the systematic review of pathogenic variants in the GPIHBP1 gene. The systematic review was registered at PROSPERO (CRD42022336232). An electronic search was performed using the keyword “GPIHBP1” in the PubMed database on 11 June 2022. Moreover, we searched for the articles related to pathogenic variants also through the search in the Human Gene Mutation Database (Stenson et al., 2003) and the Franklin by Genoox tool based on the pathogenic variant, confirmed in our patient “NM 178172.6:c.230G>A” (scope: “Gene”). By going through all of the abstracts and titles found, we included all articles meeting the following requirements: 1) articles in English published after 2002, 2) articles containing human data, 3) only articles or data from articles describing pathogenic variants in the GPIHBP1 gene, 4) reported patients were homozygous or compound heterozygous for pathogenic variants, and 5) clinical data on patients was provided.
The proband was a 5.5-year-old boy. He was born at a full-term birth weight of 3.7 kg with no known antenatal issues. The timeline of the diagnosing, treatment and management of the patient is represented in Figure 1. The parents of the patient are in a consanguineous marriage and are first-degree relatives. The patient’s mother has had an early abortion and has a 2-year-old healthy daughter. The maternal grandfather of the patient had a heart attack at 28 years of age. No family history of pancreatitis is known. The lipid profile of the proband and his family members is represented in Supplementary Figure S2.
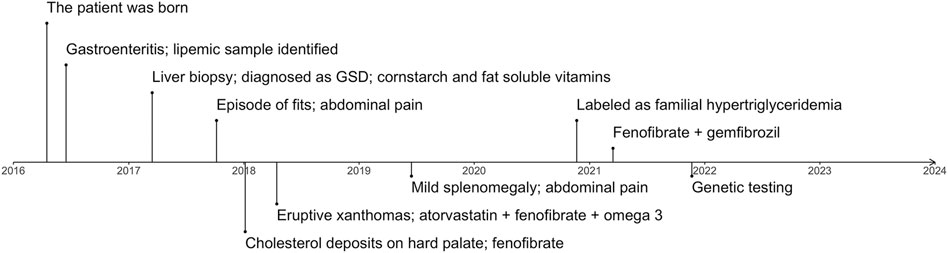
FIGURE 1. The timeline represents important complications and milestones in diagnosing and managing the patient. GSD, glycogen storage disease.
At 2 months of age, the patient had an episode of gastroenteritis needing intravenous hydration. At that time mother was first notified of “pink blood” (lipemic sample). Repeated sampling confirmed the same finding. The child was later taken to a local hospital where he was seen by a paediatric gastroenterologist who advised liver biopsy. The child had an initial liver biopsy done at 10 months of age, which showed a severely autolytic sample with Periodic acid–Schiff (PAS) stain positive in preserved areas suggestive of glycogen storage disease (GSD). At the same time, other laboratory measurements were elevated: total serum cholesterol of 850.7 mg/dl (22.0 mmol/L) and serum triglycerides of 5,137 mg/dl (58.0 mmol/L). However, fasting glucose was raised significantly (29.5 mmol/L) along with total bilirubin levels (110.0 umol/L) and ALT 550 U/L. The TC and TG levels of the patient over time are represented in Figure 2. On examination, he had pallor and a soft abdomen with no hepatomegaly. The patient was labelled as a case of GSD. Based on that, the patient was advised to start taking cornstarch, fenofibrate, allopurinol, and vitamins A, D and E.
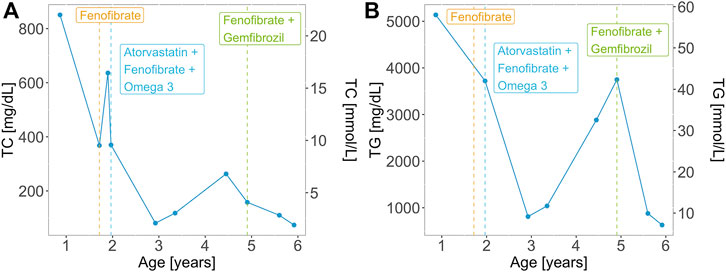
FIGURE 2. (A) Total cholesterol (TC) and (B) triglyceride (TG) levels over time for the patient with the homozygous GPIHBP1 pathogenic variant. Vertical lines represent the initiation and/or modification of the treatment.
Aged 1 year and 5 months the patient presented to the Paediatric outpatient department in Shifa Hospital for the first time with complaints of one episode of vague seizure-like activity and abdominal pain for 3 months. At age 1 year and 8 months, the patient developed hoarseness of voice. He was noted to have 2–3 yellow plaques on the hard palate. At this visit, his TC level was 368.0 mg/dl (9.5 mmol/L). Two months later he had eruptive xanthomas, a TC levels of 636.0 mg/dl (16.4 mmol/L). Aged 2 his TC levels were 370 mg/dl (9.6 mmol/L) and his TG levels were 3,720 mg/dl (42.0 mmol/L). Along with fenofibrate and omega 3 the patient was prescribed atorvastatin (5 mg) with increasing dosage (10 mg) upon a follow-up visit. Treatment of the patient over time is represented in Figure 2. The patient presented for follow-ups with lab results that repeatedly showed TG and total serum cholesterol above normal limits. He never had any documented hypoglycemia, drowsiness or seizure activity. He didn’t have hepatomegaly.
