
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet., 10 August 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.971484
This article is part of the Research TopicThe Intersection of Gene Regulation and Metabolism in Cardiovascular DiseaseView all 9 articles
 Nicholas W.S. Chew1*†
Nicholas W.S. Chew1*† Bryan Chong2
Bryan Chong2 Cheng Han Ng2
Cheng Han Ng2 Gwyneth Kong2
Gwyneth Kong2 Yip Han Chin2
Yip Han Chin2 Wang Xiao3,4,5
Wang Xiao3,4,5 Mick Lee3,4,5
Mick Lee3,4,5 Yock Young Dan2,6,7
Yock Young Dan2,6,7 Mark D. Muthiah2,6,7
Mark D. Muthiah2,6,7 Roger Foo1,2,3,4,5*
Roger Foo1,2,3,4,5*The ongoing debate on whether non-alcoholic fatty liver disease (NAFLD) is an active contributor or an innocent bystander in the development of cardiovascular disease (CVD) has sparked interests in understanding the common mediators between the two biologically distinct entities. This comprehensive review identifies and curates genetic studies of NAFLD overlapping with CVD, and describes the colinear as well as opposing correlations between genetic associations for the two diseases. Here, CVD described in relation to NAFLD are coronary artery disease, cardiomyopathy and atrial fibrillation. Unique findings of this review included certain NAFLD susceptibility genes that possessed cardioprotective properties. Moreover, the complex interactions of genetic and environmental risk factors shed light on the disparity in genetic influence on NAFLD and its incident CVD. This serves to unravel NAFLD-mediated pathways in order to reduce CVD events, and helps identify targeted treatment strategies, develop polygenic risk scores to improve risk prediction and personalise disease prevention.
Non-alcoholic fatty liver disease (NAFLD) is characterised by the condition of excess fat in the liver in the absence of excessive alcohol consumption (Ludwig et al., 1980). It is frequently associated with metabolic syndrome, affecting at least a quarter of the population worldwide (Younossi et al., 2016). NAFLD is a disease spectrum consisting of non-alcoholic fatty liver (NAFL–i.e., hepatic fat accumulation), non-alcoholic steatohepatitis (NASH), fibrosis and cirrhosis (Kleiner et al., 2005). The main risk factors for NAFLD include hyperlipidaemia, diabetes mellitus, hypertension and obesity (Rinella, 2015), which are also key risk factors for cardiovascular disease (CVD) (Liu and Lu, 2014). The causal relationship between NAFLD and CVD is therefore of particular interest, although the pathogenesis of both is widely agreed to involve the complex interplay between genetic and environmental factors (Liu and Lu, 2014). Liver fat accumulation results from an imbalance between the influx of fat which includes fatty acids from adipose tissue and de novo lipogenesis from glucose, and fat efflux which involves oxidation and synthesis of very-low-density lipoprotein (VLDL) (Brouwers et al., 2020). Even though liver fat accumulation is the prerequisite for the progression of NASH, not all NAFL individuals progress through the stages. Lipotoxicity is a main driver of NASH progression, determined by the quantity and type of accumulated lipids and the defensive capabilities of the liver against lipotoxicity (Donnelly et al., 2005; Machado and Diehl, 2016). Apart from the natural history of NASH progression, our group was also able to depict significant improvement in liver histology in the absence of pharmacological interventions, suggesting that NASH is a disease that can regress spontaneously over time with even non-pharmacological measures (Ng et al., 2022).
NAFLD appears to be directly associated with CVD, independent of traditional cardiovascular confounders such as age, sex, state of hyperglycaemia and insulin resistance (Stefan et al., 2016). It is strongly associated with an increased risk of incident CVD (Targher et al., 2010; Anstee et al., 2013; Armstrong et al., 2014; Ballestri et al., 2014; Byrne and Targher, 2015; Lonardo et al., 2015), as well as a 64% higher risk of developing non-fatal and/or fatal CVD, compared to non-NAFLD individuals (Ong et al., 2008; Targher et al., 2016). Unsurprisingly, even though patients with NAFLD are at risk of end-stage liver disease, the majority of NAFLD patients die from CVD (Targher et al., 2010; Chalasani et al., 2012; Anstee et al., 2013).
The ongoing debate of whether NAFLD is an active contributor or an innocent bystander in the development of CVD therefore encompasses the appreciation for the common mediators between the two anatomically distinct entities. Exemplifying potential shared mediators between NAFLD and CVD is the pathogenesis of atherosclerotic lesions, which is the combination of endothelial dysfunction, lipoprotein accumulation in the vessel wall, inflammatory cell infiltrates, accumulation of foam cells and proliferation of the smooth muscle cells resulting in vulnerable plaques that are prone to rupture (Brouwers et al., 2020). NAFLD could contribute theoretically to all these pathophysiological stages, through the development of dyslipidaemia (characterised by elevated plasma triacylglycerols, low levels of high-density lipoprotein (HDL) cholesterol, high levels of small-dense low-density lipoprotein (LDL) particles (Adiels et al., 2006; Adiels et al., 2008; Defilippis et al., 2013; Do et al., 2013; Holmes et al., 2017; Burgess et al., 2018), low-grade inflammation (involving IL-1β in the pathogenesis of NASH and CVD) (Ridker et al., 2017; Mirea et al., 2018), and thrombogenicity (Alessi et al., 2003; Song et al., 2017) (with high levels of plasminogen activator inhibitor type-1 [PAI-1] in NASH, that is also an important component in the fibrinolytic system). Some NAFLD susceptibility genes are associated with CVD, representing possible components mediating between both entities. A systematic analysis of the human genetics for both conditions may therefore offer important therapeutic strategies to treat NAFLD and offset the greater CVD risk. Simple steatosis indeed is not as ‘simple’ or benign as its name suggests, with fat accumulation driving both the overproduction of lipoproteins and atherosclerosis. Whilst NAFLD and CVD share similar susceptibility genes, certain genes have displayed opposing correlations between polymorphism and the 2 different diseases. For example, as both the phospholipase domain-containing protein 3 (PNPLA3) and transmembrane 6 superfamily 2 (TM6SF2) regulate VLDL particle production, the genetic polymorphism can result in increased intrahepatic triglyceride (TG) with increased risk of NAFLD, but reduced circulatory cholesterol and LDL which lends protection against coronary artery disease (CAD) (Liu et al., 2017; Simons et al., 2017). Delineating these unique pathways will be important in unravelling NAFLD-mediated pathways in order to reduce CVD events, and facilitate future studies in identifying targeted treatment strategies (Brouwers et al., 2020).
This review aims to give insight into the genetic interactions between CVD and NAFLD, and elaborate on the experimental evidence that supports a causal relationship between the two entities. We discuss the clinical implications of the findings, which may guide future therapeutic trials targeting common genetic pathways in order to simultaneously lower the risk of NAFLD and CVD. Here, CVD described in relation to NAFLD are CAD, cardiomyopathy and atrial fibrillation (AF).
Genome-wide genotyping arrays have from the start contributed to the large spectrum of genetic variants with subsequent imputation of millions of further variants leading to a wide breadth of understanding of the genetic architecture of CAD (Khera and Kathiresan, 2017; Erdmann et al., 2018). Ultra-large scale genome-wide association studies (GWASs) and meta-analyses report loci in relation to atherosclerosis, demonstrating the progressive insights for population-specific and trans-ancestry risk factors in CAD (Lu et al., 2012; Takeuchi et al., 2012; Koyama et al., 2020). This is proving utility for polygenic risk scores in CAD or myocardial infarction (MI), depending on the accuracy of predicting the effect size of risk alleles that varies with individual genetics (Gola et al., 2020). More than 200 loci have been associated with CAD and MI since 2007, with comprehensive identification of their risk alleles, and allele frequencies (Erdmann et al., 2018; Kessler and Schunkert, 2022). Initially, the impact of genetic hits lay with traditional cardiovascular risk factors: hypertension and hyperlipidaemia. Currently, approximately half of the loci explains the impact on pathophysiological pathways (such as nitric oxide signalling), and others are identified as new “drivers” of atherosclerosis and MI (Wobst et al., 2018; Dang et al., 2020). Several NAFLD susceptibility genes have a differential effect on plasma lipids and concurrent CAD risks, suggesting that plasma lipids are indeed essential mediators between NAFLD and CVD. This may guide the design of anti-NAFLD drugs targeting lipid metabolism and CAD risks (Brouwers et al., 2020). A summary of genomic variants overlapping between NAFLD and CVD is shown in Table 1.

TABLE 1. Summary of genomic variants identified in both NAFLD and CVD.
Lipoprotein lipase (LPL) is an essential enzyme for metabolising TG-rich lipoproteins. Populations harbouring common, noncoding, and rare loss of function variants at this genetic locus, have been associated with elevated CAD risk. In addition to the LPL gene, crucial endogenous regulators of LPL activity are associated with CAD, including APOC3 (Pollin et al., 2008; Willer et al., 2008; Crosby et al., 2014). APOC3, expressed in the liver, is a key regulator of plasma TG levels, shown to adversely affect cardiovascular event risks. APOC3 lowers LPL activity, and inhibits TG hydrolysis into VLDL particles and chylomicrons in the plasma, increasing plasma TG and the resultant increased risk of CAD. Rare mutations disrupting APOC3 function are associated with 39% lower plasma TG compared to noncarriers, as well as a 40% reduction in CAD risks (Crosby et al., 2014). 5% of heterozygous carriers of the null gene APOC3 mutation express half the amount of APOC3 in noncarriers. Mutation carriers have lower fasting, postprandial serum TG, as well as lower levels of LDL-cholesterol and higher HDL-cholesterol. As a result, subclinical atherosclerosis, determined by coronary artery calcification, were less common in carriers than noncarriers. This proposes that lifelong deficiency of APOC3 provides a cardioprotective impact on carriers (Pollin et al., 2008).
Polymorphisms in APOC3 are also associated with NAFLD and insulin resistance. In a cohort of Asian Indian men (Cooper, 1985; Weintraub et al., 1987; Petersen et al., 2010), carriers of APOC3 variant alleles (C-482T, T-455C, or both) demonstrated a 30% increase in fasting plasma apolipoprotein C3 concentration, compared to wild-type homozygotes. APOC3 variant carriers had 60% increased fasting plasma TG concentration. Increased plasma APOC3 concentration inhibits LPL and TG clearance, predisposing to increased fasting and postprandial hypertriglyceridemia as a result of increased chylomicron-remnant particles. Elevated concentrations of circulating chylomicron-remnant particles are taken up by the liver through a receptor-mediated process, leading to NAFLD and hepatic insulin resistance. Moreover, increased hepatocellular diacylglycerol concentrations in NAFLD result in protein kinase C epsilon isoform activation that reduces insulin signalling and hepatic insulin resistance (Samuel et al., 2004; Zhang et al., 2007). 38% of subjects with APOC3 variant alleles had NAFLD, compared to 0% among wild-type homozygotes, and those with NAFLD had marked insulin resistance. This observation was also confirmed in a second cohort of non-Asian Indian men (Petersen et al., 2010). Interestingly, other cohorts of European ancestry (Kozlitina et al., 2011; Sentinelli et al., 2011; Hyysalo et al., 2012) did not identify any significant association between APOC3 polymorphisms, NAFLD and insulin resistance.
