 Shan Chen1
Shan Chen1 Yeqing Qian
Yeqing Qian Minyue Dong
Minyue Dong- 1Laboratory of Prenatal Diagnosis, Mindong Hospital Affiliated to Fujian Medical University, Ningde, Fujian, China
- 2Women’s Hospital, School of Medicine, Zhejiang University, Hangzhou, China
- 3Key Laboratory of Reproductive Genetics, Ministry of Education (Zhejiang University), Hangzhou, China
Mutations of the Regulatory Factor X5 (RFX5) have been associated with the autosomal recessive major histocompatibility class II (MHC-II) deficiency, which is a severe immunodeficiency characterized by constitutive and interferon-gamma induced MHC II expression disorder and leads to the absence of cellular and humoral T-cell response to antigen challenge. The compound heterozygous splicing mutations of RFX5: c.353 + 6T>G (maternally inherited) and c.757 + 1G>A (paternally inherited) were identified in an infant diagnosed severe immunodeficiency. The mutation c.757 + 1G>A was classified as likely pathogenic while c.353 + 6T>G was classified as the variant of uncertain significance according to American College of Medical Genetics and Genomics (ACMG). To investigate the pathogenicity of RFX5: c.353 + 6T>G, reverse transcription PCR (RT-PCR) was conducted with the mother’s peripheral blood. An insertion of 191-bp intronic sequence (intron 6) was found in the transcripts, and this resulted in a frameshift and premature truncation of the protein, especially reduced the DNA-binding domain (DBD) of the RFX5 protein. Our data expanded the spectrum of pathogenic mutations in MHC-II deficiency and put new insights into the genetic counseling, prenatal diagnosis and preimplantation genetic testing (PGT) for the disease.
Introduction
Regulatory factor X-5 (RFX5) is essential for the regulation of major histocompatibility class II (MHC II) gene expression (Villard et al., 1997; Djidjik et al., 2012). RFX5 contains highly conserved DNA-binding domains (DBDs), located in the 90–166 residues and 407–614 residues, which bind the X box of MHC II before transcription (Clarridge et al., 2016; Farrokhi et al., 2018).
Mutations in RFX5 have been associated with the MHC-II deficiency, also named as the Bare Lymphocyte Syndrome (BLS) (OMIM:209,920) (Reith and Mach, 2001; Nekrep et al., 2002). MHC-II deficiency, a rare autosomal recessive disease, is characterized by constitutive and interferon-gamma induced MHC II expression disorder, and results in the absence of cellular and humoral T-cell response to antigen challenge, hypogammaglobulinemia and impaired antibody production (Garvie et al., 2007; Hanna and Etzioni, 2014). Over 200 cases have been reported and the prevalence varies significantly in different regions based on previous published data. Around two-thirds of the patients come from North Africa while less than 10 cases have been reported in East Asia (Djidjik et al., 2012; El Hawary et al., 2019). Children with MHC-II deficiency are extremely susceptible to a broad range of viral, bacterial and fungal, among which Pneumocystis jirovecii, Salmonella, cytomegalovirus (CMV), Cryptosporidium species and herpes simplex virus (HPV) are the most common pathogens (Hanna and Etzioni, 2014). Therefore, these patients are mainly characterized by severe and recurrent infections within the first year of life, especially involving the respiratory and gastrointestinal tract. What’s worse, the infection may be lethal (Reith and Mach, 2001; Farrokhi et al., 2018).
In the current investigation, we described a Chinese infant with MHC II deficiency caused by two novel splicing mutations, c.353 + 6T>G (maternally inherited) and c.757 + 1G>A (paternally inherited) in the RFX5 gene.
Methods
Subject
A 30-year-old healthy woman who delivered an infant (the proband) diagnosed severe immunodeficiency was referred to the Department of Reproductive Genetics, Women’s Hospital, School of Medicine Zhejiang University. Her infant presented recurrent pneumonia, reduced CD3 and CD4 positive leucocyte cell ratio, inverted CD4/CD8 ratio and reduced serum immunoglobulins levels (concentrations of IgG, IgA, and IgM) at 6 months of her age (Table 1). The infant died at 22 months of her age due to severe respiratory infection and respiratory failure. Severe immunodeficiency was diagnosed with unknown cause. The infant was born at full term to healthy un-consanguineous Chinese parents without family history of any genetic disorders.