Two years following the initial presentation, with regular follow-ups in between, the boy presented to the outpatient department with pain in the abdomen, lethargy and poor appetite. There was mild splenomegaly and upon repetition of laboratory tests, ALT (29 U/L) and serum lipase levels were normal, TC level was 81.0 mg/dl (2.1 mmol/L), LDL-C 18.0 mg/dl (0.5 mmol/L), HDL 8 mg/dl (0.2 mmol/L), triglycerides were 1,068 mg/dl (12.1 mmol/L) and 2,883 mg/dl (32.6 mmol/L) subsequently, despite receiving the treatment with atorvastatin, fenofibrate 50 mg once daily and omega 3 capsules twice a day.
The diagnosis was reviewed at age 4.5 years due to persistently high serum TG and relabeled as a case of primary/familial hypertriglyceridemia based on the lipid profile. The patient was advised to stop using cornstarch, allopurinol and atorvastatin and continue fenofibrate. On follow-up, the patient, weighing 14 kg at 4 years and 11 months of age, had his TC within normal range, however, TGs were still deranged (3749 mg/dl, 42.3 mmol/L). Response to fenofibrate (67 mg twice a day) was inadequate with persistent high TGs (>1000 mg/dl; 11.3 mmol/L), therefore gemfibrozil (300 mg twice a day) was added to the treatment regime. Aged 5 years and 7 months the triglycerides were 878.0 mg/dl (9.9 mmol/L), TC 110.0 mg/dl (2.8 mmol/L), LDL-C 10 mg/dl (0.3 mmol/L), HDL-C 0.2 mmol/L (8.0 mg/dl). His weight and height were at the 5th centile for his age and gender. At age 5 years and 11 months his lipid profile levels were: TC: 74 mg/dl (1.9 mmol/L), LDL-C: 10 mg/dl (0.3 mmol/L), HDL-C: 8 mg/dl (0.2 mmol/L) and TG: 630 mg/dl (7.1 mmol/L). At that point, he had no xanthomas.
At age 5.5 genetic testing was performed and we identified a homozygous variant c.230G>A (NM_178172.6) in exon 3 of the GPIHBP1 gene (NG_034256.1) leading to a protein change p.Cys77Tyr (NP_835466.2). The patient’s family members (father, mother and sister) were heterozygous for the c.230G>A variant (Supplementary Figures S2, S3). The variant has already been reported in the ClinVar (VCV000917845.1) (Landrum et al., 2018) as likely pathogenic in association with hyperlipoproteinemia (type ID) phenotype. The variant was classified as pathogenic by the in silico prediction tools (Revel, MetaLR, MetaSVM) (Ioannidis et al., 2016). The frequency of the variant in the gnomAD population databases (Karczewski et al., 2020) is extremely low. Following the American College of Medical Genetics and Genetics and the Association for Molecular Pathology (ACMG-AMP) criteria (Richards et al., 2015) the variant was classified as likely pathogenic.
In Table 1 we reviewed the literature on the pathogenic variants in the GPIHBP1 gene containing additional clinical information about the patients. Fifty-four patients were homozygous while seven were compound heterozygous for a pathogenic variant in the GPIHBP1 gene. One patient was heterozygous for variants in GPIHBP1 and APOC2 genes. We presented 32 unique variants in the GPIHBP1 gene. 5 pathogenic variants are located in exon 1, 3 in exon 2, 10 in exon 3 and 10 in exon 4. 4 variants represent major deletion of a whole exon/multiple exons/whole GPIHBP1 gene.

TABLE 1. Review of pathogenic variants in GPIHBP1 gene from the literature.
We presented a patient with severe hypertriglyceridemia as a consequence of a homozygous pathogenic variant in the GPIHPB1 gene. Additionally, we reviewed the literature on described cases with pathogenic variants in the GPIHBP1 gene.
Elevated TG levels have been linked to cardiovascular disease (CVD) and pancreatitis (Carrasquilla et al., 2021). For severe hypertriglyceridemia, pathogenic variants in genes LPL, APOA5, APOC2, GPIHBP1, and LMF1, associated with hyperlipidemia should be considered (Goldberg and Chait, 2020). GPIHBP1 is located on chromosome 8q24.3 and is composed of 4 exons (Liu et al., 2018). GPIHBP1 is expressed mostly in the capillary endothelial cells of the heart, brown adipose tissue and skeletal muscle, involved in energy and lipid metabolism (Cruz-Bautista et al., 2021). GPIHBP1 acts as an important partner of the LPL in plasma triglyceride metabolism (Supplementary Figure S1). The loss-of-function pathogenic variant in GPIHBP1 impairs LPL activity and its lipolytic processing of chylomicrons and very-low-density lipoproteins (Voss et al., 2011; Gin et al., 2012). LPL unfolds spontaneously and remains within the interstitial spaces. Due to the loss of its catalytic activity, triglycerides are not hydrolysed resulting into severe hypertriglyceridemia (Arora et al., 2019; Miyashita et al., 2020) (Supplementary Figure S1).
GPIHBP1 is a protein composed of 184 amino acids from the lymphocyte antigen (Ly6) family and consists of a N-terminal signal peptide region, an amino-terminal acidic domain, a Ly6 domain of ten disulfide-bonded cysteines and a highly hydrophobic carboxyl-terminal motif that is replaced within the endoplasmic reticulum by a glycosylphospatidylinostiol (GPI) anchor for tethering to the cell membrane. GPIHBP1 is distinguishable from the other Ly6 family members by the presence of a high acidic domain composed of 21 aspartates or glutamates located 12 amino acids prior to the Ly6 motif (Beigneux A. R. et al., 2009; Liu et al., 2018). Both, a high acidic and Ly6 domain are important for the GPIHBP1 and LPL interaction. Genetic modifications encoding for one of these two domains result in chylomicronemia. Most missense variants break down the disulfide bond of the Ly6 domain, resulting in the production of GPIHBP1 dimers and multimers, which are incapable of binding LPL (Beigneux et al., 2015; Fong et al., 2016).
