 Zhiying Xie1‡†
Zhiying Xie1‡† Chang Liu1‡
Chang Liu1‡ Yanyu Lu1
Yanyu Lu1 Chengyue Sun2Yilin Liu3Meng Yu1Junlong Shu1Lingchao Meng1
Chengyue Sun2Yilin Liu3Meng Yu1Junlong Shu1Lingchao Meng1 Jianwen Deng1
Jianwen Deng1 Wei Zhang1
Wei Zhang1 Zhaoxia Wang1
Zhaoxia Wang1 He Lv1*§
He Lv1*§ Yun Yuan1*§†
Yun Yuan1*§†- 1Department of Neurology, Peking University First Hospital, Beijing, China
- 2Department of Neurology, Peking University People’s Hospital, Beijing, China
- 3Department of Pathology, Peking Union Medical College Hospital, Beijing, China
The precise identification of pathogenic DMD variants is sometimes rather difficult, mainly due to complex structural variants (SVs) and deep intronic splice-altering variants. We performed genomic long-read whole DMD gene sequencing in a boy with asymptomatic hyper-creatine kinase-emia who remained genetically undiagnosed after standard genetic testing, dystrophin protein and DMD mRNA studies, and genomic short-read whole DMD gene sequencing. We successfully identified a novel pathogenic SV in DMD intron 1 via long-read sequencing. The deep intronic SV consists of a long interspersed nuclear element-1 (LINE-1) insertion/non-tandem duplication rearrangement causing partial exonization of the LINE-1, establishing a genetic diagnosis of Becker muscular dystrophy. Our study expands the genetic spectrum of dystrophinopathies and highlights the significant role of disease-causing LINE-1 insertions in monogenic diseases.
Introduction
Dystrophinopathies, caused by pathogenic DMD variants and characterized by primary involvement in skeletal and/or cardiac muscle fibers, are among the most common inherited muscular dystrophies and mainly affect male patients (Birnkrant et al., 2018). Becker muscular dystrophy (BMD) and its two allelic forms (X-linked dilated cardiomyopathy and Duchenne muscular dystrophy) are the three major phenotypes of dystrophinopathies (Muntoni, Torelli, & Ferlini, 2003). The perplexing genetic spectrum of dystrophinopathies, as a result of the high genetic complexity of the DMD gene, includes both large-scale copy number variation or structural variants (SVs) and small pathogenic DMD variants, which can occur in coding and/or non-coding regions (Neri et al., 2020; Xie et al., 2020; Waddell et al., 2021; Xie et al., 2021). Most of the pathogenic DMD variants can be identified by DNA-based standard genetic testing for dystrophinopathies, consisting of multiplex ligation-dependent probe amplification (MLPA) and short-read sequencing of 79 exons and adjacent intronic regions of DMD (Fratter et al., 2020; Neri et al., 2020; Xie et al., 2021). However, standard genetic testing cannot identify some atypical pathogenic DMD variants, including complex SVs (Xie et al., 2020; Gonçalves et al., 2021; Waddell et al., 2021), deep intronic DMD variants (Gonçalves et al., 2017; Waddell et al., 2021; Xie et al., 2021), and synonymous variants which cause abnormal splicing of DMD pre-mRNA and result in abnormal dystrophin protein (Juan-Mateu et al., 2013). The detection and construction of these atypical pathogenic DMD variants require the application of various DNA- and RNA-based genetic testing techniques and in silico splicing analysis (Xie et al., 2021).
Herein, we performed standard genetic testing in a boy with a highly suspected BMD based on his clinical and pathological characteristics, which failed to detect pathogenic variant(s) in him. In order to discover potential deep intronic splice-altering variants in DMD, muscle biopsy-based dystrophin protein and DMD mRNA studies, as well as genomic short-read whole DMD gene sequencing, were performed on him. We successfully identified a novel deep intronic SV in DMD via genomic long-read whole DMD gene sequencing of the patient. The novel pathogenic SV missed by short-read whole DMD gene sequencing is a ∼6-kb-long interspersed nuclear element-1 (LINE-1) insertion/non-tandem duplication rearrangement, causing partial exonization of the LINE-1 in addition to the normal splicing of DMD, which ultimately established a genetic diagnosis of BMD in the patient.