In the current therapeutic pipeline, selective antisense APOC3 inhibitor, Volanesorsen, produces a dose-dependent 31–71% decrease in TG, used for targeting TG reduction in familial chylomicronemia syndrome (Pollin et al., 2008; Willer et al., 2008; Crosby et al., 2014; Gaudet et al., 2015). A randomised, placebo-controlled phase 2 trial demonstrated dose-dependent and prolonged decrease in levels of plasma APOC3 when the drug was administered as a monotherapy or as an add-on to fibrates. Studies are needed to examine whether TG reduction via antisense APOC3 inhibition translates also to improved liver-related outcomes given the close links between hepatic lipoprotein metabolism and NAFLD (Heeren and Scheja, 2021).
The TRIB1 gene encodes for the Tribbles homolog 1 protein, which is part of a family of phosphoproteins that play a role in cell function regulation. The single nucleotide polymorphisms (SNPs) in or near TRIB1 (such as rs17321515) have demonstrated significant associations with increased circulatory triglycerides and CAD. Another promising target with associations for cardiovascular traits is the recently identified TRIB1 locus (Teslovich et al., 2010; IBC 50K CAD Consortium, 2011; Deloukas et al., 2013). Regulatory mechanisms associated with TRIB1 on circulatory lipids and immune cells support the notion that TRIB1 is associated with the development of CVD. Atherosclerosis develops from vascular wall damage, endothelial cell dysfunction or death, inflammatory cytokine and chemokine production, oxidised LDL particles, formation of foam cells, and subsequently plaque formation (Jaipersad et al., 2014; Grootaert et al., 2018). In atherosclerosis pathophysiology, macrophages undergo M2 to M1 transdifferentiation (Deloukas et al., 2013). TRIB1 has been associated with M2 macrophage transition, offering an important role in homeostatic maintenance and repair of damaged tissue (Khallou-Laschet et al., 2010; Baitsch et al., 2011; Satoh et al., 2013). Hence TRIB1 knockout impairs M2 macrophage mediated repair, leading to metabolic disease and CVD. Furthermore, mitogen-activated protein kinase (MAPK) mediates inflammatory factor-mediated migration and vascular smooth muscle cell proliferation. TRIB1 negatively regulates MAPK activity by binding to MAPK kinase, thus reducing the chemotaxis of inflammatory factors and inhibiting vascular smooth muscle cell migration and proliferation (Manichaikul et al., 2012; Wang et al., 2015; Pan, 2017; Reustle and Torzewski, 2018).
Data from GWA meta-analyses (Zeggini et al., 2008; Newton-Cheh et al., 2009; Teslovich et al., 2010) demonstrated that the risk allele at the TRIB1 locus is associated with higher TG and lower HDL-cholesterol. TRIB1 affects lipid metabolism with an inverse correlation between hepatocellular TRIB1 expression and lipogenic gene expression. Thus TRIB1 expression reduces the risk of CVD through the reduction of TG, maintenance of steady-state phagocytosis, percentages of M2 macrophages, and inhibiting the chemotaxis of inflammatory factors and vessel wall damage (Zhang et al., 2021). Animal studies show that TRIB1 overexpression in wild-type mice lowered circulating TG levels (Burkhardt et al., 2010); conversely, targeted TRIB1 deletion in mice led to higher circulating TG. The risk of CAD is thus potentially downstream of TRIB1, which is likely to be mediated by the regulation of TRIB1 expression resulting in adverse lipid profiles (The, 2011). A study in the Chinese Han population (Liu et al., 2019a) demonstrated that TRIB1 rs17321515 AA + GA genotypes were significantly associated with CAD in the general population as well as in the NAFLD population, despite adjusting for important confounders. Moreover, TRIB1 rs17321515-A carriers displayed worse lipid profiles than non-carriers. TRIB1 rs17321515 GA + AA genotypes and TRIB1 rs2954029 TA + AA genotypes also independently increased NAFLD risk in the Chinese Han population (Liu et al., 2019b).
TRIB1 overexpression in mouse liver decreases plasma cholesterol, TG and VLDL secretion (Bauer et al., 2015; Soubeyrand et al., 2016). Together with Sin3A associated protein 18 (SAP18) and mSin3A, TRIB1 activates the transcription of microsomal triglyceride transporter protein (Makishima et al., 2015) (MTTP). Hence TRIB1 knockout in human hepatic cells reduces the expression of MTTP and APOB (Burkhardt et al., 2010; Makishima et al., 2015; Nagiec et al., 2015) (which is the main apolipoprotein component of VLDL and LDL). Conversely, TRIB1 has a functional role in lipogenesis, where TRIB1 overexpression downregulates the carbohydrate response element binding protein (ChREBP), which is a glucose-sensitive molecule important for hepatic lipogenesis (Iwamoto et al., 2015; Softic et al., 2017). TRIB1 knockout increases the expression of liver C/EBPα protein, thus increasing the production and accumulation of hepatic fat, resulting in liver cell damage, liver steatosis and NAFLD (Bauer et al., 2015; Kahali et al., 2015). Further studies on this treatment approach being transferred to human treatment can be the next important step (Kessler and Schunkert, 2021).
Apolipoprotein E (ApoE) is a constituent of lipoproteins with large variations due to cysteine-arginine exchanges. APOE gene variants give rise to apoE isoforms with six permutations of the apoE or ε variants–E2/2, E2/3, E2/4, E3/3, E3/4, E4/4 (Mahley, 2001). Common apoE variants account for approximately 4% of the variance in plasma cholesterol. Another variant of apoE (rs35136575) also affects LDL concentrations (Boerwinkle and Utermann, 1988).
ApoE facilitates the clearance of TG-rich (apoB-containing) lipoprotein remnants from the bloodstream into the liver. It affects vascular function through mechanisms such as inflammatory responses, platelet aggregation and inflammatory effects involving M2 macrophage phenotype transformation and proliferation of lymphocytes and T helper cells (Riddell et al., 1997; Baitsch et al., 2011; Zhang et al., 2011). As such, the apoE isoforms influence CVD risk from birth–for example, the hypolipidemic effect of apoE2 can be seen from childhood with lower LDL and higher HDL concentrations (Kallio et al., 1997; Isasi et al., 2000). The apoE4 isoform is associated with increased carotid intima-media thickness, LDL, Lp(a) and apoB levels (Granér et al., 2008; Luo et al., 2017). The cardiovascular effect of common variants is observed from its influence on the lipid profile with the potential of more severe and pathological sequelae. Dysbetalipoproteinemia arises in middle-aged males and post-menopausal females, and remnant lipoproteins accumulate due to impaired clearance or overproduction of lipoproteins. TG accumulation in dysbetalipoporteinemia increases overall atherogenicity. Dominantly inherited mutations offer the diagnosis of familial hypercholesterolemia (Marais, 2019).
In NASH patients, there was an increased prevalence of the APOE e3 allele compared to healthy controls: APOE polymorphism was significantly associated with NASH, particularly the APOE3/3 genotype (Sazci et al., 2008). In animal studies, loss of ApoE reduced susceptibility to obesity and NAFLD (Naik et al., 2013). The APOE e3 allele (Sazci et al., 2008) is also significantly more prevalent in biopsy-proven NASH patients compared to controls, whereas the APOE e2 allele appears protective against NAFLD (Demirag et al., 2007). Currently, the APOE3/3 genotype is recognised to play a role in the aetiopathogenesis of NASH (Sazci et al., 2008). Larger studies are needed to examine this association across different ethnicities.
Arising from the above, some have proposed genetic testing of common variants for clinical screening, with apoE2 protective against and apoE4 at increased risk of vascular disease (Jacobsen et al., 2010; Zhao et al., 2017). ApoE may provide novel therapeutic targets for the treatment of atherosclerosis. The apoE mimetic peptide EpK enhances cholesterol efflux from cells and has beneficial effects in animal model of atherosclerosis (Zhao et al., 2011). Another hepatic-expressed peptide, hEp, lowers lipoprotein levels and also protects against atherosclerosis (Xu et al., 2016). An individual’s apoE status may indeed provide valuable insight into cardiovascular health, on the backdrop of hyperlipidaemia and CVD. Furthermore, the APOE genotype may also affect response to statin therapy, with the largest HDL increase in apoE2 carriers. Fibrates produces the largest decrease in TG in apoE2 carriers, and the slowest effect in apoE4 carriers (Buckley et al., 2004).
Many CAD susceptibility genes are associated with concentrations of circulating lipid species. One locus, phosphatidylethanolamine N-methyltransferase (PEMT), encodes for the enzyme for lipid biosynthesis (Fernandez et al., 2013). PEMT catalyses all three methylation steps in the conversion of phosphatidyethanolamine to phosphatidylcholine, using S-adenosylmethionine as a methyl group donor. PEMT is largely expressed in the liver and accounts for 30% of liver phosphatidylcholine production (Vance, 2014). A functional SNP can lead to the loss of function with amino acid replacement Val175Met in PEMT which alters the catalytic function for the conversion of phosphatidyethanolamine to phosphatidylcholine. Phosphatidylcholine is important for VLDL formation for hepatic TG secretion. The V175M results in loss of function, associated with diminished PEMT activity, with increased propensity to lipid accumulation (Noga et al., 2002). Carriers of the PEMT risk allele indeed show decreased levels of multiple glycerophospholipids, such as the cardioprotective lipid species LPC 16:0 and/or LPC 20:4 (Fernandez et al., 2013). Here, integrative analysis of genomics with lipidomics advanced the insights for underlying mechanisms and pathogenesis of CVD.