Amino acids at positions 24-50 (exon 2) compose the acidic domain and amino acids at positions 63-148 (exons 3 and 4) compose the Ly6 domain of the GPIHBP1 protein (Figure 3). Amino acids at positions 27-50 (exon 2) are important for LPL transport to the lumenal surface of endothelial cells. Amino acids at positions 103-109 (exon 4) are important for interaction with LPL (UniProt, Q8IV16). The majority of pathogenic variants disrupt the Ly6 domain’s folding, resulting in multimerized and defective GPIHBP1 molecules on the cell surface (Holmes and Cox, 2012; Mysling et al., 2016).
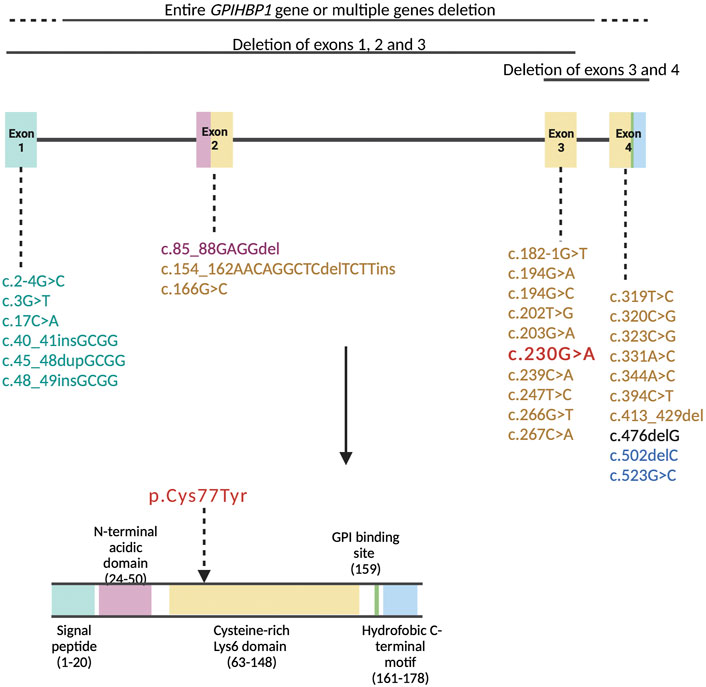
FIGURE 3. Pathogenic variants in the GPIHBP1 gene based on the literature review represented in Table 1 are organized by exon numbers. The colour of the exon denotes the protein domain affected by the pathogenic variation. The variant of our patient (c.230G > A) is in bold red colour.
Patients with GPIHBP1 deficiency express a similar phenotype as patients with LPL deficiency, such as severe chylomicronemia (plasma TG levels above 1,500 mg/dl (16.9 mmol/L) presented already in childhood and high risk of pancreatitis (Miyashita et al., 2020). Highest reported TG and TC levels for the patient at age 10 months were 5,137 mg/dl (58.0 mmol/L) and 850.7 mg/dl (22.0 mmol/L) respectively. Because hyperbilirubinemia was detected during this examination, extreme TC levels could be the consequence of biliary congestion. Hyperbilirubinemia was not detected on further examinations, yet increased TC and TG levels remained.
Moreover, as the recessive manner of inheritance of the disorder, the parents and the younger sister of the patient did not express the extreme phenotype associated with the disorder, although the father has elevated TC and TG levels (Supplementary Figure S1), similarly, as reported by Wang and Hegele, 2007 for heterozygous family members of a proband with a homozygous variant in the GPIHBP1 gene. Likewise, our patient’s father may have an extra genetic variant that contributes to his phenotype but is not present in the patient’s mother or sister.
The variant p.Cys77Tyr of our patient is located in exon 3, encoding for the Ly6 domain of the GPIHBP1 protein (Figure 3). Cysteine residues of Ly6 domain are essential for the 3-fingered structural motif formation. Interfering with any of the disulfide links is expected to cause significant structural changes in the protein (Rabacchi et al., 2016). Beigneux et al. (2009b) report that cells expressing the cysteine mutants in GPIHBP1 are unable to bind and transport LPL from the subendothelial space to the endoluminal surface of the endothelial cells. Furthermore, several genetic variants involving cysteine alterations to another amino acid have previously been documented. Eight patients with homozygous and four with compound heterozygous variants p.Cys65Tyr, p.Cys68Gly, p.Cys83Arg, p.Cys83Arg, p.Cys89Phe and p.Cys89Ter (Olivecrona et al., 2010; Charrière et al., 2011; Iacocca et al., 2019) with severe elevation in TG (1,000–6,480 mg/dl, 11.3–73.2 mmol/L) and episodes of acute pancreatitis have previously been described. Moreover, Lima et al. (2021) present a series of twelve cases with an intronic variant c.182-1G>C, resulting in the skipping of cysteine-rich exon 3. These patients likewise had extremely elevated TG levels (885–40,141 mg/dl, 10–453.2 mmol/L), but interestingly only four suffered from acute pancreatitis, none of them had hepatosplenomegaly and four of them had eruptive xanthomas. Additionally, twelve patients with deletion of exon 3 and 4 or entire gene deletion had TG values between 957 and 37,284 mg/dl (10.8 and 421.0 mmol/L) and seven of them were affected by acute pancreatitis (Rios et al., 2012; Berge et al., 2014; Chokshi et al., 2014; Patni et al., 2016; Iacocca et al., 2019). Table 1 contains additional information about these patients.