Methods and results
Clinical and pathological features
The patient enrolled in the study is a 3.5-year-old boy with an asymptomatic hyper-creatine kinase-emia phenotype. He presented to Peking University First Hospital Neuromuscular Center at the age of 3.5 years because of an incidental finding of hyper-creatine kinase-emia. His serum creatine kinase was markedly elevated in every test (range 2,897–4,717 IU/L; normal 25–195 IU/L). No one else in his family had hyper-creatine kinase-emia. He had no delayed motor milestones and no exercise-induced muscle pain or cramps. Physical examination confirmed that he had mild calf hypertrophy and no muscle weakness, which was further validated by his muscle MRI examinations, showing no muscle fatty infiltration of the pelvis and thigh muscles. His muscle biopsy revealed myopathic changes, including several clusters of necrotic and regenerating muscle fibers; a few hypertrophic, atrophic, and hypercontracted muscle fibers; and a small number of internal nuclei (Figure 1E). The immunohistochemical staining showed a severe reduction of dystrophin-N, a slight reduction of dystrophin-C, and a positive expression of dystrophin-R (Figures 1F–H), which established a highly suspected diagnosis of BMD in the patient.
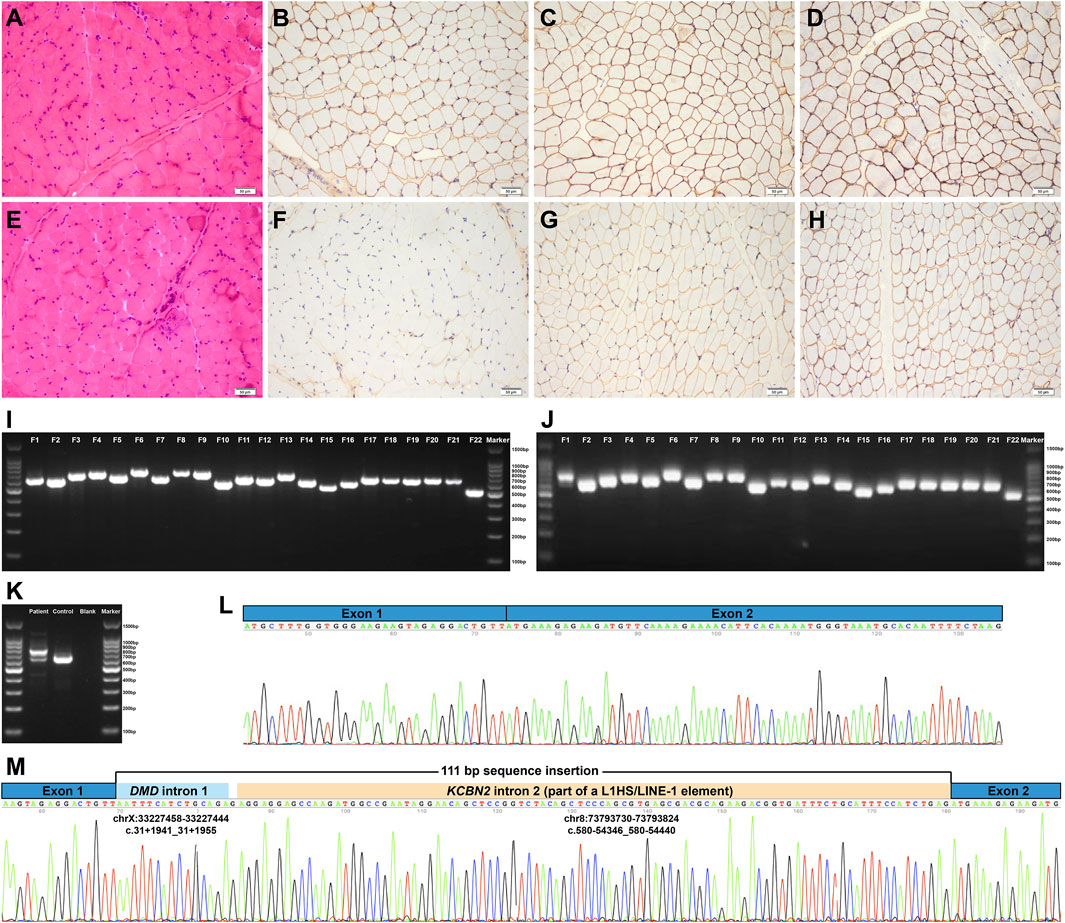
FIGURE 1. Pathologic features and DMD mRNA studies of the patient. (A–D) Normal control showing no pathological changes and positive expression of dystrophin-N, dystrophin-C, and dystrophin-R (200× magnification). (E) Hematoxylin–eosin staining revealed myopathic changes in the patient. (F–H) Immunohistochemical staining showing severe reduction of dystrophin-N, slight reduction of dystrophin-C, and positive expression of dystrophin-R. (I) Gel electrophoresis analysis of the amplified cDNA fragments confirmed that the 22 overlapping cDNA fragments were successfully amplified in the normal control I and the patient J. (J) Two different bands were found in F1, indicating the aberrant DMD transcripts. (K) Further RT-PCR analysis of the aberrant DMD transcripts revealed that the upper band was longer than the normal band, whereas the lower band was the same size as the normal band, that is, the wild-type transcript. (L) and (M) Sanger sequencing of the aberrant DMD transcripts revealed a 111-bp sequence insertion between DMD exons 1 and 2 in addition to the normal splicing of DMD exons 1 to 2. RT-PCR, reverse transcription–polymerase chain reaction; F, fragment.