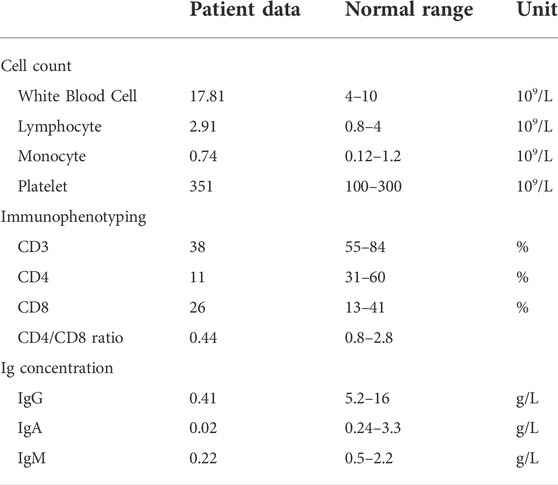
TABLE 1. Abnormal immunological findings of the infant with the associated normal range.
The use of medical records of this family is was approved by the Institutional Review Board of the Women’s Hospital, School of Medicine, Zhejiang University and the participants provided their written informed consents.
WES and bioinformatic analysis
To determine the cause for severe immunodeficiency, the whole exome sequencing (WES) was provided. Genomic DNA from all the family members was extracted by a QIAamp DNA blood midi kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol and then was fragmented by Covaris LE220 (Massachusetts, United States) to generate a paired-end library (200–250 bp). All amplified libraries were performed on the BGISEQ-500 platform (BGI, Shenzhen, China), the single-strand DNA was mixed with MGIEasy™ DNA Library Prep Kit V1 (BGI, Shenzhen, China) and then sequenced using 100SR chemistry with BGISEQ-500RS high-throughput sequencing Kit (BGI, Shenzhen, China).
Splice AI (https://spliceailookup.broadinstitute.org/) was used to predict the effect of variants. Pathogenic variants are assessed according to the protocol issued by the American College of Medical Genetics and Genomics (ACMG) (Richards et al., 2015). DECIPHER (http://decipher.sanger.ac.uk), OMIM (http://omim.org/), PubMed (http://www.ncbi.nlm.nih.gov/pubmed), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and HGMD (http://www. hgmd. cf.ac.uk/ac/index.php) databases were used to investigate the clinical relevance of the mutations.
Sanger sequencing validation
Sanger sequencing was carried out to confirm the variants in RFX5 gene. The primers used for c.757 + 1G>A were as follows: RFX5-E9F, TAGCTGAGGCAGAGGATGAAGA; and RFX5-E9R, GGTGAGGAGGAAACTGAGGAAT. The primers used for c.353 + 6T>G were as follows: RFX5-E5F, GTTAGGGTCTTAGTAATGCTTGTTCC; and RFX5-E5R, CCTTCGAGCTTTGATGTCAGG. The primers were designed using Oligo Primer Designer (Rychlik 2007). The DNA was amplified using the following procedure: 94°C for 10 min; 35 cycles at 94°C for 30 s, 60°C for 30 s, 72°C for 30 s; 72°C for 10 min. Sequencing was performed by an ABI 3130 DNA analyzer.
RNA extraction, real time-PCR, and sequencing
Total RNAs of the mother’s peripheral blood cells was extracted using TRIzol (Takara, Japan). Extracted total RNAs was reverse-transcribed using RT Kit (Takara, Japan) following the manufacturer’s instructions. RT-PCR was performed using GoldStar Best Master Mix (CWBIO, Beijing). Sequences of primers used were as follows: RFX5-spF, GAAGATGAGCCTGATGCTAAGAG; and RFX5-spR, GGCGACCTCAACGATGGAAC. The procedure of the PCR was as follows: 94°C for 10 min followed by 35 cycles at 94°C for 30 s, 60°C for 30 s, 72°C for 30 s, and a final extension step at 72°C for 10 min. Sequencing was performed by an ABI 3130 DNA analyzer.
Results
Identification of the compound heterozygous mutations in RFX5
Compound heterozygous splicing mutations in RFX5: c.353 + 6 T>G and c.757 + 1 G>A were identified by WES and confirmed by Sanger sequencing (Figure 1). These mutations have never been reported in any database (gnomAD, ClinVar or HGMD) or literature. Splice AI was used to predict the effects of the RFX5: c.353 + 6T>G and c.757 + 1G>A on splicing. The delta score of donor loss were 0.79 and 0.85, respectively. The result indicates that both of the mutations affect the splicing. The mutation RFX5: c.757 + 1G>A was inherited from her father and classified as likely pathogenic, while the mutation RFX5: c.353 + 6T>G was maternally inherited and classified as variant of uncertain significance (VUS) according to ACMG recommendations.