European Atherosclerosis Society provided practical clinical guidelines for rare dyslipidaemia management for patients with extreme LDL-C, TG and HDL-C levels (Hegele et al., 2020). Early detection of rare dyslipidemias in a pre-clinical stage is possible with an effective FH screening program capable of detecting other dyslipidemia than FH (Groselj et al., 2018, 2022; Marusic et al., 2020; Sustar et al., 2022) or with a gene panel applied as a part of a newborn screening program (Remec et al., 2021). It is important to implement a worldwide registry for rare dyslipidemias, comparable to what already exists for FH/homozygous FH (Tromp et al., 2022; Vallejo-Vaz et al., 2018, 2021).
Statin therapy substantially reduced the CVD risk in patients with high LDL-C levels (Silverman et al., 2016). Nevertheless, other factors, such as triglycerides or triglyceride-rich lipoproteins (TRLs), contribute to the CVD risk with low-grade inflammation, as a part of atherosclerosis (Nordestgaard, 2016). The goal of the treatment is to reduce plasma TG levels to less than 500–1,000 mg/dl to prevent acute pancreatitis (Okazaki et al., 2021). To prevent abdominal pain and acute pancreatitis, patients should be on a low-fat diet of total dietary fat intake of <10–15% of daily calories (<15–20 g per day) and treated with common lipid-lowering drugs (fibrates, omega-3 fatty acids, statins) (Esan and Wierzbicki, 2020; Navarro Hermoso and Valdivielso, 2021). Volanesorsen, an antisense oligonucleotide inhibitor of apoC3 is a promising medicine for the reduction of TG levels by 70–80% (Esan and Wierzbicki, 2020). Furthermore, gemfibrozil is a useful medication for the reduction of TGs in patients with very high TG serum levels. The mechanism of action of gemfibrozil is based on the activation of nuclear transcription factors for up-regulation of LPL transcription and down-regulation of the LPL inhibitor apo C-III, resulting in a decrease in triglyceride levels and an increase in HDL. Moreover, gemfibrozil reduces hepatic triglyceride synthesis by inhibiting peripheral lipolysis and decreasing hepatic removal of free fatty acids. It inhibits the synthesis and increases the clearance of very low-density lipoprotein (VLDL) (Goldberg and Hegele, 2012). Our patient was managed with statins (atorvastatin) and later with a combination of fibrates (fenofibrate and gemfibrozil). Combination therapy with fenofibrate and gemfibrozil has helped in lowering TG levels (<1000 mg/dl) which was not achieved by monotherapy with fenofibrate. Statins were stopped when the diagnosis of GSD was dismissed, and the patient was treated as a case of primary HTG.
In conclusion, genetic testing for rare dyslipidaemias should be considered early in cases of severe elevations of plasma cholesterol and/or triglycerides, to enable adequate and precise management of the patient. Besides LPL, there are other genes involved in primary hypertriglyceridemia phenotypes (most notably, APOC2, APOA5, LMF1, and GPIHBP1). Our paediatric patient had a homozygous pathogenic GPIHBP1 variant, causing severe hypertriglyceridemia, cholesterol deposits at the hard palate, eruptive xanthomas, lethargy, poor appetite, and mild splenomegaly. Combination treatment with fenofibrate and gemfibrozil was shown to help reduce TG levels.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Ethics Committee Pakistan (033-523-2019). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
UG and FS designed this study. FS, SS, SK, and US collected and analysed the data. US drafted the manuscript. All authors contributed to the manuscript and approved the final version for submission.
The study was funded by the 1) Slovenian Research Agency Program (P3-0343) and Project (J3-2536) and 2) Higher Education Commission, Pakistan (Award No 20-15760).