Standard genetic testing and DMD mRNA analysis
We performed standard genetic testing for dystrophinopathies based on his suspected diagnosis of BMD, including MLPA-analysis of exonic deletion/duplication in DMD (copy number variations) and a short-read sequencing panel for inherited neuromuscular disorders (Xie et al., 2021). However, the standard genetic testing did not detect any disease-causing variants. Hence, we performed muscle-derived DMD mRNA analysis to identify potential abnormal DMD transcripts.
We extracted total mRNA from the remaining muscle biopsy sample and amplified 22 overlapping cDNA fragments of the full-length DMD mRNA (NM_004006.2, the primary full-length transcript in skeletal muscle) using the reverse transcription-polymerase chain reaction (RT-PCR) approach (Xie et al., 2021). Gel electrophoresis analysis of the 22 amplified cDNA fragments revealed aberrant splicing transcripts in the first cDNA fragment, showing two different bands; the upper band was longer than the normal band, whereas the lower band was the same size as the normal band, that is, the wild-type transcript (Figures 1J,K). Sanger sequencing of the aberrant DMD transcripts revealed a 111-bp sequence insertion between DMD exons 1 and 2 in addition to the normal splicing of DMD exons 1 to 2 (Figures 1L and M). The Human BLAT Search tool was adopted to search genomic sequences (GRCh37/hg19) that were homologous to the aberrant 111-bp sequence insertion. The homology search confirmed that the aberrant insertion consisted of a 15-bp sequence originating from DMD intron 1 (NM_004006.2:c.31+1941_31+1955), a single guanine nucleotide, and a 95-bp sequence homologous to a deep intronic region in KCBN2 intron 2 (NM_004770.2:c.580-54346_580-54440; Figure 1M). The 95-bp sequence was part of an L1HS/LINE-1 element annotated by the RepeatMasker Web Server with default conditions (http://www.repeatmasker.org/cgi-bin/WEBRepeatMasker), that is, the partial exonization of LINE-1. According to the Human Genome Variation Society nomenclature (den Dunnen et al., 2016), the aberrant insertion transcript was described as follows: NM_004006.2:r.[=,31_32ins[NM_004006.2:r.31+1941_31+1955;g;NM_004770.2:r.580-54346_580-54440]]. The aberrant insertion transcript encoded a premature termination codon (NP_003997.1:p.[=,Tyr11*]; Figure 2I), which was targeted for degradation by nonsense-mediated decay. The aberrant insertion transcript and the normally spliced DMD transcript resulted in the decreased expression of dystrophin protein (Figures 1F–H).