In the liver, the PEMT catalyses three methylation steps in the conversion of phosphatidyethanolamine to phosphatidylcholine (Browning and Horton, 2004). The fast onset of liver cell damage in a methionine and choline deficient diet, and liver-specific expression of PEMT may be attributed to the high demand for choline and phosphatidylcholine (Browning and Horton, 2004) that is important for the maintenance of normal liver function (Vance and Ridgway, 1988; Vance et al., 1997). The PEMT and CDP-choline pathways are two important regulatory pathways in maintaining phosphatidylcholine homeostasis in hepatocytes. Phosphatidylcholine homeostasis is necessary as it is the primary phospholipid of all classes in humans, necessary for VLDL secretion (Vance and Vance, 1985; Yao and Vance, 1988, 1989; Vermeulen et al., 1997; Dong et al., 2007). The extent and rate of TG accumulation in hepatocytes is determined by hepatocyte efficacy for excreting VLDL. In addition, the single nucleotide polymorphism G433T in microsomal TG transfer protein (MTTP) gene also influences the degree and rate of fat deposition in hepatocytes (Namikawa et al., 2004). TG, together with cholesterol and phospholipids, assemble together to form apolipoprotein B in hepatocytes and is secreted as VLDL. As such, phosphatidylcholine deficiency and MTTP activity impairment (Yao and Vance, 1988, 1989; Noga et al., 2002) adversely affects VLDL secretion from hepatocytes, causing accumulation in hepatocytes, which is at the core of NASH pathophysiology. Furthermore, aberrant sterol regulatory element-binding proteins (SREBPs) activity can lead to excess stored fat and obesity, through the activation of genes involved in lipid synthesis, trafficking and homeostasis. A study suggested that the maturation of nuclear, transcriptionally active SREBP-1 is influenced by phosphatidylcholine (Walker et al., 2011). Therefore, genetic conditions (such as PEMT Val175Met) that limit phosphatidylcholine production can in turn activate SREBP-1, and as a consequence increasing the size and amount of lipid droplets, and exacerbating the risk of developing metabolic diseases (Walker et al., 2011). A separate study revealed that in PEMT knockout mice, the ratio of phosphatidylcholine/phosphatidylethanolamine is decreased, leading to loss of membrane integrity, which has important clinical implications in the progression of NAFLD (Li et al., 2006). Overall, the V175M loss-of-function mutation in the human PEMT gene confers susceptibility to NASH (Song et al., 2005), and this has been shown in both the Chinese (Zhou et al., 2010) and Japanese population (Dong et al., 2007). In the NASH group, Val175Met carriers have significantly lower body mass index (BMI) with more non-obese patients, compared to homozygotes of wild type PEMT (Dong et al., 2007). The PEMT Val175Met variant may be a potential molecular target for novel NASH therapy.
The insulin receptor substrate-1 (IRS-1) gene encodes a protein which is phosphorylated by insulin receptor tyrosine kinase and plays an important role in the insulin-stimulated signal transduction pathway. Both hyperglycaemia and insulin resistance can downregulate IRS-1, which is a pivotal intermediary in insulin/IGF-1 signalling (Xi et al., 2019). IRS-1 is essential for maintaining vascular smooth muscle cell differentiation. Hyperglycaemia or insulin resistance-induced IRS-1 downregulation decreased p53/Kruppel like factor 4 (KLF4) association and increased dedifferentiation and proliferation of vascular smooth muscle cells. Therefore, enhancing IRS-1 dependent p53 stabilisation may retard atherosclerosis progression, particularly in individuals with hyperglycaemic or insulin resistance states (Xi et al., 2019). Conversely, genetic variations near IRS-1 (such as the major allele of rs2972146 (Teslovich et al., 2010), rs2943641 (Rung et al., 2009), rs2943634 (Samani et al., 2007)), resulting in decreased IRS-1 expression, are associated with impaired metabolic profile, such as an increased visceral to subcutaneous fat ratio, insulin resistance, hyperlipidaemia, decreased adiponectin levels, thus enhancing the risk of diabetes and CAD (Samani et al., 2007; Rung et al., 2009). A GWA meta-analysis explained that there is reduced IRS-1 expression, associated with the genetic variations near IRS-1, in major insulin target tissues such as adipose tissue and muscle. IRS-1 genetic variations have a deleterious effect on insulin resistance, reduced ability to store subcutaneous fat, and disrupted insulin signalling in liver and muscle, resulting in ectopic deposition of lipids (Stumvoll and Jacob, 1999; Arner, 2002).
Insulin resistance, common co-existing with NAFLD, is contributed by inflammatory factors binding to IRS for ubiquitin-mediated degradation via the activation of Suppressors of Cytokine Signalling 3 (SOCS3). This leads to insulin desensitisation. Studies have shown that the frequency of the Arg allele of the Gly972Arg polymorphism of IRS-1 gene was significantly increased in NAFLD. Gly972Arg carriers are at significantly higher risk of NAFLD (Bhatt and Guleria, 2021). The association between IRS-1 gene polymorphism and incident type 2 diabetes has been demonstrated in both the Asian and Caucasian populations (Dongiovanni et al., 2010; Li et al., 2016). The IRS-1 (Gly972Arg) polymorphism also affects insulin receptor activity, predisposing to hepatocyte injury and decreased hepatic insulin signalling in NAFLD individuals (Dongiovanni et al., 2010). In terms of treatment, diabetic NAFLD patients are treated with PPARγ-agonists thiazolidinedione, which acts as an insulin sensitizer that reduces lipid release and increases lipid uptake, storage and reduces hepatic gluconeogenesis (Vanni et al., 2010). However, polymorphisms in IRS-1 can affect insulin receptor activity, and can significantly predispose biopsy-proven NAFLD individuals to worse disease severity (Dongiovanni et al., 2010).
GCKR is a NAFLD susceptibility gene that encodes liver-specific glucokinase regulatory protein (GKRP), which plays an important role in de novo lipogenesis and development of NAFLD (Donnelly et al., 2005; Brouwers et al., 2015). Several studies have demonstrated that gene variants (rs1260326, rs780094, rs780093) are associated with CAD (Simons et al., 2018), higher serum triacylglycerols, lower serum HDL cholesterol and the presence of small dense LDL particles (Sookoian and Pirola, 2011; Brouwers et al., 2015; Tabas et al., 2015; Lauridsen et al., 2018). This lipid profile is an example of vertical pleiotropy or mediation, in which the genetic effect on lipids is via the liver given that this lipid phenotype is reported to be the consequence of NAFLD (Defilippis et al., 2013; Brouwers et al., 2015). Furthermore, the metabolic effect of these common variants of GCKR can be different in patients with type 2 diabetes, as glucokinase has been shown to be more active when plasma glucose levels are within the diabetic range. This may allude to the difference in effect size of the GCKR allele variant on plasma TG being more pronounced in diabetic patients compared to non-diabetic individuals (Agius, 2008; Liu et al., 2012). In fact, Nynke Simons and colleagues (Simons et al., 2016) have reported that rs1260326 interacts with indices of glucose metabolism (such as fasting plasma glucose, Hba1c, glucose tolerance states) which are prominent components of diabetic dyslipidaemia (Taskinen, 2003). Those with moderately controlled diabetes (Hba1c 8.0%) carrying 2 T alleles tended to have higher plasma TG levels compared to homozygous carriers of the C allele, whilst no differences were noted in healthy individuals (Simons et al., 2016).
The molecular pathways that give rise to fatty liver involves excessive hepatic glucose levels and increased lipogenesis (Beer et al., 2009). GCKR rs1260326-T associates with decreased GCKR activity to inhibit glucokinase, leading to increased hepatic glucose uptake, decreased fatty acid oxidation and enhanced lipogenesis. Hepatic fatty acids can either be converted to TG and stored in hepatic lipid droplets, or secreted in VLDL particles (Beer et al., 2009; Hodson and Frayn, 2011). Therefore, decreased GCKR activity contributes to the progression of steatosis or levels of circulating lipids. In line with this observation, GCKR rs1260326-T increases NAFLD risk and increases the concentrations of apolipoprotein B which contains lipoprotein particles and TG. Moreover, GCKR rs1260326-T is associated with increased glycolysis-related metabolites, circulating fatty acids and elevated fatty acid saturation, in combination with the increased overall glycolytic and lipogenic activities (Beer et al., 2009; Rees et al., 2012; Santoro et al., 2015). Moreover, in a large GWAS, GCKR rs789904-T was associated with hepatic steatosis diagnosed by computed tomography and biopsy-proven NASH involving lobular inflammation and fibrosis (Speliotes et al., 2011). Moreover, GCKR rs1260326-T was recognised as an important factor for inter-individual variation in liver fat (Santoro et al., 2012; Dongiovanni et al., 2015b; Goffredo et al., 2016).
There is always a theoretical concern regarding the development of drugs, with the potential adverse effect of worsening NAFLD. The common GCKR variant has a more pronounced effect on hepatic fat accumulation and plasma triacylglycerols in individuals with obesity (Simons et al., 2016; Stender et al., 2017) and hyperglycaemia. It is plausible that individuals with obesity or poorly controlled diabetes may be more prone to adverse effects of liver-specific glucokinase activators, which increase hepatic glucose intake, leading to increased accumulation of hepatic adiposity through enhanced de novo lipogenesis (Lloyd et al., 2013; Hodson and Brouwers, 2018; Zhu et al., 2018). Studies are needed to provide greater clarity on these potential adverse effects due to the intertwined effects of NAFLD, plasma lipid levels and CAD risks.
Another NAFLD susceptibility gene PNPLA3 has been shown to associate with all stages of NAFLD (Romeo et al., 2008; Sookoian and Pirola, 2011). The common variant PNPLA3 rs738409 was studied in a Mendelian randomisation (MR) study to evaluate the causal relationship between NAFLD and CAD (Lauridsen et al., 2018). Several studies demonstrated that the rs738409-G allele, which predisposes to NAFLD, confers modest protection from CAD (Liu et al., 2017; Simons et al., 2017). A plausible explanation for this apparent paradox may be the function of the gene product. It has been proposed that the true function of PNPLA3 is in lipid droplet remodelling and VLDL production (Trépo et al., 2016), and PNPLA3 variants are associated with lower plasma lipid concentrations for both triacylglycerols and LDL cholesterol (Liu et al., 2017), explaining the negative correlation between the polymorphism and CAD. The rs738409-G polymorphism for PNPLA3 (Ile148Met) reduces hydrolyse activity, impairing intrahepatic TG breakdown which hampers VLDL particle production and secretion (Anstee and Day, 2015). Therefore, the PNPLA3 minor allele is correlated with reduced plasma lipids (Tang et al., 2015), and the negative association between PNPLA3 and CAD.
However as indicated, the same I148M polymorphism is associated with the risk of developing NASH, as well as advanced fibrosis and cirrhosis. The possible mechanisms for this association are that the wild-type PNPLA3 protein hydrolyses TG and retinyl esters, whilst the rs738409-G variant results in a loss of function, causing TG accumulation and retinyl esters in lipid droplets in hepatic stellate cells and hepatocytes (Huang et al., 2011; Pingitore et al., 2014; Pirazzi et al., 2014). This predisposes the liver to cellular injury, and hinders extracellular protein release in the hepatic stellate cells that has beneficial effects in preventing fibrosis progression, portal hypertension and tumorigenesis (Pingitore et al., 2016). Moreover, hepatic stellate cells carrying PNPLA3 variant demonstrated activated Yap/Hedgehog pathways, enhancing the altered anaerobic glycolysis and synthesis of Hedgehog and Yap signalling (Bruschi et al., 2020). PNPLA3 I148M isoform can also further prevent the binding of α/β hydrolase domain-containing 5 (ABHD5) and Adipose Triglyceride Lipase (ATGL) as it escapes ubiquitylation, that plays a role in TG hydrolysis (Basu Ray, 2019). With PNPLA3-I148M overexpression, the lysophosphatidic acid acyl transferase activity increases, suggestive of a gain-of-function mutation that promotes lipid synthesis (Mcmahon et al., 2014). This variant has also been positively correlated with alcoholic liver diseases, chronic hepatitis C-related cirrhosis and hepatocellular carcinoma (Bruschi et al., 2017). The significant association between PNPLA3 rs738409-G and NAFLD was first reported by Romeo et al. (2008). The frequency of PNPLA3 I148M is higher in the Hispanic population, and lower in European Americans and African Americans (Bruschi et al., 2017). It is also associated with hepatic fat, independently replicated in several studies of various ancestries and geographical disparate cohorts (Trépo et al., 2016). Sookoian and colleagues confirmed the strong link between I148M PNPLA3 and NAFLD severity, determined by liver biopsy, after adjusting for important confounders such as body mass index, age, sex and insulin sensitivity. This genetic variant was not only associated with the increased risk of simple steatosis, but also NASH, advanced fibrosis and cirrhosis in the Japanese, Italians, Malaysians and Americans populations (Valenti et al., 2010a; Valenti et al., 2010b; Hotta et al., 2010; Rotman et al., 2010; Zain et al., 2012). A further interesting observation was that the recurrence of NASH post-transplantation was significantly linked to the donor genotype, rather than the host. With higher prevalence of NASH recurrences in liver recipients who received the homozygous I148M allele, this suggests that PNPLA3 exerts its core physiological function primarily in the liver (Miyaaki et al., 2018). Reports also highlight differences in the lipid profiles between “metabolic NAFLD” and “PNPLA3 NAFLD”, with higher levels of polyunsaturated TGs in the genetic “PNPLA3 NAFLD” compared to the metabolic-related NAFLD, thus proposing that these two entities may be distinct drivers of fatty liver diseases.