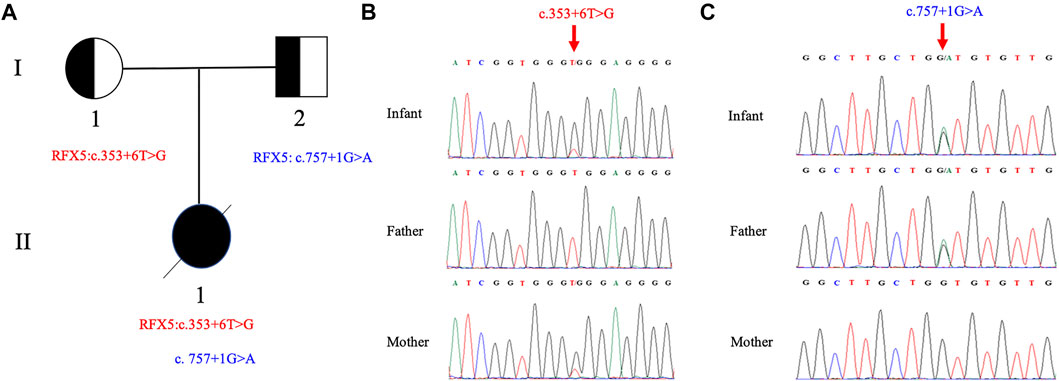
FIGURE 1. Pedigree of the family and Sanger sequence analysis. (A) II1(proband) have two compound splicing variants in RFX5 gene. One variant (c.353+6T > G) is from I1 (mother) and the other (c.757+1G > A) is from I2 (father). Chromatograms of (B) c.353+6T > G and (C) c.757+1G > A are identified by Sanger sequencing, respectively. The top chromatograms are from the infant, the middle two are from the father, and the bottom two are from the mother (red arrows indicate the mutation).
Pathogenicity of RFX5: c.353 + 6T>G
Based on the genotype–phenotype correlation, we hypothesized that RFX5: c.353 + 6T>G may affect the splicing. To confirm this hypothesis, mRNA was extracted from the women’s peripheral blood cells. RT-PCR was performed with the primers (RFX5-spF/RFX5-spR) designed to amplify exons three to nine of RFX5. It was found that the woman (I1) and the controls (C1 and C2) shared the band of PCR products at 675 bp, while I1 had another bigger band of 866 bp (Figure 2A). The Sanger sequencing of the bigger band (866 bp) showed that 191-bp intron six sequences were retained from the transcripts of the mother, compared with the 675 bp band (Figures 2B,C). The mutation (c.353 + 6T>G) introduced an insertion of 191-bp intron six sequences, which may cause a truncated RFX5 protein by a frameshift and creation of a premature stop codon. As is showed in Figure 2D, wild-type deduced RFX5 protein has three domains, among which two domains are DNA-binding domains (DBDs). However, the deduced RFX5 protein of c.353 + 6T>G splicing mutation only has one truncated DBD. The truncated DBD may damage the ability of RFX5 to bind X box of the MHC II promoter and then reduce the expression of MHC II molecular at the transcriptional level.