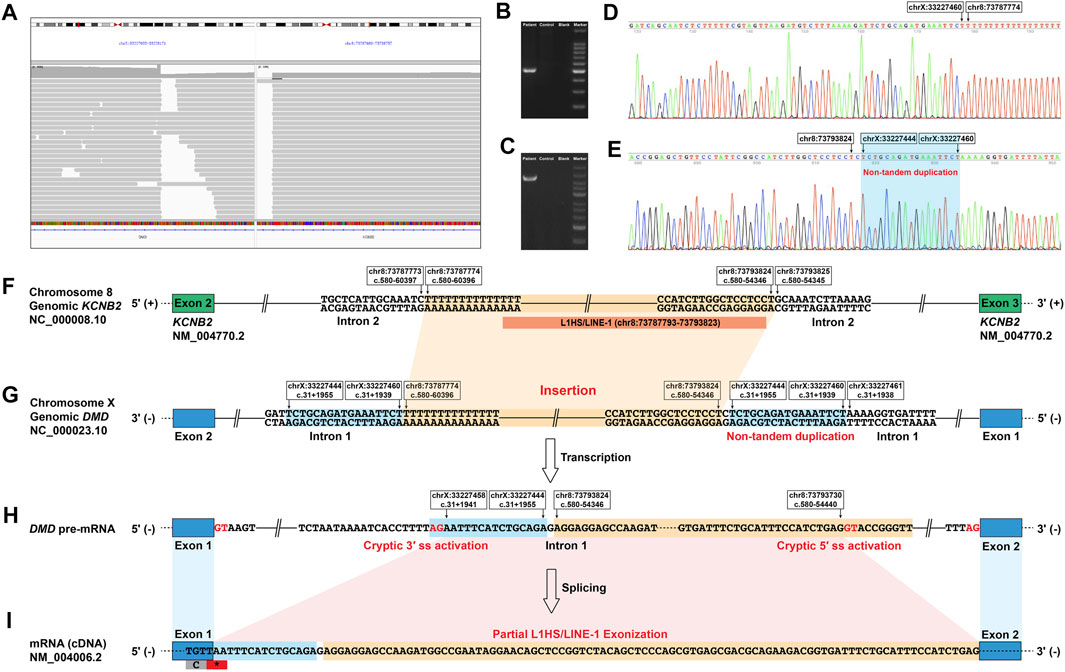
FIGURE 2. Identification of a novel deep intronic structural variant in DMD and its aberrant splicing effect on DMD pre-mRNA. (A) The IGV screenshot of long-read whole DMD gene sequencing of the patient indicated an insertion of a ∼6-kb sequence from chromosome 8 in the deep intronic region of DMD intron 1. Successful PCR amplification of the genomic 3′ (B) and 5′ (C) breakpoint regions using primers designed for the deep intronic DMD variant detected by the long-read whole DMD gene sequencing. Successful Sanger sequencing of the genomic 3′ (D) and 5′ (E) breakpoint regions confirmed the exact sequences of the breakpoint regions. (F–I) Schematic diagram of the abnormal DMD splicing event caused by the deep intronic structural variant in the patient. (F) and (G) The structural variant located in the deep intronic region of DMD intron 1, insertion of a 6,051-bp sequence originating from a deep intronic region in KCNB2 intron 2 flanked by a non-tandem duplication of a 17-bp sequence originating from DMD intron 1. Most of the inserted sequence was annotated as an L1HS element belonging to the LINE-1 family. (H) The structural variant activated a cryptic 5′ ss in the LINE-1 element and a cryptic 3′ ss in the deep intronic region of DMD intron 1. (I) The cryptic 5′ ss and 3′ ss activation resulted in the 111-bp sequence insertion between DMD exons 1 and 2, consisting of a 15-bp sequence of DMD intron 1, a single guanine nucleotide, and the partial exonization of LINE-1 (95 bp). IGV, Integrative Genomics Viewer; L1 or LINE-1, long interspersed nuclear element-1; 5′ ss, donor splice site; 3′ ss, acceptor splice site; *, stop codon.