The rs58542926-T allele of the TM6SF2 gene has cardioprotective effects (Dongiovanni et al., 2015a; Simons et al., 2017). TM6SF2 is involved in VLDL production, and variants of the gene are associated with reduced plasma LDL-cholesterol and triacylglycerols (Liu et al., 2017). Similar to the PNPLA3 polymorphism, the TM6SF2 SNP has a negative relationship with CAD.
Previous GWAS reported TM6SF2 SNPs associated with increased risk of NAFLD. TM6SF2 acts as a liver fat metabolism regulator, affecting TG secretion and hepatic fat droplet contents (Mahdessian et al., 2014). The TM6SF2 rs5854926 coding variant (TM6SF2 E167K) is associated with reduced TM6SF2 expression in the liver, higher serum alanine aminotransferase levels and TG content, as well as reduced total cholesterol and LDL (Holmen et al., 2014; Kozlitina et al., 2014; Dongiovanni et al., 2015b; Grandone et al., 2016). As NAFLD is caused by hepatic accumulation of TG, its elevated levels in the liver likely mediates the increased risk for the rs58542926-T allele (E167K) (Donnelly et al., 2005). Indeed, this allele is also an independent risk factor for liver steatosis (Kozlitina et al., 2014; Sookoian et al., 2015; Grandone et al., 2016). A meta-analysis revealed that the TM6SF2 rs5854926 was significantly associated with NAFLD in both the Asian and Caucasian populations (Chen et al., 2019).
The tumour necrosis factor-α (TNF-a) encodes a multifunctional proinflammatory cytokine that is part of the TNF superfamily. Its encoding gene is located in the short arm of chromosome 5 in the major histocompatibility complex class III region. Mostly secreted by macrophages, TNF is involved in the regulation of a large range of biological processes such as cell proliferation, differentiation, apoptosis and lipid metabolism (El-Tahan et al., 2016). The role of TNF-a in the pathophysiology of congestive heart disease (Levine et al., 1990; Mcmurray et al., 1991; Dutka et al., 1993; Katz et al., 1994). Has been reported in several studies. As genetic polymorphisms in TNF locus is related to inflammatory disease processes, the TNF-a gene polymorphisms and their associations with dilated cardiomyopathy (DCM) are of emerging interest (Kubota et al., 1998; Ito et al., 2000; Tiret et al., 2000). However, to date, there are conflicting data on the association between TNF-a polymorphisms and DCM. Alikasifoglu et al. (2003) examined Turkish patients with DCM but was not able to demonstrate any associations between TNF-a/-238 and -308 (G/A) polymorphisms and DCM, in parallel with several other findings (Kubota et al., 1998; Tiret et al., 2000). There were also no associations between TNF-a polymorphisms and ischemic heart disease (Herrmann et al., 1998). On the contrary, a recent study reported that the TNF-a promoter variant 2 (TNF2) (TNF-a/-308 A allele) was enriched in patients with end-stage non-ischemic DCM (Densem et al., 2002). Another report on heart transplant patients with severe symptomatic myocardial dysfunction found a higher proportion of TNF2 allele carriers in those with non-ischemic aetiologies, compared to those with ischemic cardiomyopathy. Those with non-ischemic cardiomyopathy had higher prevalence of TNF2 compared to healthy individuals (Densem et al., 2002). These preliminary findings propose a genetic predisposition in a small subgroup of patients who may potentially respond favourably to anti-TNF-a therapy (Densem et al., 2002).
A postulated reason for this discrepancy may be the different inclusion criteria of cardiomyopathy, with several studies that reported the lack of association between TNF-a polymorphisms and DCM mainly included patients with New York Heart Association (NYHA) class II or III, with only a small number of patients with end-stage heart failure. Moreover, due to the high mortality associated with end-stage heart failure, a sizeable proportion of patients may not have reached the hospital and therefore opportunities were lost in screening for the TNF-a polymorphism (Alikasifoglu et al., 2003). Another explanation may be the variability of TNF-a polymorphisms associated with DCM genesis in the different ethnic cohorts. For instance, Ito and colleagues reported that the TNF-a/-308 A allele was more prevalent in Japanese patients with idiopathic DCM (Ito et al., 2000), which was not found in the Turkish cohort (Alikasifoglu et al., 2003). Similarly, TNF-a/-238 (G/A) allele frequencies in the Turkish cohort differed from those in the France and Northern Ireland cohorts (Herrmann et al., 1998). Ethnic factors may have an important role in the variability of results, and further studies are needed to clarify this hypothesis.
Increased levels of TNF-α have also been associated with increased risk of NAFLD progression to NASH (Antuna-Puente et al., 2008; Marra and Bertolani, 2009), due to its correlation with insulin resistance and inflammatory milieu. The cytokine has a broad role in inflammation, autoimmunity, tumour apoptosis and metabolic dysregulation (Pfeffer, 2003). Candidate gene studies have identified several polymorphisms in the TNF-α gene promotor (Qidwai and Khan, 2011). The higher prevalence of the TNF-α/-238 promotor polymorphism in NAFLD or NASH patients, compared to controls, are reported in the Italian (Valenti et al., 2002), Mexican and Chinese (Hu et al., 2009; Zhou et al., 2010; Trujillo-Murillo et al., 2011) cohorts. In the Japanese, two other polymorphisms have instead been identified, -1031C and -863A (Tokushige et al., 2007). Of interest, there was an inverse effect on regulation of glucose and lipid homeostasis associated with the TNF-α/-238 polymorphism, demonstrating increased risk of impaired insulin sensitivity, but lower LDL-cholesterol and BMI (Naik et al., 2013). A genomic meta-analysis of 8 studies also confirmed the association between -238 polymorphisms in the TNF-α gene and NAFLD (Wang et al., 2012). The difference in the prevalence of various polymorphisms across the unique cohorts are likely underpinned by ethnic differences, the frequency of variations or lack of statistical power across different studies (Wong et al., 2008). Furthermore, TNF-a mRNA overexpression was also more prominent in patients with more significant fibrosis, compared to those with mild or non-existent fibrosis, thus elucidating the important role of the TNF-a system in the pathogenesis of NASH (Crespo et al., 2001).
The interleukin-6 (IL6) gene encodes a cytokine that plays an important role in inflammation and the maturation of B cells. The encoded protein is an endogenous pyrogen that is primarily produced in areas at acute and chronic inflammation. The functioning of this gene is implicated in a large range of inflammatory diseases (Marcus et al., 2008). In fact, the mechanism by which inflammation and stimulation of C-reactive protein secretion occurring in AF remains unclear. Nevertheless, animal studies have suggested that IL6 stimulates matrix metalloproteinase-2, mediating atrial remodelling in AF (Xu et al., 2004; Luckett and Gallucci, 2007). IL6 has reported significant correlation with increased left atrial size (Psychari et al., 2005), an important predictor for new onset AF. Moreover, polymorphisms in the promotor region of IL6 correlated with postoperative AF (Gaudino et al., 2003; Bittar et al., 2005). Cohort studies have shown that the CC genotype of the IL6–174 G/C IL6 polymorphism is associated with at least 2 fold increase in AF incidence, compared to GC and GG genotypes. Moreover, the same genotype (CC) is correlated with significant elevation of IL6 levels.
The IL6–174C genetic polymorphism was more prevalent in NAFLD, compared to controls. C carriers in NAFLD patients had higher homeostatic model assessment for insulin resistance (HOMA-IR) and fasting insulin compared to G carriers. Cohort studies of a Caucasian population demonstrated that the prevalence of IL6-174C variant was higher in NASH than NAFLD patients, and was associated with increased insulin resistance (Carulli et al., 2009). This is in line with several reports that support the association of the C allele with diabetes, insulin resistance and other manifestations of metabolic syndrome (Carulli et al., 2009). This provides a better understanding of the genetic susceptibility and pathogenesis of NASH, with the IL6-174C polymorphism being an independent predictor of both NAFLD and NASH, and also involved in inflammation and insulin resistance (Carulli et al., 2009).
The knowledge of genetic CVD risk may offer preventive and treatment strategies to targeted patient groups. Individuals with high genetic risks can have substantial risk reduction through pre-emptive improvement in lifestyle measures with regular moderate exercise, healthy diet and abstinence from smoking (Khera et al., 2016). The UK Biobank which examined individuals with a poor lifestyle, showed that those with higher genetic risk had more than 4-fold increase in CAD risk, compared to those with lower genetic risk. Importantly, those with high genetic risk but healthy lifestyle had lower CAD risk compared to those with low genetic risk but poor lifestyle (Said et al., 2018). Similarly, in the case of NASH, not all obese individuals with fatty liver progress to NASH, whilst conversely, some lean individuals with fatty liver do progress to NASH, emphasizing the important interactions between environmental risk factors and heritable factors (Younossi et al., 2016). However, the beneficial influence that genetic risk scores has on behavioural modifications remains lacking, as reported by a study that genetic risk information failed to influence smoking, cessation physical activity or dietary habits (Hollands et al., 2016; Jouni et al., 2017). Future prospective studies are warranted to investigate whether the knowledge of genetic risk can be translated to decrease CVD risks (Kessler and Schunkert, 2021). Figure 1 represents the interactions between environmental factors, the individual’s metabolic profile, and the genetic predisposition for NAFLD and CVD.