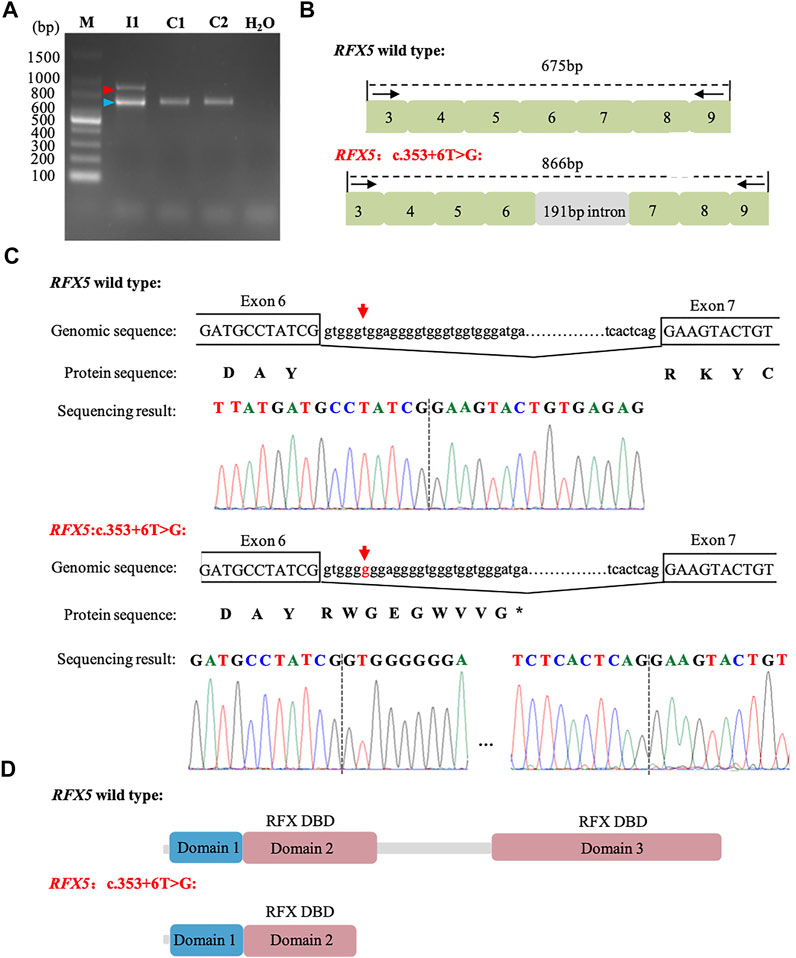
FIGURE 2. Analysis of c. 353 + 6T>G splicing mutation in RFX5. (A): RT-PCR analysis of RFX5 cDNA from peripheral blood samples. Agarose gel (1.5%) electrophoresis of RT-PCR products generated from I1 (mother), C1 and C2 (normal controls). Amplicons resulting from aberrantly spliced mRNA and normal spliced mRNA are marked by red and blue arrowheads, respectively. (B): Schematic representation of exon three to nine and intron six organization in RFX5. (C): Sequence analysis of the RT-PCR products from the mother. The arrows indicate the position of the c. 353 + 6T>G mutation. (D): The structure with domains of wild-type and c. 353 + 6T>G splicing mutation RFX5 protein. Wild-type RFX5 protein consists of three domains, among which domain two and domain three are the important DBDs while the splicing mutation leads to the premature of the protein and significantly damages the protein structure.
Discussion
In the current investigation, we described a Chinese infant with MHC class II deficiency due to the compound heterozygous splicing mutations of the RFX5 gene for the first time. The paternally inherited mutation c.757 + 1G>A of RFX5 was likely pathogenic and the maternally inherited mutation c.353 + 6T>G was proved to affect splicing, which may result in frameshift and truncation of the protein. Both of the mutations have never been reported in any database or literature, indicating our findings expand the spectrum of the diagnose for the MHC II deficiency and provide insight and information for genetic counseling.
Major histocompatibility complex (MHC) II deficiency is a primary immunodeficiency with an autosomal recessive inheritance pattern and is characterized by the early onset of severe and recurrent respiratory and gastrointestinal infections, developmental delay, and death in early life (Plaeger-Marshall et al., 1988; Lum et al., 2020). Almost all of the patients suffer from recurrent pneumonia and prolonged diarrhea (Clement, 1990; Ben-Mustapha et al., 2013). In the present study, the infant presented typically clinical and immunological features, like severe pneumonia, reduced CD3 and CD4 positive leucocyte cell ratio, inverse CD4/CD8 ratio and reduced serum immunoglobulin levels (concentrations of IgG, IgA, and IgM). The infant eventually died at 22 months of age due to acute respiratory infection and respiratory failure.