We would like to acknowledge Prof. Munir Iqbal Malik for help with patient management and Quratul Ain and Madeeha Khan for collecting blood samples from the hospital and isolating DNA. Figure 3, Supplementary Figures S1, S2 were created with BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.983283/full#supplementary-material
Ahmad, Z., and Wilson, D. P. (2014). Familial chylomicronemia syndrome and response to medium-chain triglyceride therapy in an infant with novel mutations in GPIHBP1. J. Clin. Lipidol. 8, 635–639. doi:10.1016/j.jacl.2014.08.010
Álvarez-López, H., Ruiz-Gastélum, E., and Díaz-Aragón, A. (2021). Tratamiento actual de la hipertrigliceridemia. Cardiovasc. Metab. Sci. 32, 242–246. doi:10.35366/100805
Ariza, M. J., Martínez-Hernández, P. L., Ibarretxe, D., Rabacchi, C., Rioja, J., Grande-Aragón, C., et al. (2016). Novel mutations in the GPIHBP1 gene identified in 2 patients with recurrent acute pancreatitis. J. Clin. Lipidol. 10, 92–100. doi:10.1016/j.jacl.2015.09.007
Ariza, M. J., Rioja Villodres, J., Ibarretxe, D., Camacho, A., Díaz-Díaz, J. L., Mangas, A., et al. (2018). Molecular basis of the familial chylomicronemia syndrome in patients from the national dyslipidemia registry of the Spanish atherosclerosis society. J. Clin. Lipidol. 12, 1482–1492. e3. doi:10.1016/j.jacl.2018.07.013
Arora, R., Nimonkar, A. v., Baird, D., Wang, C., Chiu, C. H., Horton, P. A., et al. (2019). Structure of lipoprotein lipase in complex with GPIHBP1. Proc. Natl. Acad. Sci. U. S. A. 116, 10360–10365. doi:10.1073/pnas.1820171116
Basit, A., Sabir, S., Riaz, M., Fawwad, A., Abro, M. U. R., Ahmed, K. I., et al. (2020). Ndsp 05: Prevalence and pattern of dyslipidemia in urban and rural areas of Pakistan; a sub analysis from second National Diabetes Survey of Pakistan (NDSP) 2016–2017. J. Diabetes Metab. Disord. 19, 1215–1225. doi:10.1007/s40200-020-00631-z
Beigneux, A. P., Fong, L. G., Bensadoun, A., Davies, B. S. J., Oberer, M., Gårdsvoll, H., et al. (2015). GPIHBP1 missense mutations often cause multimerization of GPIHBP1 and thereby prevent lipoprotein lipase binding. Circ. Res. 116, 624–632. doi:10.1161/CIRCRESAHA.116.305085
Beigneux, A. P., Franssen, R., Bensadoun, A., Gin, P., Melford, K., Peter, J., et al. (2009a). Chylomicronemia with a mutant GPIHBP1 (Q115P) that cannot bind lipoprotein lipase. Arterioscler. Thromb. Vasc. Biol. 29, 956–962. doi:10.1161/ATVBAHA.109.186577
Beigneux, A. P., Gin, P., Davies, B. S. J., Weinstein, M. M., Bensadoun, A., Fong, L. G., et al. (2009b). Highly conserved cysteines within the Ly6 domain of GPIHBP1 are crucial for the binding of lipoprotein lipase. J. Biol. Chem. 284, 30240–30247. doi:10.1074/jbc.M109.046391
Beigneux, A. P., Miyashita, K., Ploug, M., Blom, D. J., Ai, M., Linton, M. F., et al. (2017). Autoantibodies against GPIHBP1 as a cause of hypertriglyceridemia. N. Engl. J. Med. 376, 1647–1658. doi:10.1056/nejmoa1611930
Beigneux, A. R., Weinstein, M. M., Davies, B. S. J., Gin, P., Bensadoun, A., Fong, L. G., et al. (2009c). GPIHBP1 and lipolysis: An update. Curr. Opin. Lipidol. 20, 211–216. doi:10.1097/MOL.0b013e32832ac026
Berge, K. E., Retterstøl, K., Romeo, S., Pirazzi, C., and Leren, T. P. (2014). Type 1 hyperlipoproteinemia due to a novel deletion of exons 3 and 4 in the GPIHBP1 gene. Atherosclerosis 234, 30–33. doi:10.1016/j.atherosclerosis.2014.02.005
Brown, E. E., Sturm, A. C., Cuchel, M., Braun, L. T., Duell, P. B., Underberg, J. A., et al. (2020). Genetic testing in dyslipidemia: A scientific statement from the national lipid association. J. Clin. Lipidol. 14, 398–413. doi:10.1016/j.jacl.2020.04.011
Buonuomo, P. S., Bartuli, A., Rabacchi, C., Bertolini, S., and Calandra, S. (2015). A 3-day-old neonate with severe hypertriglyceridemia from novel mutations of the GPIHBP1 gene. J. Clin. Lipidol. 9, 265–270. doi:10.1016/j.jacl.2014.10.003
Carrasquilla, G. D., Christiansen, M. R., and Kilpeläinen, T. O. (2021). The genetic basis of hypertriglyceridemia. Curr. Atheroscler. Rep. 23, 39. doi:10.1007/s11883-021-00939-y
Charrière, S., Peretti, N., Bernard, S., di Filippo, M., Sassolas, A., Merlin, M., et al. (2011). GPIHBP1 C89F neomutation and hydrophobic C-terminal domain G175R mutation in two pedigrees with severe hyperchylomicronemia. J. Clin. Endocrinol. Metab. 96, 1675–1679. doi:10.1210/jc.2011-1444
Chokshi, N., Blumenschein, S. D., Ahmad, Z., and Garg, A. (2014). Genotype-phenotype relationships in patients with type i hyperlipoproteinemia. J. Clin. Lipidol. 8, 287–295. doi:10.1016/j.jacl.2014.02.006
Chyzhyk, V., Kozmic, S., Brown, A. S., Hudgins, L. C., Starc, T. J., Davila, A. D., et al. (2019). Extreme hypertriglyceridemia: Genetic diversity, pancreatitis, pregnancy, and prevalence. J. Clin. Lipidol. 13, 89–99. doi:10.1016/j.jacl.2018.09.007
Coca-Prieto, I., Kroupa, O., Gonzalez-Santos, P., Magne, J., Olivecrona, G., Ehrenborg, E., et al. (2011). Childhood-onset chylomicronaemia with reduced plasma lipoprotein lipase activity and mass: Identification of a novel GPIHBP1 mutation. J. Intern. Med. 270, 224–228. doi:10.1111/j.1365-2796.2011.02361.x
Cruz-Bautista, I., Huerta-Chagoya, A., Moreno-Macías, H., Rodríguez-Guillén, R., Ordóñez-Sánchez, M. L., Segura-Kato, Y., et al. (2021). Familial hypertriglyceridemia: An entity with distinguishable features from other causes of hypertriglyceridemia. Lipids Health Dis. 20, 14. doi:10.1186/s12944-021-01436-6
De Jesus, J. M. (2011). Expert panel on integrated guidelines for cardiovascular health and risk reduction in children and adolescents: Summary report. Pediatrics 128, S213–S256. doi:10.1542/peds.2009-2107C
Desvignes, J., Bartoli, M., Krahn, M., Miltgen, M., Christophe, B., Salgado, D., et al. (2018). VarAFT : A variant annotation and filtration system for human next generation sequencing data. Nucleic Acids Res. 46, W545–W553. doi:10.1093/nar/gky471
Esan, O., and Wierzbicki, A. S. (2020). Volanesorsen in the treatment of familial chylomicronemia syndrome or hypertriglyceridaemia: Design, development and place in therapy. Drug Des. devel. Ther. 14, 2623–2636. doi:10.2147/DDDT.S224771
Fong, L. G., Young, S. G., Beigneux, A. P., Bensadoun, A., Oberer, M., Jiang, H., et al. (2016). GPIHBP1 and plasma triglyceride metabolism. Trends Endocrinol. Metab. 27, 455–469. doi:10.1016/j.tem.2016.04.013
Franklin by Genoox (2022). Available at: https://franklin.genoox.com (Accessed May 30, 2022).