Genomic short- and long-read whole DMD gene sequencing
Genomic DNA was isolated from the peripheral blood of the patient, and short-read whole DMD gene sequencing was done as previously described (Xie et al., 2021) to detect possible variant(s) that could cause the aberrant insertion transcript. However, no possible aberrant splicing-causing variants were detected. Thus, we performed genomic long-read whole DMD gene sequencing (Xie et al., 2021) using the Nanopore PromethION (Oxford Nanopore, Oxford, United Kingdom) sequencer (average read depth 306X; average read length 3942 bp) to detect possible SVs that missed by the short-read whole DMD gene sequencing. Ninety-two reads with an average length of 4,362 bp indicating a ∼6-kb insertion originating from chromosome 8 in a deep intronic region of DMD intron 1 was identified. Exact sequences of the breakpoint regions were confirmed by Sanger sequencing of the genomic 3′ (Figures 2B,D) and 5′ (Figures 2C,E) breakpoint regions using primers designed for the deep intronic SV in DMD according to the genetic information provided by the 92 reads. The deep intronic SV in DMD intron 1 was an insertion/non-tandem duplication rearrangement, an insertion of a 6,051-bp sequence originating from a deep intronic region in KCNB2 intron 2 (chr8:73787774-73793824) flanked by a non-tandem duplication of 17 bp sequence originating from DMD intron 1 (chrX:33227444-33227460), which was finally described as follows (Figures 2F,G): NC_000023.10:g.33227460_33227461ins[NC_000008.10:g.73787774_73793824; C;NC_000023.10:g.33227444_33227460]; NM_004006.2:c.31+1955_31+1956ins[G; NM_004770.2:c.580-54346_c.580-60396; NM_004006.2:c.31+1939_31+1955]. Most of the inserted sequence (chr8:73787793-73793823) was annotated as an L1HS/LINE-1 element (Figure 2F). The deep intronic SV identified in this study is classified as a novel pathogenic DMD variant according to the standard guidelines (Richards et al., 2015), which is not reported in the literature and absent from population and disease-specific databases, including the Genome Aggregation Database, Database of Genomic Variants, ClinVar, and Leiden Open Variation Database.
The novel deep intronic SV activated a cryptic donor splice site [GAG|GTACCG with a Human Splicing Finder score (Desmet et al., 2009) of 78.55 and a Maximum Entropy score (Yeo & Burge, 2004) of 8.5] in the LINE-1 element and a cryptic acceptor splice site (ATCACCTTTTAG|AA with a Human Splicing Finder score of 80.69 and a Maximum Entropy score of 6.31) in the deep intronic region of DMD intron 1, resulting in the aberrant splicing of DMD pre-mRNA (Figures 2H,I).
Discussion
As the largest gene identified in the human genome, the DMD gene with a genomic size of over 2.5 Mb has numerous constitutive exons and very long introns. Common fragile sites and transposable elements, including DNA transposons and retrotransposons that can induce large and complex genomic rearrangements, are not uncommon in the vast intronic sequences of DMD (Muntoni et al., 2003). The accurate genetic counseling of dystrophinopathies relies primarily on the identification of underlying disease-causing DMD variants in the affected patients and the determination of the carrier status in the family (Kubota et al., 2022). The precise identification of pathogenic DMD variants is important to ensure that correct candidates are subjected to the different emerging personalized therapies.
In addition to exonic DMD variants, which directly cause alterations in dystrophin protein sequences and constitute the majority of pathogenic DMD variants, deep intronic variants that can cause aberrant DMD splicing and subsequent defects in dystrophin protein are increasingly found in patients with dystrophinopathies (Xie et al., 2021). The precise identification of deep intronic DMD variants demands various DNA- and RNA-based genetic testing approaches due to the genetic complexity of DMD. In the present study, we performed DMD mRNA studies in a boy with asymptomatic hyper-creatine kinase-emia who remained without pathogenic variants after standard genetic testing and detected an aberrant DMD splicing event. Genomic short-read whole DMD gene sequencing was then performed to detect possible variant(s) causing the aberrant splicing event. As no potential splice-altering variants were detected through short-read whole DMD gene sequencing, genomic long-read whole DMD gene sequencing was performed in the patient, which detected a novel pathogenic SV in the deep intronic region of DMD intron 1. The novel deep intronic SV is a ∼6-kb LINE-1 insertion/non-tandem duplication rearrangement identified through long-read sequencing, which further indicates that short reads are not enough to cover variants involving repetitive elements or large-scale SVs (Xie et al., 2020). The pathogenic DMD variants involving repetitive elements, such as the novel genomic SV causing partial exonization of the LINE-1 identified in our patient, might be a significant cause of genetically unsolved dystrophinopathies. This study highlights the importance of long-read sequencing in the detection and construction of pathogenic DMD variants involving repetitive elements. Precise detection and construction of deep intronic splice-altering variants in DMD have potential implications for the genetic therapy targeting normal dystrophin expression.