The underlying cause of MHC class II deficiency lies in the absence or reduced expression of MHC class II molecules which are regulated by MHC II enhanceosome (a cell-specific multiprotein complex) (Masternak et al., 2000; Hanna and Etzioni, 2014). The MHC class II molecules, also referred to as human leukocyte antigens (HLAs), are multigenic and highly polymorphic glycoproteins that aggregate to form heterodimers of a and β chains (Hanna and Etzioni, 2014). Moreover, HLAs are usually divided into three molecules, HLA-DR, -DP, and -DQ, which are located on the surface of antigen-presenting cells (APCs) such as dendritic cells and macrophages (Waldburger et al., 2000). HLAs present antigens endocytosed by APCs to the receptor of CD4+ helper T cells, directing the T cell activation, differentiation, and proliferation (Villard et al., 2000). It is reported that MHC class II gene mutations might damage cellular and humoral immunity by affecting CD4 T-cell development and reducing the Th-cell-dependent antibody production (Nonoyama et al., 1998; Sage et al., 2014; Aluri et al., 2018). Class II transactivator (CIITA), RFX-associated protein (RFXAP), regulatory factor X-5 (RFX5), and RFXAP-containing ankyrin repeat (RFXANK) are widely recognized key enhanceosome of MHC class II molecules so far. Accordingly, based on the four different transcript factors, MHC II deficiency is divided into four groups from group A to D, which are summarized by the deficiency of CIITA, RFXANK, RFX5, and RFXAP, respectively (Steimle et al., 1995; Scholl et al., 1997). In our study, two splicing mutations (c. 757 + 1G>A and c. 353 + 6T>G) in RFX5 gene were found in the proband, who belongs to group C. The former mutation was located at the classical splicing site, and the latter mutation was proven to lead to a stop codon after amino acid 126, leading to a loss of more than 50% of the protein including the highly conserved DBD. Therefore, the ability of RFX5 to bind X box could be affected and the expression of MHC II molecular reduced. Taken together, the compound heterozygous mutations in proband might explain the cause for immune deficiency.
Up to now, 19 pathogenic/uncertain significance mutations have been reported in RFX5(HGMD Professional 2022.2). Among them, are five missense mutations (two pathogenic mutations and three uncertain significance mutations), four nonsense mutations (all pathogenic mutations), five splicing mutations (all pathogenic mutations), four small deletions mutations (three pathogenic mutations and one uncertain significance mutation) and one small insertions mutation (pathogenic mutation). The five splicing mutations are c.116 + 1G>A, c.151-1G>A, c.234-1G>A, c.556–2A>G and c.116 + 5G>A, respectively. Four of them are of the classical splice site variants, the last one (c.116 + 5 G>A) is a point mutation in a splice donor site, which results in 10 nucleotide upstream in exon two deletion in RFX5 mRNA (Villard et al., 1997). The splicing mutations reported in this study are novel.
Hematopoietic stem cell transplantation (HSCT) is currently the only available curative treatment for MHC-II deficiency. However, the success rate is reported to be poor in MHC class II-deficient patients (Small et al., 2013; Posovszky et al., 2019). On the basis of a previous study, two patients did not survive although they underwent HSCT after diagnosis. One patient died of diarrhea and Gram-negative sepsis within 8 days of transplant procedure and the other died post-HSCT due to lung damage and systemic candidiasis (Aluri et al., 2018). Even though the patients with MHC II deficiency do not express MHC II required for the rejection process, the residual host immunity is sufficient to cause rejection even with immunosuppression. In a recent study, it was suggested that the low survival rate in these patients may lie in the presentation of donor antigens by donor antigen-presenting cells to recipient Th cells leading to graft rejection. (Kallen and Pullarkat, 2015). Apart from poor engraftment, a high rate of post-HSCT death can be caused by diagnosis and/or treatment delays, multiorgan failure and persistent viral infections. More importantly, pregnant women with immunodeficiency fetuses mostly experience normal prenatal examination in imaging (ultrasound or MRI) and laboratory tests, like the mother in our study. Therefore, it is of great value to carry out prenatal diagnosis or PGD in such families.
In summary, we reported two novel splicing mutations (c.353 + 6T>G and c.757 + 1G>A) in RFX5 which are associated with MHC class II deficiency. The mutations were predicted to affect the RFX5 protein translation and even result in the premature of the protein. In addition, our study validates that the RT-PCR is necessary if the genotype–phenotype correlation was very consistent while only one classical splicing site gene mutation of autosomal recessive disease was detected. It contributed to a new genetic foundation for prenatal diagnosis and prenatal diagnosis of MHC class II deficiency.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding authors.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee of Women’s Hospital, School of Medicine Zhejiang University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
MD conceived of the study, participated in its design and revised the manuscript; ZL analyzed the clinical data; SC collected clinical data and drafted the manuscript; YX carried out the RT-PCR and revised the manuscript; YQ extracted the genomic DNA and designed primers; All authors have read and approved the final manuscript.