Franssen, R., Young, S. G., Peelman, F., Hertecant, J., Sierts, J. A., Schimmel, A. W. M., et al. (2010). Chylomicronemia with low postheparin lipoprotein lipase levels in the setting of GPIHBP1 defects. Circ. Cardiovasc. Genet. 3, 169–178. doi:10.1161/CIRCGENETICS.109.908905
Gin, P., Goulbourne, C. N., Adeyo, O., Beigneux, A. P., Davies, B. S. J., Tat, S., et al. (2012). Chylomicronemia mutations yield new insights into interactions between lipoprotein lipase and GPIHBP1. Hum. Mol. Genet. 21, 2961–2972. doi:10.1093/hmg/dds127
Goldberg, A. S., and Hegele, R. A. (2012). Severe hypertriglyceridemia in pregnancy. J. Clin. Endocrinol. Metab. 97, 2589–2596. doi:10.1210/jc.2012-1250
Goldberg, R. B., and Chait, A. (2020). A comprehensive update on the chylomicronemia syndrome. Front. Endocrinol. 11, 593931. doi:10.3389/fendo.2020.593931
Gonzaga-Jauregui, C., Mir, S., Penney, S., Jhangiani, S., Midgen, C., Finegold, M., et al. (2014). Whole-exome sequencing reveals GPIHBP1 mutations in infantile colitis with severe hypertriglyceridemia. J. Pediatr. Gastroenterol. Nutr. 59, 17–21. doi:10.1097/MPG.0000000000000363
Groselj, U., Kovac, J., Sustar, U., Mlinaric, M., Fras, Z., Podkrajsek, K. T., et al. (2018). Universal screening for familial hypercholesterolemia in children: The Slovenian model and literature review. Atherosclerosis 277, 383–391. doi:10.1016/j.atherosclerosis.2018.06.858
Groselj, U., Wiegman, A., and Gidding, S. S. (2022). Screening in children for familial hypercholesterolaemia: Start now. Eur. Heart J., ehac224. doi:10.1093/eurheartj/ehac224
Hegele, R. A., Berberich, A. J., Ban, M. R., Wang, J., Digenio, A., Alexander, V. J., et al. (2018). Clinical and biochemical features of different molecular etiologies of familial chylomicronemia. J. Clin. Lipidol. 12, 920–927. e4. doi:10.1016/j.jacl.2018.03.093
Hegele, R. A., Borén, J., Ginsberg, H. N., Arca, M., Averna, M., Binder, C. J., et al. (2020). Rare dyslipidaemias, from phenotype to genotype to management: A European atherosclerosis society task force consensus statement. Lancet. Diabetes Endocrinol. 8, 50–67. doi:10.1016/S2213-8587(19)30264-5
Holmes, R. S., and Cox, L. A. (2012). Comparative studies of glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1: Evidence for a eutherian mammalian origin for the GPIHBP1 gene from an LY6-like gene. 3 Biotech. 2, 37–52. doi:10.1007/s13205-011-0026-4
Iacocca, M. A., Dron, J. S., and Hegele, R. A. (2019). Progress in finding pathogenic DNA copy number variations in dyslipidemia. Curr. Opin. Lipidol. 30, 63–70. doi:10.1097/MOL.0000000000000581
Ioannidis, N. M., Rothstein, J. H., Pejaver, V., Middha, S., McDonnell, S. K., Baheti, S., et al. (2016). Revel: An ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 99, 877–885. doi:10.1016/j.ajhg.2016.08.016
Jung, M. K., Jin, J., Kim, H. O., Kwon, A., Chae, H. W., Kang, S. J., et al. (2017). A 1-month-old infant with chylomicronemia due to GPIHBP1 gene mutation treated by plasmapheresis. Ann. Pediatr. Endocrinol. Metab. 22, 68–71. doi:10.6065/apem.2017.22.1.68
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., et al. (2020). The mutational constraint spectrum quantified from variation in 141, 456 humans. Nature 581, 434–443. doi:10.1038/s41586-020-2308-7
Kersten, S. (2021). Role and mechanism of the action of angiopoietin-like protein ANGPTL4 in plasma lipid metabolism. J. Lipid Res. 62, 100150. doi:10.1016/J.JLR.2021.100150
Landrum, M. J., Lee, J. M., Benson, M., Brown, G. R., Chao, C., Chitipiralla, S., et al. (2018). ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067. doi:10.1093/nar/gkx1153
Lima, J. G., Helena C Nobrega, L., Moura Bandeira, F. T., Pires Sousa, A. G., Medeiros de Araujo Macedo, T. B., Cavalcante Nogueira, A. C., et al. (2021). A novel GPIHBP1 mutation related to familial chylomicronemia syndrome: A series of cases. Atherosclerosis 322, 31–38. doi:10.1016/j.atherosclerosis.2021.02.020