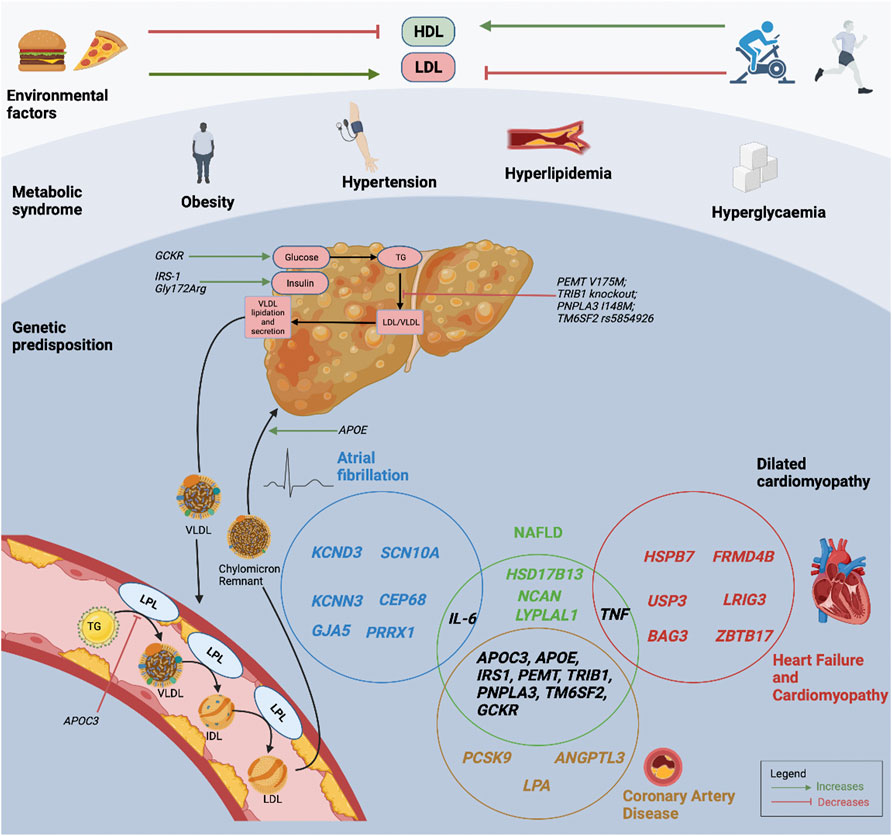
FIGURE 1. Interactions between environmental factors, metabolic profiles, and an individual’s genetic predisposition for NAFLD and CVD. The Venn diagram demonstrates the interactions between NAFLD and CVD susceptibility genes. HDL, high-density lipoprotein cholesterol; LDL, low-density lipoprotein cholesterol; LPL, lipoprotein lipase; NAFLD, non-alcoholic fatty liver disease; TG, triglyceride; VLDL, very low-density lipoprotein cholesterol.
Obesity exposes the association of PNPLA3 I148M to elevated hepatic fat levels and risk of NASH, with more pronounced impact of hepatic injury in obese individuals compared to lean individuals, and confers genetic susceptibility from a young age (Romeo et al., 2010a; Romeo et al., 2010b; Giudice et al., 2011; Palmer et al., 2012). The effect of the PNPLA3 I148M allele on hepatic fat levels also drastically increase in patients with high visceral abdominal fat levels (Graff et al., 2013) and BMI (Stender et al., 2017). The effect of high BMI in enhancing NASH risk in PNPLA3 I148M carriers may be mediated by insulin resistance (Barata et al., 2019). The prevalence of NASH ranged from 9% in lean 148-Ile homozygotes to 84% in obese 148-Met homozygotes (Stender et al., 2017). The obesogenic environment transforms PNPLA3 I148M into a major determinant in NAFLD and NASH pathophysiology, predisposing these individuals to CVD events. Evidence also suggests that PNPLA3 I148M may modify treatment response, with an effect on body weight and liver fat reduction in NAFLD patients (Carlsson et al., 2020) (i.e., lifestyle modification, bariatric surgery, omega-3 fatty acids). The interactions between obesity and TM6SF2 E167K and GCKR have also been described (Azuma et al., 2009; Stender et al., 2017).
Insulin resistance and type 2 diabetes, important risk factors for both CVD and NASH, have interactions with gene function that associate with different lipid profiles. NAFLD associated with PNPLA3 I148M had higher levels of hepatic polyunsaturated triacylglycerols, compared to NAFLD associated with insulin resistance which had higher levels of saturated and mono-unsaturated triacylglycerols, free fatty acids and ceramides. This can lead to different lipid compositions in the liver, especially in PNPLA3 I148M carriers, with reduced polyunsaturated triglyceride levels in VLDL particles.
PNPLA3 I148M carriers also have an increased risk of type 2 diabetes, as reported in a large GWAS study and fine-mapping meta-analysis (Fuchsberger et al., 2016; Dongiovanni et al., 2018; Mahajan et al., 2018). In a MR study, the genetic risk score for hepatic fat accumulation revealed a casual relationship with insulin resistance, but the relationship was no longer present when the model was adjusted for liver fibrosis (Dongiovanni et al., 2018). With these findings, the authors proposed that insulin resistance is not determined by the genetically-associated high hepatic fat levels per se, but rather the insulin resistance develops as a consequence of the progression of liver disease, which may be mediated by the inflammatory and pro-fibrotic environment (Dongiovanni et al., 2018). The causes and consequences of hepatic steatosis and inflammation in NASH patients may differ between PNPLA3 I148M carriers and those without the variant (Carlsson et al., 2020).
The genetic cross-talk between CVD and NAFLD is therefore best exemplified with the example of the PNPLA3 I148M polymorphism. Despite epidemiological data that advanced NAFLD is associated with increased risk of CAD, there is little evidence that hepatic fat accumulation causes atherosclerosis (Santos et al., 2019). The PNPLA3 I148M variant correlates to only a small reduction in ischemic heart disease risk (Liu et al., 2017; Lauridsen et al., 2018) but was strongly associated with liver-related and all-cause mortality (Unalp-Arida and Ruhl, 2020). Moreover, circulating TG and LDL cholesterol concentrations can be lower or unchanged in PNPLA3 I148M carriers compared to noncarriers (Speliotes et al., 2010; Hyysalo et al., 2014; Liu et al., 2017; Sliz et al., 2018). Nonetheless, NAFLD in itself bears increased mortality (Stender and Loomba, 2020). Therefore, the complex interplay between NAFLD and CAD is ultimately a combination of shared underlying risk factors, and putative overlapping genetic background that determines the cause and consequence of NAFLD on CVD outcomes (Carlsson et al., 2020).
The progressive role of genetics in CVD and NAFLD has added knowledge to the pathophysiology of the disease entities, and offers novel therapeutic targets. It also offers the chance of identifying individuals at risk with greater precision than relying only on conventional risk scores. Polygenic risk scores have the ability of predicting those at risk in early stages before other risk factors or imaging modalities can be applied effectively. Genetics may guide prevention strategies before the development of conventional risk factors or the disease manifestation (Kessler and Schunkert, 2021). Polygenic risk scores, calculated as the summation of the number of genetic variants weighted for their effect estimate, can also be used to study the inter-relationship between the various CVD and NAFLD, and perhaps help to predict NAFLD individuals who will progress to develop CVD manifestations. Polygenic risk scores have indeed performed better for predicting CAD or MI, compared to traditional risk factors (such as smoking or hyperlipidaemia) in cohorts of various ancestry (Tada et al., 2015; Inouye et al., 2018; Khera et al., 2018; Khera et al., 2019; Wang et al., 2020).
There are several challenges with polygenic risk scores which include the transferability of results from individuals of European ancestry to other ethnicities. The concept of “missing heritability” in CVD remains an important issue, with the heritability of CAD that can be explained by currently recognised risk variants, believed to be less than approximately 30%. The feasibility of identifying variants that can explain the large portion of heritability of complex traits is still unclear. The genetic basis of complex CVD may be seen as probabilistic rather than deterministic. Moreover, the risk of CVD that is often thought to be determined by monogenic risk variants, is likely modulated by polygenic risk (Fahed et al., 2020). Guidelines on genetic risk scores in the prevention and treatment of CVD is needed to address the indication, implementation and adequate genetic counselling before these scores can be used routinely in the clinical setting.
Moreover, the potential impact of genetic variants on the treatment of NASH can be encapsulated by the example of PNPLA3 I148M variant and the response to treatment (Sanyal et al., 2015; Wattacheril et al., 2018). At present, there are no pharmacological therapies for NASH treatment, but guidelines recommend weight reduction through lifestyle measures. However, lifestyle interventions are often limited, short-term and ineffective (Baran and Akyüz, 2014; Gitto et al., 2015). Metabolic bariatric surgery decreases hepatic steatosis, steatohepatitis and fibrosis (Laursen et al., 2019; Lee et al., 2019). Hence, it has been noted that PNPLA3 I148M carriers had more effective reduction in hepatic fat levels with lifestyle interventions and bariatric surgery, compared to non-carriers (Shen et al., 2015; Krawczyk et al., 2016a; Krawczyk et al., 2016b). However, omega-3 fatty acid treatment was reportedly less effective in reducing hepatic fat levels in PNPLA3 I148M carriers, compared to noncarriers in randomised trials (Nobili et al., 2013; Scorletti et al., 2015; Eriksson et al., 2018; Oscarsson et al., 2018).
Overall, genetic information is key for precision medicine. The goal of polygenic risk scores is to assist individuals with changing their lifestyle and modifiable behaviour, as well as to make informed decisions on preventive pharmacological (e.g., lipid-lowering) or surgical (e.g., bariatric) therapy in the effort for primary prevention (Kessler and Schunkert, 2021).
Over the past decade, tremendous effort has been taken to elucidate the genetics of CVD and NAFLD. This has provided exciting insights to CVD and NAFLD pathophysiology, and the increasing awareness of the genetic cross-talk between the two pathologies. Indeed our understanding of the overlap remains incomplete. More novel ground-breaking treatment targets such as proprotein convertase subtilisin/kexin type 9 (PCSK9) may be forthcoming, and a breakthrough in NAFLD and/or CVD prevention and reduction will be the next important milestone. The emergence of polygenic risk scores reflects the anticipation surrounding the power of genetics and precision medicine, in improving risk prediction, personalising prevention and treatment strategies.
• There are several non-alcoholic fatty liver disease (NAFLD) susceptibility genes that have colinear correlations with cardiovascular diseases including coronary artery disease, cardiomyopathy and atrial fibrillation
• Certain NAFLD susceptibility genes, such as phospholipase domain-containing protein 3 and transmembrane 6 superfamily 2 that regulate VLDL particle production, have cardioprotective effects for coronary artery disease.
• Genetic and environmental risk factors have complex interactions that lead to disparity in genetic influence on NAFLD and incident cardiovascular disease.