Funding
This work was supported by the Key Research and Development Program of Zhejiang Province (grant numbers 2019C03025), Zhejiang Provincial Natural Science Foundation of China (grant numbers LY22H110004) and the National Natural Science Foundation of China (grant numbers 81901382).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aluri, J., Gupta, M., Dalvi, A., Mhatre, S., Kulkarni, M., Hule, G., et al. (2018). Clinical, immunological, and molecular findings in five patients with major histocompatibility complex class II deficiency from India. Front. Immunol. 9, 188. doi:10.3389/fimmu.2018.00188
Ben-Mustapha, I., Ben-Farhat, K., Guirat-Dhouib, N., Dhemaied, E., Larguèche, B., Ben-Ali, M., et al. (2013). Clinical, immunological and genetic findings of a large tunisian series of major histocompatibility complex class II deficiency patients. J. Clin. Immunol. 33, 865–870. doi:10.1007/s10875-013-9863-8
Clarridge, K., Leitenberg, D., Loechelt, B., Picard, C., and Keller, M. (2016). Major histocompatibility complex class II deficiency due to a novel mutation in RFXANK in a child of Mexican descent. J. Clin. Immunol. 36, 4–5. doi:10.1007/s10875-015-0219-4
Clement, L. T. (1990). The class II major histocompatibility complex antigen deficiency syndrome: Consequences of absent class II major histocompatibility antigens for lymphocyte differentiation and function. J. Invest. Dermatol. 94, 118S–121s. doi:10.1111/1523-1747.ep12876078
Djidjik, R., Messaoudani, N., Tahiat, A., Meddour, Y., Chaib, S., Atek, A., et al. (2012). Clinical, immunological and genetic features in eleven Algerian patients with major histocompatibility complex class II expression deficiency. Allergy Asthma Clin. Immunol. 8, 14. doi:10.1186/1710-1492-8-14
El Hawary, R. E., Mauracher, A. A., Meshaal, S. S., Eldash, A., Abd Elaziz, D. S., Alkady, R., et al. (2019). MHC-II deficiency among Egyptians: Novel mutations and unique phenotypes. J. Allergy Clin. Immunol. Pract. 7, 856–863. doi:10.1016/j.jaip.2018.07.046
Farrokhi, S., Shabani, M., Aryan, Z., Zoghi, S., Krolo, A., Boztug, K., et al. (2018). MHC class II deficiency: Report of a novel mutation and special review. Allergol. Immunopathol. 46, 263–275. doi:10.1016/j.aller.2017.04.006
Garvie, C. W., Stagno, J. R., Reid, S., Singh, A., Harrington, E., and Boss, J. M. (2007). Characterization of the RFX complex and the RFX5(l66a) mutant: Implications for the regulation of MHC class II gene expression. Biochemistry 46, 1597–1611. doi:10.1021/bi6023868
Hanna, S., and Etzioni, A. (2014). MHC class I and II deficiencies. J. Allergy Clin. Immunol. 134, 269–275. doi:10.1016/j.jaci.2014.06.001
Kallen, M. E., and Pullarkat, S. T. (2015). Type II bare lymphocyte syndrome: Role of peripheral blood flow cytometry and utility of stem cell transplant in treatment. J. Pediatr. Hematol. Oncol. 37, e245–e249. doi:10.1097/MPH.0000000000000278
Lum, S. H., Anderson, C., Mcnaughton, P., Engelhardt, K. R., Mackenzie, B., Watson, H., et al. (2020). Improved transplant survival and long-term disease outcome in children with MHC class II deficiency. Blood 135, 954–973. doi:10.1182/blood.2019002690
Masternak, K., Muhlethaler-Mottet, A., Villard, J., Zufferey, M., Steimle, V., and Reith, W. (2000). CIITA is a transcriptional coactivator that is recruited to MHC class II promoters by multiple synergistic interactions with an enhanceosome complex. Genes Dev. 14, 1156–1166. doi:10.1101/gad.14.9.1156
Nekrep, N., Jabrane-Ferrat, N., Wolf, H. M., Eibl, M. M., Geyer, M., and Peterlin, B. M. (2002). Mutation in a winged-helix DNA-binding motif causes atypical bare lymphocyte syndrome. Nat. Immunol. 3, 1075–1081. doi:10.1038/ni840