Lin, M. H., Tian, X. H., Hao, X. L., Fei, H., Yin, J. L., Yan, D. D., et al. (2020). Management of a pregnant patient with chylomicronemia from a novel mutation in GPIHBP1: A case report. BMC Pregnancy Childbirth 20, 272. doi:10.1186/s12884-020-02965-1
Liu, C., Li, L., Guo, D., Lv, Y., Zheng, X. L., Mo, Z., et al. (2018). Lipoprotein lipase transporter GPIHBP1 and triglyceride-rich lipoprotein metabolism. Clin. Chim. Acta. 487, 33–40. doi:10.1016/j.cca.2018.09.020
Liu, S., Wang, Z., Zheng, X., Zhang, Y., Wei, S., OuYang, H., et al. (2022). Case report: Successful management of a 29-day-old infant with severe hyperlipidemia from a novel homozygous variant of GPIHBP1 gene. Front. Pediatr. 10, 792574. doi:10.3389/fped.2022.792574
Marusic, T., Sustar, U., Sadiq, F., Kotori, V., Mlinaric, M., Kovac, J., et al. (2020). Genetic and clinical characteristics of patients with homozygous and compound heterozygous familial hypercholesterolemia from three different populations: Case series. Front. Genet. 11, 572176. doi:10.3389/fgene.2020.572176
Miyashita, K., Lutz, J., Hudgins, L. C., Toib, D., Ashraf, A. P., Song, W., et al. (2020). Chylomicronemia from GPIHBP1 autoantibodies. J. Lipid Res. 61, 1365–1376. doi:10.1194/jlr.R120001116
Mysling, S., Kristensen, K. K., Larsson, M., Beigneux, A. P., Gårdsvoll, H., Loren, F. G., et al. (2016). The acidic domain of the endothelial membrane protein GPIHBP1 stabilizes lipoprotein lipase activity by preventing unfolding of its catalytic domain. Elife 5, e12095. doi:10.7554/eLife.12095
Navarro Hermoso, A., and Valdivielso, P. (2021). Treatment of chylomicronemia. Clin. Investig. Arterioscler. 33, 75–79. doi:10.1016/j.arteri.2021.01.004
Nordestgaard, B. G. (2016). Triglyceride-rich lipoproteins and atherosclerotic cardiovascular disease: New insights from epidemiology, genetics, and biology. Circ. Res. 118, 547–563. doi:10.1161/CIRCRESAHA.115.306249
Okazaki, H., Gotoda, T., Ogura, M., Ishibashi, S., Inagaki, K., Daida, H., et al. (2021). Current diagnosis and management of primary chylomicronemia. J. Atheroscler. Thromb. 28, 883–904. doi:10.5551/jat.RV17054
Olivecrona, G., Ehrenborg, E., Semb, H., Makoveichuk, E., Lindberg, A., Hayden, M. R., et al. (2010). Mutation of conserved cysteines in the Ly6 domain of GPIHBP1 in familial chylomicronemia. J. Lipid Res. 51, 1535–1545. doi:10.1194/jlr.M002717
Paquette, M., Hegele, R. A., Paré, G., and Baass, A. (2018). A novel mutation in GPIHBP1 causes familial chylomicronemia syndrome. J. Clin. Lipidol. 12, 506–510. doi:10.1016/j.jacl.2018.01.011
Parhofer, K. G., and Laufs, U. (2019). The diagnosis and treatment of hypertriglyceridemia. Dtsch. Arztebl. Int. 116, 825–832. doi:10.3238/arztebl.2019.0825
Patni, N., Brothers, J., Xing, C., and Garg, A. (2016). Type 1 hyperlipoproteinemia in a child with large homozygous deletion encompassing GPIHBP1. J. Clin. Lipidol. 10, 1035–1039. e2. doi:10.1016/j.jacl.2016.04.001
Plengpanich, W., Young, S. G., Khovidhunkit, W., Bensadoun, A., Karnman, H., Ploug, M., et al. (2014). Multimerization of glycosylphosphatidylinositol-anchored high density lipoprotein-binding protein 1 (GPIHBP1) and familial chylomicronemia from a serine-to-cysteine substitution in GPIHBP1 Ly6 domain. J. Biol. Chem. 289, 19491–19499. doi:10.1074/jbc.M114.558528
Rabacchi, C., D’Addato, S., Palmisano, S., Lucchi, T., Bertolini, S., Calandra, S., et al. (2016). Clinical and genetic features of 3 patients with familial chylomicronemia due to mutations in GPIHBP1 gene. J. Clin. Lipidol. 10, 915–921. e4. doi:10.1016/j.jacl.2016.03.009
Remec, Z. I., Trebusak Podkrajsek, K., Repic Lampret, B., Kovac, J., Groselj, U., Tesovnik, T., et al. (2021). Next-generation sequencing in newborn screening: A review of current state. Front. Genet. 12, 662254. doi:10.3389/fgene.2021.662254
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular Pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Rios, J. J., Shastry, S., Jasso, J., Hauser, N., Garg, A., Bensadoun, A., et al. (2012). Deletion of GPIHBP1 causing severe chylomicronemia. J. Inherit. Metab. Dis. 35, 531–540. doi:10.1007/s10545-011-9406-5