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by NC and BC. The first draft of the manuscript was written by NC and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adiels, M., Olofsson, S. O., Taskinen, M. R., and Borén, J. (2008). Overproduction of very low-density lipoproteins is the hallmark of the dyslipidemia in the metabolic syndrome. Atvb 28, 1225–1236. doi:10.1161/atvbaha.107.160192
Adiels, M., Taskinen, M. R., Packard, C., Caslake, M. J., Soro-Paavonen, A., Westerbacka, J., et al. (2006). Overproduction of large VLDL particles is driven by increased liver fat content in man. Diabetologia 49, 755–765. doi:10.1007/s00125-005-0125-z
Agius, L. (2008). Glucokinase and molecular aspects of liver glycogen metabolism. Biochem. J. 414, 1–18. doi:10.1042/bj20080595
Alessi, M. C., Bastelica, D., Mavri, A., Morange, P., Berthet, B., Grino, M., et al. (2003). Plasma PAI-1 levels are more strongly related to liver steatosis than to adipose tissue accumulation. Atvb 23, 1262–1268. doi:10.1161/01.atv.0000077401.36885.bb
Alikasifoglu, M., Tokgözoglu, L., Acil, T., Atalar, E., Oto, M. A., Kes, S. S., et al. (2003). Tumor necrosis factor-α polymorphism in Turkish patients with dilated cardiomyopathy. Eur. J. heart Fail. 5, 161–163. doi:10.1016/s1388-9842(02)00238-6
Anstee, Q. M., and Day, C. P. (2015). The genetics of nonalcoholic fatty liver disease: Spotlight on PNPLA3 and TM6SF2. Seminars liver Dis. 35, 270–290. doi:10.1055/s-0035-1562947
Anstee, Q. M., Targher, G., and Day, C. P. (2013). Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 10, 330–344. doi:10.1038/nrgastro.2013.41
Antuna-Puente, B., Feve, B., Fellahi, S., and Bastard, J. P. (2008). Adipokines: The missing link between insulin resistance and obesity. Diabetes & Metabolism 34, 2–11. doi:10.1016/j.diabet.2007.09.004
Armstrong, M. J., Adams, L. A., Canbay, A., and Syn, W.-K. (2014). Extrahepatic complications of nonalcoholic fatty liver disease. Hepatology 59, 1174–1197. doi:10.1002/hep.26717
Arner, P. (2002). Insulin resistance in type 2 diabetes: Role of fatty acids. Diabetes Metab. Res. Rev. 18, S5–S9. doi:10.1002/dmrr.254
Azuma, K., Kadowaki, T., Cetinel, C., Kadota, A., El-Saed, A., Kadowaki, S., et al. (2009). Higher liver fat content among Japanese in Japan compared with non-Hispanic whites in the United States. Metabolism 58, 1200–1207. doi:10.1016/j.metabol.2009.03.021
Baitsch, D., Bock, H. H., Engel, T., Telgmann, R., Müller-Tidow, C., Varga, G., et al. (2011). Apolipoprotein E induces antiinflammatory phenotype in macrophages. Atvb 31, 1160–1168. doi:10.1161/atvbaha.111.222745
Ballestri, S., Lonardo, A., Bonapace, S., Byrne, C. D., Loria, P., and Targher, G. (2014). Risk of cardiovascular, cardiac and arrhythmic complications in patients with non-alcoholic fatty liver disease. Wjg 20, 1724–1745. doi:10.3748/wjg.v20.i7.1724
Baran, B., and Akyüz, F. (2014). Non-alcoholic fatty liver disease: What has changed in the treatment since the beginning? Wjg 20, 14219–14229. doi:10.3748/wjg.v20.i39.14219
Barata, L., Feitosa, M. F., Bielak, L. F., Halligan, B., Baldridge, A. S., Guo, X., et al. (2019). Insulin resistance exacerbates genetic predisposition to nonalcoholic fatty liver disease in individuals without diabetes. Hepatol. Commun. 3, 894–907. doi:10.1002/hep4.1353
Basu Ray, S. (2019). PNPLA3-I148M: A problem of plenty in non-alcoholic fatty liver disease. Adipocyte 8, 201–208. doi:10.1080/21623945.2019.1607423
Bauer, R. C., Yenilmez, B. O., and Rader, D. J. (2015). Tribbles-1: A novel regulator of hepatic lipid metabolism in humans. Biochem. Soc. Trans. 43, 1079–1084. doi:10.1042/bst20150101
Beer, N. L., Tribble, N. D., McCulloch, L. J., Roos, C., Johnson, P. R., Orho-Melander, M., et al. (2009). The P446L variant in GCKR associated with fasting plasma glucose and triglyceride levels exerts its effect through increased glucokinase activity in liver. Hum. Mol. Genet. 18, 4081–4088. doi:10.1093/hmg/ddp357
Bhatt, S. P., and Guleria, R. (2021). Association of IRS1 (Gly972Arg) and IRS2 (Gly1057Asp) genes polymorphisms with OSA and NAFLD in asian Indians. PLOS ONE 16, e0245408. doi:10.1371/journal.pone.0245408
Bittar, M. N., Carey, J. A., Barnard, J., Fildes, J. E., Pravica, V., Yonan, N., et al. (2005). Interleukin 6 G-174C polymorphism influences outcome following coronary revascularization surgery. heart Surg. forum 8, E140–E145. doi:10.1532/hsf98.20041120
Boerwinkle, E., and Utermann, G. (1988). Simultaneous effects of the apolipoprotein E polymorphism on apolipoprotein E, apolipoprotein B, and cholesterol metabolism. Am. J. Hum. Genet. 42, 104–112.
Brouwers, M. C. G. J., Simons, N., Stehouwer, C. D. A., and Isaacs, A. (2020). Non-alcoholic fatty liver disease and cardiovascular disease: Assessing the evidence for causality. Diabetologia 63, 253–260. doi:10.1007/s00125-019-05024-3
Brouwers, M., Jacobs, C., Bast, A., Stehouwer, C. D. A., and Schaper, N. C. (2015). Modulation of glucokinase regulatory protein: A double-edged sword? Trends Mol. Med. 21, 583–594. doi:10.1016/j.molmed.2015.08.004
Browning, J. D., and Horton, J. D. (2004). Molecular mediators of hepatic steatosis and liver injury. J. Clin. Invest. 114, 147–152. doi:10.1172/jci200422422
Bruschi, F. V., Tardelli, M., Claudel, T., and Trauner, M. (2017). PNPLA3 expression and its impact on the liver: Current perspectives. Hmer 9, 55–66. doi:10.2147/hmer.s125718
Bruschi, F. V., Tardelli, M., Einwallner, E., Claudel, T., and Trauner, M. (2020). PNPLA3 I148M up-regulates Hedgehog and Yap signaling in human hepatic stellate cells. Int. J. Mol. Sci. 21, 8711. doi:10.3390/ijms21228711
Buckley, R., Shewring, B., Turner, R., Yaqoob, P., and Minihane, A. M. (2004). Circulating triacylglycerol and apoE levels in response to EPA and docosahexaenoic acid supplementation in adult human subjects. Br. J. Nutr. 92, 477–483. doi:10.1079/bjn20041235
Burgess, S., Foley, C. N., and Zuber, V. (2018). Inferring causal relationships between risk factors and outcomes from genome-wide association study data. Annu. Rev. Genom. Hum. Genet. 19, 303–327. doi:10.1146/annurev-genom-083117-021731
Burkhardt, R., Toh, S.-A., Lagor, W. R., Birkeland, A., Levin, M., Li, X., et al. (2010). Trib1 is a lipid- and myocardial infarction-associated gene that regulates hepatic lipogenesis and VLDL production in mice. J. Clin. Invest. 120, 4410–4414. doi:10.1172/jci44213
Byrne, C. D., and Targher, G. (2015). Nafld: A multisystem disease. J. Hepatology 62, S47–S64. doi:10.1016/j.jhep.2014.12.012
Carlsson, B., Lindén, D., Brolén, G., Liljeblad, M., Bjursell, M., Romeo, S., et al. (2020). Review article: The emerging role of genetics in precision medicine for patients with non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 51, 1305–1320. doi:10.1111/apt.15738
Carulli, L., Canedi, I., Rondinella, S., Lombardini, S., Ganazzi, D., Fargion, S., et al. (2009). Genetic polymorphisms in non-alcoholic fatty liver disease: Interleukin-6−174G/C polymorphism is associated with non-alcoholic steatohepatitis. Dig. Liver Dis. 41, 823–828. doi:10.1016/j.dld.2009.03.005
Chalasani, N., Younossi, Z., Lavine, J. E., Diehl, A. M., Brunt, E. M., Cusi, K., et al. (2012). The diagnosis and management of non-alcoholic fatty liver disease: Practice guideline by the American association for the study of liver diseases, American college of gastroenterology, and the American gastroenterological association. Hepatology 55, 2005–2023. doi:10.1002/hep.25762
Chen, X., Zhou, P., De, L., Li, B., and Su, S. (2019). The roles of transmembrane 6 superfamily member 2 rs58542926 polymorphism in chronic liver disease: A meta-analysis of 24,147 subjects. Mol. Genet. Genomic Med. 7, e824. doi:10.1002/mgg3.824
Cooper, A. D. (1985). Role of the liver in the degradation of lipoproteins. Gastroenterology 88, 192–205. doi:10.1016/s0016-5085(85)80155-4
Crespo, J., Cayn, A., Fernndez-Gil, P., Hernndez-Guerra, M., Mayorga, M., Domnguez-Dez, A., et al. (2001). Gene expression of tumor necrosis factor [alpha ] and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology 34, 1158–1163. doi:10.1053/jhep.2001.29628
Crosby, J., Peloso, G. M., Auer, P. L., Crosslin, D. R., Stitziel, N. O., and Lange, L. A. The Tg and Hdl Working Group of the Exome Sequencing Project, N.H., Lung, and Blood Institute (2014). Loss-of-Function mutations in APOC3, triglycerides, and coronary disease. N. Engl. J. Med. 371, 22–31. doi:10.1056/NEJMoa1307095
Dang, T. A., Schunkert, H., and Kessler, T. (2020). cGMP signaling in cardiovascular diseases. J. Cardiovasc. Pharmacol. 75, 516–525. doi:10.1097/fjc.0000000000000744