Long interspersed nuclear elements, typically spanning about 6 kb in length, belong to the retrotransposons, which cover about 17% of the human genome (Kim, Lee, & Han, 2012). LINE-1s, which are difficult to detect with the short-read sequencing technique such as the novel DMD SV identified in our patient, can cause inherited diseases through various pathogenic mechanisms, including de novo LINE-1 insertions, LINE-1 insertion-mediated deletions, and genomic rearrangements (Kim et al., 2012). To our knowledge, only eight patients with dystrophinopathy have been reported to be caused by pathogenic DMD variants involving LINE-1s. One of the eight patients is caused by a LINE-1 insertion in the 5′ untranslated region of DMD, causing instability of the mature DMD mRNA (Yoshida, Nakamura, Yazaki, Ikeda, & Takeda, 1998), one is caused by a LINE-1-meditated large-scale SV in DMD causing multiple exons-skipping (Xie et al., 2020), two are caused by deep intronic LINE-1s insertions causing partial exonization of LINE-1s (Gonçalves et al., 2017; Xie et al., 2021), and four are caused by exonic LINE-1 insertions causing exon-skipping (Narita et al., 1993; Holmes, Dombroski, Krebs, Boehm, & Kazazian, 1994; Musova et al., 2006; Awano et al., 2010). Our study is the third to report on a deep intronic LINE-1 insertion causing partial exonization of the LINE-1, expanding the spectrum of pathogenic DMD variants and highlighting the significant role of disease-causing LINE-1 insertions in monogenic diseases.
Data availability statement
The structural variant data presented in the study have been submitted to Leiden Open Variation Database, accession number Variant #0000872856.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee at Peking University First Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
ZX, CL, HL, and YY conceived and planned this study and took full responsibility for the paper. ZX, CL, and HL contributed to the methodology of the study. ZX, CL, CS, YL, YL, MY, JS, LM, JD, and WZ conducted the clinical study. ZX, CL, CS, YL, YL, and ZW conducted genetic research. ZX and YY checked all the genetic analyses. ZX and CL took the lead in writing the manuscript. HL and YY supervised this study.
Funding
This work was supported by a grant from the Beijing Municipal Science and Technology Commission (Grant no. Z191100006619034).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Awano, H., Malueka, R. G., Yagi, M., Okizuka, Y., Takeshima, Y., and Matsuo, M. (2010). Contemporary retrotransposition of a novel non-coding gene induces exon-skipping in dystrophin mRNA. J. Hum. Genet. 55 (12), 785–790. doi:10.1038/jhg.2010.111
Birnkrant, D. J., Bushby, K., Bann, C. M., Apkon, S. D., Blackwell, A., Brumbaugh, D., et al. (2018). Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 17 (3), 251–267. doi:10.1016/s1474-4422(18)30024-3
den Dunnen, J. T., Dalgleish, R., Maglott, D. R., Hart, R. K., Greenblatt, M. S., McGowan-Jordan, J., et al. (2016). HGVS recommendations for the description of sequence variants: 2016 update. Hum. Mutat. 37 (6), 564–569. doi:10.1002/humu.22981
Desmet, F. O., Hamroun, D., Lalande, M., Collod-Béroud, G., Claustres, M., and Béroud, C. (2009). Human splicing finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 37 (9), e67. doi:10.1093/nar/gkp215
Fratter, C., Dalgleish, R., Allen, S. K., Santos, R., Abbs, S., Tuffery-Giraud, S., et al. (2020). EMQN best practice guidelines for genetic testing in dystrophinopathies. Eur. J. Hum. Genet. 28 (9), 1141–1159. doi:10.1038/s41431-020-0643-7
Gonçalves, A., Fortuna, A., Ariyurek, Y., Oliveira, M. E., Nadais, G., Pinheiro, J., et al. (2021). Integrating whole-genome sequencing in clinical genetics: A novel disruptive structural rearrangement identified in the dystrophin gene (DMD). Int. J. Mol. Sci. 23 (1). doi:10.3390/ijms23010059
Gonçalves, A., Oliveira, J., Coelho, T., Taipa, R., Melo-Pires, M., Sousa, M., et al. (2017). Exonization of an intronic LINE-1 element causing becker muscular dystrophy as a novel mutational mechanism in dystrophin gene. Genes (Basel) 8 (10). doi:10.3390/genes8100253
Holmes, S. E., Dombroski, B. A., Krebs, C. M., Boehm, C. D., and Kazazian, H. H. (1994). A new retrotransposable human L1 element from the LRE2 locus on chromosome 1q produces a chimaeric insertion. Nat. Genet. 7 (2), 143–148. doi:10.1038/ng0694-143
Juan-Mateu, J., González-Quereda, L., Rodríguez, M. J., Verdura, E., Lázaro, K., Jou, C., et al. (2013). Interplay between DMD point mutations and splicing signals in Dystrophinopathy phenotypes. PLoS One 8 (3), e59916. doi:10.1371/journal.pone.0059916
Kim, Y. J., Lee, J., and Han, K. (2012). Transposable elements: No more 'junk DNA. Genomics Inf. 10 (4), 226–233. doi:10.5808/gi.2012.10.4.226
Kubota, A., Ishiura, H., Porto, K. J. L., Tanaka, M., Mitsui, J., Unuma, A., et al. (2022). DMD exon 2 duplication due to a complex genomic rearrangement is associated with a somatic mosaicism. Neuromuscul. Disord. 32 (3), 263–269. doi:10.1016/j.nmd.2021.12.004
Muntoni, F., Torelli, S., and Ferlini, A. (2003). Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2 (12), 731–740.