Nonoyama, S., Etzioni, A., Toru, H., Ruggerie, D. P., Lewis, D., Pollack, S., et al. (1998). Diminished expression of CD40 ligand may contribute to the defective humoral immunity in patients with MHC class II deficiency. Eur. J. Immunol. 28, 589–598. doi:10.1002/(SICI)1521-4141(199802)28:02<589::AID-IMMU589>3.0.CO;2-J
Plaeger-Marshall, S., Haas, A., Clement, L. T., Giorgi, J. V., Chen, I. S., Quan, S. G., et al. (1988). Interferon-induced expression of class II major histocompatibility antigens in the major histocompatibility complex (MHC) class II deficiency syndrome. J. Clin. Immunol. 8, 285–295. doi:10.1007/BF00916557
Posovszky, C., Sirin, M., Jacobsen, E., Lorenz, M., Schwarz, K., Schmidt-Choudhury, A., et al. (2019). Persisting enteropathy and disturbed adaptive mucosal immunity due to MHC class II deficiency. Clin. Immunol. 203, 125–133. doi:10.1016/j.clim.2019.04.012
Reith, W., and Mach, B. (2001). The bare lymphocyte syndrome and the regulation of MHC expression. Annu. Rev. Immunol. 19, 331–373. doi:10.1146/annurev.immunol.19.1.331
Sage, A. P., Murphy, D., Maffia, P., Masters, L. M., Sabir, S. R., Baker, L. L., et al. (2014). MHC Class II-restricted antigen presentation by plasmacytoid dendritic cells drives proatherogenic T cell immunity. Circulation 130, 1363–1373. doi:10.1161/CIRCULATIONAHA.114.011090
Scholl, T., Mahanta, S. K., and Strominger, J. L. (1997). Specific complex formation between the type II bare lymphocyte syndrome-associated transactivators CIITA and RFX5. Proc. Natl. Acad. Sci. U. S. A. 94, 6330–6334. doi:10.1073/pnas.94.12.6330
Small, T. N., Qasim, W., Friedrich, W., Chiesa, R., Bleesing, J. J., Scurlock, A., et al. (2013). Alternative donor SCT for the treatment of MHC class II deficiency. Bone Marrow Transpl. 48, 226–232. doi:10.1038/bmt.2012.140
Steimle, V., Durand, B., Barras, E., Zufferey, M., Hadam, M. R., Mach, B., et al. (1995). A novel DNA-binding regulatory factor is mutated in primary MHC class II deficiency (bare lymphocyte syndrome). Genes Dev. 9, 1021–1032. doi:10.1101/gad.9.9.1021
Villard, J., Peretti, M., Masternak, K., Barras, E., Caretti, G., Mantovani, R., et al. (2000). A functionally essential domain of RFX5 mediates activation of major histocompatibility complex class II promoters by promoting cooperative binding between RFX and NF-Y. Mol. Cell. Biol. 20, 3364–3376. doi:10.1128/mcb.20.10.3364-3376.2000
Villard, J., Reith, W., Barras, E., Gos, A., Morris, M. A., Antonarakis, S. E., et al. (1997). Analysis of mutations and chromosomal localisation of the gene encoding RFX5, a novel transcription factor affected in major histocompatibility complex class II deficiency. Hum. Mutat. 10, 430–435. doi:10.1002/(SICI)1098-1004(1997)10:6<430::AID-HUMU3>3.0.CO;2-H
Keywords: RFX5, MHC-II deficiency, splicing mutation, DNA-binding domain, whole exome sequencing
Citation: Chen S, Xu Y, Qian Y, Li Z and Dong M (2022) Case Report: Novel splicing mutations in RFX5 causing MHC class II deficiency. Front. Genet. 13:978688. doi: 10.3389/fgene.2022.978688
Received: 26 June 2022; Accepted: 08 September 2022;
Published: 07 October 2022.
Edited by:
Fatma Savran Oguz, Istanbul University, TurkeyReviewed by:
Afagh Alavi, University of Social Welfare and Rehabilitation Sciences, IranKlara Dalva, Tissue Typing Laboratory, Turkey
Copyright © 2022 Chen, Xu, Qian, Li and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Minyue Dong, ZG9uZ215QHpqdS5lZHUuY24=; Zhaohui Li, bGk2MzM2QDE2My5jb20=
†These authors have contributed equally to this work