Silverman, M. G., Ference, B. A., Im, K., Wiviott, S. D., Giugliano, R. P., Grundy, S. M., et al. (2016). Association between lowering ldl-C and cardiovascular risk reduction among different therapeutic interventions: A systematic review and meta-analysis. JAMA - J. Am. Med. Assoc. 316, 1289–1297. doi:10.1001/jama.2016.13985
Stenson, P. D., Ball, E. v., Mort, M., Phillips, A. D., Shiel, J. A., Thomas, N. S. T., et al. (2003). Human gene mutation database (HGMD): 2003 update. Hum. Mutat. 21, 577–581. doi:10.1002/humu.10212
Surendran, R. P., Visser, M. E., Heemelaar, S., Wang, J., Peter, J., Defesche, J. C., et al. (2012). Mutations in LPL, APOC2, APOA5, GPIHBP1 and LMF1 in patients with severe hypertriglyceridaemia. J. Intern. Med. 272, 185–196. doi:10.1111/j.1365-2796.2012.02516.x
Sustar, U., Groselj, U., Trebusak Podkrajsek, K., Mlinaric, M., Kovac, J., Thaler, M., et al. (2022). Early discovery of children with lysosomal acid lipase deficiency with the universal familial hypercholesterolemia screening program. Front. Genet. IN PRESS. doi:10.3389/fgene.2022.936121
Tromp, T. R., Hartgers, M. L., Hovingh, G. K., Vallejo-Vaz, A. J., Ray, K. K., Soran, H., et al. (2022). Worldwide experience of homozygous familial hypercholesterolaemia: Retrospective cohort study. Lancet 399, 719–728. doi:10.1016/S0140-6736(21)02001-8
UniProt (2022). UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 49 (D1), D480–D489. Available at: https://www.uniprot.org/uniprot/Q8IV16#family_and_domains (Accessed May 2, 2022). doi:10.1093/nar/gkaa11008
Vallejo Vaz, A. J., Stevens, C. A. T., Lyons, A. R. M., Dharmayat, K. I., Freiberger, T., Hovingh, G. K., et al. (2021). Global perspective of familial hypercholesterolaemia : A cross-sectional study from the EAS familial hypercholesterolaemia studies collaboration ( FHSC ). Lancet 6736, 1713–1725. doi:10.1016/S0140-6736(21)01122-3
Vallejo-Vaz, A. J., Marco, M. De, Stevens, C. A. T., Akram, A., Freiberger, T., Hovingh, G. K., et al. (2018). Overview of the current status of familial hypercholesterolaemia care in over 60 countries - the EAS Familial Hypercholesterolaemia Studies Collaboration (FHSC). Atherosclerosis 277, 234–255. doi:10.1016/j.atherosclerosis.2018.08.051
Voss, C. v., Davies, B. S. J., Tat, S., Gin, P., Fong, L. G., Pelletier, C., et al. (2011). Mutations in lipoprotein lipase that block binding to the endothelial cell transporter GPIHBP1. Proc. Natl. Acad. Sci. U. S. A. 108, 7980–7984. doi:10.1073/pnas.1100992108
Wang, J., and Hegele, R. A. (2007). Homozygous missense mutation (G56R) in glycosylphosphatidylinositol- anchored high-density lipoprotein-binding protein 1 (GPI-HBP1) in two siblings with fasting chylomicronemia (MIM 144650). Lipids Health Dis. 6, 23–27. doi:10.1186/1476-511X-6-23
Wu, S. A., Kersten, S., and Qi, L. (2021). Lipoprotein lipase and its regulators: An unfolding story. Trends Endocrinol. Metab. 32, 48–61. doi:10.1016/j.tem.2020.11.005
Keywords: hypertriglyceridemia, triglycerides, glycosylphosphatiliylinositol-anchored high-density lipoprotein-binding protein 1, hyperlipidemia, GPIHBP1
Citation: Sustar U, Groselj U, Khan SA, Shafi S, Khan I, Kovac J, Bizjan BJ, Battelino T and Sadiq F (2022) A homozygous variant in the GPIHBP1 gene in a child with severe hypertriglyceridemia and a systematic literature review. Front. Genet. 13:983283. doi: 10.3389/fgene.2022.983283
Received: 30 June 2022; Accepted: 18 July 2022;
Published: 16 August 2022.
Edited by:
Hayato Tada, Kanazawa University, JapanCopyright © 2022 Sustar, Groselj, Khan, Shafi, Khan, Kovac, Bizjan, Battelino and Sadiq. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Urh Groselj, dXJoLmdyb3NlbGpAa2Nsai5zaQ==; Fouzia Sadiq, ZGlyZWN0b3IucmVzZWFyY2hAc3RtdS5lZHUucGs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.