Musova, Z., Hedvicakova, P., Mohrmann, M., Tesarova, M., Krepelova, A., Zeman, J., et al. (2006). A novel insertion of a rearranged L1 element in exon 44 of the dystrophin gene: Further evidence for possible bias in retroposon integration. Biochem. Biophys. Res. Commun. 347 (1), 145–149. doi:10.1016/j.bbrc.2006.06.071
Narita, N., Nishio, H., Kitoh, Y., Ishikawa, Y., Ishikawa, Y., Minami, R., et al. (1993). Insertion of a 5' truncated L1 element into the 3' end of exon 44 of the dystrophin gene resulted in skipping of the exon during splicing in a case of Duchenne muscular dystrophy. J. Clin. Invest. 91 (5), 1862–1867. doi:10.1172/jci116402
Neri, M., Rossi, R., Trabanelli, C., Mauro, A., Selvatici, R., Falzarano, M. S., et al. (2020). The genetic landscape of dystrophin mutations in Italy: A nationwide study. Front. Genet. 11, 131. doi:10.3389/fgene.2020.00131
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Waddell, L. B., Bryen, S. J., Cummings, B. B., Bournazos, A., Evesson, F. J., Joshi, H., et al. (2021). WGS and RNA studies diagnose noncoding DMD variants in males with high creatine kinase. Neurol. Genet. 7 (1), e554. doi:10.1212/nxg.0000000000000554
Xie, Z., Sun, C., Liu, Y., Yu, M., Zheng, Y., Meng, L., et al. (2021). Practical approach to the genetic diagnosis of unsolved dystrophinopathies: A stepwise strategy in the genomic era. J. Med. Genet. 58 (11), 743–751. doi:10.1136/jmedgenet-2020-107113
Xie, Z., Sun, C., Zhang, S., Liu, Y., Yu, M., Zheng, Y., et al. (2020). Long-read whole-genome sequencing for the genetic diagnosis of dystrophinopathies. Ann. Clin. Transl. Neurol. 7 (10), 2041–2046. doi:10.1002/acn3.51201
Yeo, G., and Burge, C. B. (2004). Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 11 (2-3), 377–394. doi:10.1089/1066527041410418
Keywords: DMD, LINE-1, aberrant splicing, deep-intronic structural variant, long-read sequencing
Citation: Xie Z, Liu C, Lu Y, Sun C, Liu Y, Yu M, Shu J, Meng L, Deng J, Zhang W, Wang Z, Lv H and Yuan Y (2022) Exonization of a deep intronic long interspersed nuclear element in Becker muscular dystrophy. Front. Genet. 13:979732. doi: 10.3389/fgene.2022.979732
Received: 28 June 2022; Accepted: 25 July 2022;
Published: 25 August 2022.
Edited by:
Matthew E. R. Butchbach, Nemours Children’s Health Delaware, United StatesReviewed by:
Rosário Santos, Porto Hospital Center, PortugalJorge Oliveira, Universidade do Porto, Portugal
Copyright © 2022 Xie, Liu, Lu, Sun, Liu, Yu, Shu, Meng, Deng, Zhang, Wang, Lv and Yuan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yun Yuan, eXVhbnl1bjIwMDJAMTI2LmNvbQ==; He Lv, Y3JhbmVoZWJtdUBzaW5hLmNvbQ==
†ORCID: Zhiying Xie, orcid.org/0000-0003-2102-6026; Yun Yuan, orcid.org/0000-0003-3282-9123
‡These authors share first authorship
§These authors share senior authorship