DeFilippis, A. P., Blaha, M. J., Martin, S. S., Reed, R. M., Jones, S. R., Nasir, K., et al. (2013). Nonalcoholic fatty liver disease and serum lipoproteins: The multi-ethnic study of atherosclerosis. Atherosclerosis 227, 429–436. doi:10.1016/j.atherosclerosis.2013.01.022
Deloukas, P., Kanoni, S., Kanoni, C., Willenborg, M., Farrall, T. L., Assimes, J. R., et al. fnm (2013). Large-scale association analysis identifies new risk loci for coronary artery disease. Nat. Genet. 45, 25–33.doi:10.1038/ng.2480
Demirag, M. D., Onen, H. I., Karaoguz, M. Y., Dogan, I., Karakan, T., Ekmekci, A., et al. (2007). Apolipoprotein E gene polymorphism in nonalcoholic fatty liver disease. Dig. Dis. Sci. 52, 3399–3403. doi:10.1007/s10620-007-9740-5
Densem, C. G., Hutchinson, I. V., Yonan, N., and Brooks, N. H. (2002). Tumour necrosis factor α gene polymorphism: A predisposing factor to non-ischaemic myocardial dysfunction? Heart 87, 153–155. doi:10.1136/heart.87.2.153
Do, R., Willer, C. J., Schmidt, E. M., Sengupta, S., Gao, C., Peloso, G. M., et al. (2013). Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat. Genet. 45, 1345–1352. doi:10.1038/ng.2795
Dong, H., Wang, J., Li, C., Hirose, A., Nozaki, Y., Takahashi, M., et al. (2007). The phosphatidylethanolamine N-methyltransferase gene V175M single nucleotide polymorphism confers the susceptibility to NASH in Japanese population. J. Hepatology 46, 915–920. doi:10.1016/j.jhep.2006.12.012
Dongiovanni, P., Petta, S., Maglio, C., Fracanzani, A. L., Pipitone, R., Mozzi, E., et al. (2015a). Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology 61, 506–514. doi:10.1002/hep.27490
Dongiovanni, P., Romeo, S., and Valenti, L. (2015b). Genetic factors in the pathogenesis of nonalcoholic fatty liver and steatohepatitis. Biomed. Res. Int. 2015, 460190. doi:10.1155/2015/460190
Dongiovanni, P., Stender, S., Pietrelli, A., Mancina, R. M., Cespiati, A., Petta, S., et al. (2018). Causal relationship of hepatic fat with liver damage and insulin resistance in nonalcoholic fatty liver. J. Intern Med. 283, 356–370. doi:10.1111/joim.12719
Dongiovanni, P., Valenti, L., Rametta, R., Daly, A. K., Nobili, V., Mozzi, E., et al. (2010). Genetic variants regulating insulin receptor signalling are associated with the severity of liver damage in patients with non-alcoholic fatty liver disease. Gut 59, 267–273. doi:10.1136/gut.2009.190801
Donnelly, K. L., Smith, C. I., Schwarzenberg, S. J., Jessurun, J., Boldt, M. D., and Parks, E. J. (2005). Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investigation 115, 1343–1351. doi:10.1172/jci23621
Dutka, D. P., Elborn, J. S., Delamere, F., Shale, D. J., and Morris, G. K. (1993). Tumour necrosis factor alpha in severe congestive cardiac failure. Br. Heart J. 70, 141–143. doi:10.1136/hrt.70.2.141
El-Tahan, R. R., Ghoneim, A. M., and El-Mashad, N. (2016). TNF-α gene polymorphisms and expression. SpringerPlus 5, 1508. doi:10.1186/s40064-016-3197-y
Erdmann, J., Kessler, T., Munoz Venegas, L., and Schunkert, H. (2018). A decade of genome-wide association studies for coronary artery disease: The challenges ahead. Cardiovasc. Res. 114, 1241–1257. doi:10.1093/cvr/cvy084
Eriksson, J. W., Lundkvist, P., Jansson, P. A., Johansson, L., Kvarnström, M., Moris, L., et al. (2018). Effects of dapagliflozin and n-3 carboxylic acids on non-alcoholic fatty liver disease in people with type 2 diabetes: A double-blind randomised placebo-controlled study. Diabetologia 61, 1923–1934. doi:10.1007/s00125-018-4675-2
Fahed, A. C., Wang, M., Homburger, J. R., Patel, A. P., Bick, A. G., Neben, C. L., et al. (2020). Polygenic background modifies penetrance of monogenic variants for tier 1 genomic conditions. Nat. Commun. 11, 3635. doi:10.1038/s41467-020-17374-3
Fernandez, C., Sandin, M., Sampaio, J. L., Almgren, P., Narkiewicz, K., Hoffmann, M., et al. (2013). Plasma lipid composition and risk of developing cardiovascular disease. PLOS ONE 8, e71846. doi:10.1371/journal.pone.0071846
Fuchsberger, C., Flannick, J., Teslovich, T. M., Mahajan, A., Agarwala, V., Gaulton, K. J., et al. (2016). The genetic architecture of type 2 diabetes. Nature 536, 41–47. doi:10.1038/nature18642
Gaudet, D., Alexander, V. J., Baker, B. F., Brisson, D., Tremblay, K., Singleton, W., et al. (2015). Antisense inhibition of apolipoprotein C-III in patients with hypertriglyceridemia. N. Engl. J. Med. 373, 438–447. doi:10.1056/nejmoa1400283
Gaudino, M., Andreotti, F., Zamparelli, R., Castelnuovo, A. D., Nasso, G., Burzotta, F., et al. (2003). The −174G/C interleukin-6 polymorphism influences postoperative interleukin-6 levels and postoperative atrial fibrillation. Is atrial fibrillation an inflammatory complication? Circulation 108, 195–199. doi:10.1161/01.cir.0000087441.48566.0d
Gitto, S., Vitale, G., Villa, E., and Andreone, P. (2015). Treatment of nonalcoholic steatohepatitis in adults: Present and future. Gastroenterology Res. Pract. 2015, 732870. doi:10.1155/2015/732870
Giudice, E. M., Grandone, A., Cirillo, G., Santoro, N., Amato, A., Brienza, C., et al. (2011). The association of PNPLA3 variants with liver enzymes in childhood obesity is driven by the interaction with abdominal fat. PLoS One 6, e27933. doi:10.1371/journal.pone.0027933
Goffredo, M., Caprio, S., Feldstein, A. E., D'adamo, E., Shaw, M. M., Pierpont, B., et al. (2016). Role of TM6SF2 rs58542926 in the pathogenesis of nonalcoholic pediatric fatty liver disease: A multiethnic study. Hepatology 63, 117–125. doi:10.1002/hep.28283
Gola, D., Erdmann, J., Läll, K., Mägi, R., Müller-Myhsok, B., Schunkert, H., et al. (2020). Population bias in polygenic risk prediction models for coronary artery disease. Circulation Genomic Precis. Med. 13, e002932. doi:10.1161/CIRCGEN.120.002932
Graff, M., North, K. E., Franceschini, N., Reiner, A. P., Feitosa, M., Carr, J. J., et al. (2013). PNPLA3 gene-by-visceral adipose tissue volume interaction and the pathogenesis of fatty liver disease: The NHLBI family heart study. Int. J. Obes. (Lond) 37, 432–438. doi:10.1038/ijo.2012.65
Grandone, A., Cozzolino, D., Marzuillo, P., Cirillo, G., Di Sessa, A., Ruggiero, L., et al. (2016). TM6SF2 Glu167Lys polymorphism is associated with low levels of LDL-cholesterol and increased liver injury in obese children. Pediatr. Obes. 11, 115–119. doi:10.1111/ijpo.12032
Granér, M., Kahri, J., Varpula, M., Salonen, R. M., Nyyssönen, K., Jauhiainen, M., et al. (2008). Apolipoprotein E polymorphism is associated with both carotid and coronary atherosclerosis in patients with coronary artery disease. Nutr. Metabolism Cardiovasc. Dis. 18, 271–277. doi:10.1016/j.numecd.2007.01.003
Grootaert, M. O. J., Moulis, M., Roth, L., Martinet, W., Vindis, C., Bennett, M. R., et al. (2018). Vascular smooth muscle cell death, autophagy and senescence in atherosclerosis. Cardiovasc Res. 114, 622–634. doi:10.1093/cvr/cvy007
Heeren, J., and Scheja, L. (2021). Metabolic-associated fatty liver disease and lipoprotein metabolism. Mol. Metab. 50, 101238. doi:10.1016/j.molmet.2021.101238
Herrmann, S. M., Ricard, S., Nicaud, V., Mallet, C., Arveiler, D., Evans, A., et al. (1998). Polymorphisms of the tumour necrosis factor-alpha gene, coronary heart disease and obesity. Eur. J. Clin. investigation 28, 59–66. doi:10.1046/j.1365-2362.1998.00244.x
Hodson, L., and Brouwers, M. (2018). Non-alcoholic fatty liver disease concerns with glucokinase activators. Lancet Diabetes Endocrinol. 6, 684–685. doi:10.1016/s2213-8587(18)30196-7
Hodson, L., and Frayn, K. N. (2011). Hepatic fatty acid partitioning. Curr. Opin. Lipidol. 22, 216–224. doi:10.1097/mol.0b013e3283462e16
Hollands, G. J., French, D. P., Griffin, S. J., Prevost, A. T., Sutton, S., King, S., et al. (2016). The impact of communicating genetic risks of disease on risk-reducing health behaviour: Systematic review with meta-analysis. BMJ 352, i1102. doi:10.1136/bmj.i1102
Holmen, O. L., Zhang, H., Fan, Y., Hovelson, D. H., Schmidt, E. M., Zhou, W., et al. (2014). Systematic evaluation of coding variation identifies a candidate causal variant in TM6SF2 influencing total cholesterol and myocardial infarction risk. Nat. Genet. 46, 345–351. doi:10.1038/ng.2926
Holmes, M. V., Ala-Korpela, M., and Smith, G. D. (2017). Mendelian randomization in cardiometabolic disease: Challenges in evaluating causality. Nat. Rev. Cardiol. 14, 577–590. doi:10.1038/nrcardio.2017.78
Hotta, K., Yoneda, M., Hyogo, H., Ochi, H., Mizusawa, S., Ueno, T., et al. (2010). Association of the rs738409 polymorphism in PNPLA3 with liver damage and the development of nonalcoholic fatty liver disease. BMC Med. Genet. 11, 172. doi:10.1186/1471-2350-11-172
Hu, Z.-W., Luo, H.-B., Xu, Y.-M., Guo, J.-W., Deng, X.-L., Tong, Y.-W., et al. (2009). Tumor necrosis factor-alpha gene promoter polymorphisms in Chinese patients with nonalcoholic fatty liver diseases. Acta gastro-enterologica Belg. 72, 215–221.
Huang, Y., Cohen, J. C., and Hobbs, H. H. (2011). Expression and characterization of a PNPLA3 protein isoform (I148M) associated with nonalcoholic fatty liver disease. J. Biol. Chem. 286, 37085–37093. doi:10.1074/jbc.m111.290114
Hyysalo, J., Gopalacharyulu, P., Bian, H., Hyötyläinen, T., Leivonen, M., Jaser, N., et al. (2014). Circulating triacylglycerol signatures in nonalcoholic fatty liver disease associated with the I148M variant in PNPLA3 and with obesity. Diabetes 63, 312–322. doi:10.2337/db13-0774
Hyysalo, J., Stojkovic, I., Kotronen, A., Hakkarainen, A., Sevastianova, K., Makkonen, J., et al. (2012). Genetic variation in PNPLA3 but not APOC3 influences liver fat in non-alcoholic fatty liver disease. J. Gastroenterology Hepatology 27, 951–956. doi:10.1111/j.1440-1746.2011.07045.x
IBC 50K CAD Consortium (2011). Large-scale gene-centric analysis identifies novel variants for coronary artery disease. PLOS Genet. 7, e1002260. doi:10.1371/journal.pgen.1002260
Inouye, M., Abraham, G., Nelson, C. P., Wood, A. M., Sweeting, M. J., Dudbridge, F., et al. (2018). Genomic risk prediction of coronary artery disease in 480,000 adults. J. Am. Coll. Cardiol. 72, 1883–1893. doi:10.1016/j.jacc.2018.07.079
Isasi, C. R., Shea, S., Deckelbaum, R. J., Couch, S. C., Starc, T. J., Otvos, J. D., et al. (2000). Apolipoprotein ε2 allele is associated with an anti-atherogenic lipoprotein profile in children: The columbia university BioMarkers study. Pediatrics 106, 568–575. doi:10.1542/peds.106.3.568
Ito, M., Takahashi, H., Fuse, K., Hirono, S., Washizuka, T., Kato, K., et al. (2000). Polymorphisms of tumor necrosis factor-α and interleukin-10 genes in Japanese patients with idiopathic dilated cardiomyopathy. Jpn. Heart J. 41, 183–191. doi:10.1536/jhj.41.183
Iwamoto, S., Boonvisut, S., Makishima, S., Ishizuka, Y., Watanabe, K., and Nakayama, K. (2015). The role of TRIB1 in lipid metabolism; from genetics to pathways. Biochem. Soc. Trans. 43, 1063–1068. doi:10.1042/bst20150094
Jacobsen, R., Martinussen, T., Christiansen, L., Jeune, B., Andersen-Ranberg, K., Vaupel, J. W., et al. (2010). Increased effect of the ApoE gene on survival at advanced age in healthy and long-lived Danes: Two nationwide cohort studies. Aging Cell 9, 1004–1009. doi:10.1111/j.1474-9726.2010.00626.x
Jaipersad, A. S., Lip, G. Y., Silverman, S., and Shantsila, E. (2014). The role of monocytes in angiogenesis and atherosclerosis. J. Am. Coll. Cardiol. 63, 1–11. doi:10.1016/j.jacc.2013.09.019
Jouni, H., Haddad, R. A., Marroush, T. S., Brown, S.-A., Kruisselbrink, T. M., Austin, E. E., et al. (2017). Shared decision-making following disclosure of coronary heart disease genetic risk: Results from a randomized clinical trial. J. Investigative Med. 65, 681–688. doi:10.1136/jim-2016-000318
Kahali, B., Halligan, B., and Speliotes, E. K. (2015). Insights from genome-wide association analyses of nonalcoholic fatty liver disease. Semin. Liver Dis. 35, 375–391. doi:10.1055/s-0035-1567870
Kallio, M. J., Salmenperä, L., Siimes, M. A., Perheentupa, J., Gylling, H., and Miettinen, T. A. (1997). Apoprotein E phenotype determines serum cholesterol in infants during both high-cholesterol breast feeding and low-cholesterol formula feeding. J. Lipid Res. 38, 759–764. doi:10.1016/s0022-2275(20)37242-4
Katz, S. D., Rao, R., Berman, J. W., Schwarz, M., Demopoulos, L., Bijou, R., et al. (1994). Pathophysiological correlates of increased serum tumor necrosis factor in patients with congestive heart failure. Relation to nitric oxide-dependent vasodilation in the forearm circulation. Circulation 90, 12–16. doi:10.1161/01.cir.90.1.12
Kessler, T., and Schunkert, H. (2021). Coronary artery disease genetics enlightened by genome-wide association studies. JACC Basic Transl. Sci. 6, 610–623. doi:10.1016/j.jacbts.2021.04.001
Kessler, T., and Schunkert, H. (2022). Genomic strategies toward identification of novel therapeutic targets. Handb. Exp. Pharmacol. 270, 1–34. doi:10.1007/164_2020_360
Khallou-Laschet, J., Varthaman, A., Fornasa, G., Compain, C., Gaston, A. T., Clement, M., et al. (2010). Macrophage plasticity in experimental atherosclerosis. PLoS One 5, e8852. doi:10.1371/journal.pone.0008852
Khera, A. V., Chaffin, M., Aragam, K. G., Haas, M. E., Roselli, C., Choi, S. H., et al. (2018). Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 50, 1219–1224. doi:10.1038/s41588-018-0183-z
Khera, A. V., Chaffin, M., Zekavat, S. M., Collins, R. L., Roselli, C., Natarajan, P., et al. (2019). Whole-genome sequencing to characterize monogenic and polygenic contributions in patients hospitalized with early-onset myocardial infarction. Circulation 139, 1593–1602. doi:10.1161/circulationaha.118.035658
Khera, A. V., Emdin, C. A., Drake, I., Natarajan, P., Bick, A. G., Cook, N. R., et al. (2016). Genetic risk, adherence to a healthy lifestyle, and coronary disease. N. Engl. J. Med. 375, 2349–2358. doi:10.1056/nejmoa1605086
Khera, A. V., and Kathiresan, S. (2017). Genetics of coronary artery disease: Discovery, biology and clinical translation. Nat. Rev. Genet. 18, 331–344. doi:10.1038/nrg.2016.160
Kleiner, D. E., Brunt, E. M., Van Natta, M., Behling, C., Contos, M. J., Cummings, O. W., et al. (2005). Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41, 1313–1321. doi:10.1002/hep.20701
Koyama, S., Ito, K., Terao, C., Akiyama, M., Horikoshi, M., Momozawa, Y., et al. (2020). Population-specific and trans-ancestry genome-wide analyses identify distinct and shared genetic risk loci for coronary artery disease. Nat. Genet. 52, 1169–1177. doi:10.1038/s41588-020-0705-3
Kozlitina, J., Boerwinkle, E., Cohen, J. C., and Hobbs, H. H. (2011). Dissociation between APOC3 variants, hepatic triglyceride content and insulin resistance. Hepatology 53, 467–474. doi:10.1002/hep.24072
Kozlitina, J., Smagris, E., Stender, S., Nordestgaard, B. G., Zhou, H. H., Tybjærg-Hansen, A., et al. (2014). Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 46, 352–356. doi:10.1038/ng.2901
Krawczyk, M., Jiménez-Agüero, R., Alustiza, J. M., Emparanza, J. I., Perugorria, M. J., Bujanda, L., et al. (2016a). PNPLA3 p.I148M variant is associated with greater reduction of liver fat content after bariatric surgery. Surg. Obes. Relat. Dis. 12, 1838–1846. doi:10.1016/j.soard.2016.06.004
Krawczyk, M., Stachowska, E., Milkiewicz, P., Lammert, F., and Milkiewicz, M. (2016b). Reduction of caloric intake might override the prosteatotic effects of the PNPLA3 p.I148M and TM6SF2 p.E167K variants in patients with fatty liver: Ultrasound-based prospective study. Digestion 93, 139–148. doi:10.1159/000441185
Kubota, T., Mcnamara, D. M., Wang, J. J., Trost, M., Mctiernan, C. F., Mann, D. L., et al. (1998). Effects of tumor necrosis factor gene polymorphisms on patients with congestive heart failure. Circulation 97, 2499–2501. doi:10.1161/01.cir.97.25.2499
Lauridsen, B. K., Stender, S., Kristensen, T. S., Kofoed, K. F., Køber, L., Nordestgaard, B. G., et al. (2018). Liver fat content, non-alcoholic fatty liver disease, and ischaemic heart disease: Mendelian randomization and meta-analysis of 279 013 individuals. Eur. Heart J. 39, 385–393. doi:10.1093/eurheartj/ehx662
Laursen, T. L., Hagemann, C. A., Wei, C., Kazankov, K., Thomsen, K. L., Knop, F. K., et al. (2019). Bariatric surgery in patients with non-alcoholic fatty liver disease - from pathophysiology to clinical effects. World J. Hepatol. 11, 138–149. doi:10.4254/wjh.v11.i2.138
Lee, Y., Doumouras, A. G., Yu, J., Brar, K., Banfield, L., Gmora, S., et al. (2019). Complete resolution of nonalcoholic fatty liver disease after bariatric surgery: A systematic review and meta-analysis. Clin. Gastroenterol. Hepatol. 17, 1040–1060. e1011. doi:10.1016/j.cgh.2018.10.017
Levine, B., Kalman, J., Mayer, L., Fillit, H. M., and Packer, M. (1990). Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N. Engl. J. Med. 323, 236–241. doi:10.1056/nejm199007263230405
Li, Q., Qiao, Y., Wang, C., Zhang, G., Zhang, X., and Xu, L. (2016). Associations between two single-nucleotide polymorphisms (rs1801278 and rs2943641) of insulin receptor substrate 1 gene and type 2 diabetes susceptibility: A meta-analysis. Endocrine 51, 52–62. doi:10.1007/s12020-015-0770-z
Li, Z., Agellon, L. B., Allen, T. M., Umeda, M., Jewell, L., Mason, A., et al. (2006). The ratio of phosphatidylcholine to phosphatidylethanolamine influences membrane integrity and steatohepatitis. Cell Metab. 3, 321–331. doi:10.1016/j.cmet.2006.03.007
Liu, D. J., Peloso, G. M., Yu, H., Butterworth, A. S., Wang, X., Mahajan, A., et al. (2017). Exome-wide association study of plasma lipids in >300,000 individuals. Nat. Genet. 49, 1758–1766. doi:10.1038/ng.3977
Liu, H., and Lu, H.-Y. (2014). Nonalcoholic fatty liver disease and cardiovascular disease. World J. gastroenterology 20, 8407–8415. doi:10.3748/wjg.v20.i26.8407
Liu, Q., Liu, S.-S., Zhao, Z.-Z., Zhao, B.-T., Du, S.-X., Jin, W.-W., et al. (2019a). TRIB1 rs17321515 gene polymorphism increases the risk of coronary heart disease in general population and non-alcoholic fatty liver disease patients in Chinese Han population. Lipids Health Dis. 18, 165. doi:10.1186/s12944-019-1108-2
Liu, Q., Xue, F., Meng, J., Liu, S.-S., Chen, L.-Z., Gao, H., et al. (2019b). TRIB1 rs17321515 and rs2954029 gene polymorphisms increase the risk of non-alcoholic fatty liver disease in Chinese Han population. Lipids Health Dis. 18, 61. doi:10.1186/s12944-019-1001-z
Liu, S., Ammirati, M. J., Song, X., Knafels, J. D., Zhang, J., Greasley, S. E., et al. (2012). Insights into mechanism of glucokinase activation: Observation of multiple distinct protein conformations. J. Biol. Chem. 287, 13598–13610. doi:10.1074/jbc.m111.274126
Lloyd, D. J., St Jean, D. J., Kurzeja, R. J., Wahl, R. C., Michelsen, K., Cupples, R., et al. (2013). Antidiabetic effects of glucokinase regulatory protein small-molecule disruptors. Nature 504, 437–440. doi:10.1038/nature12724
Lonardo, A., Ballestri, S., Marchesini, G., Angulo, P., and Loria, P. (2015). Nonalcoholic fatty liver disease: A precursor of the metabolic syndrome. Dig. Liver Dis. 47, 181–190. doi:10.1016/j.dld.2014.09.020
Lu, X., Wang, L., Chen, S., He, L., Yang, X., Shi, Y., et al. (2012). Genome-wide association study in Han Chinese identifies four new susceptibility loci for coronary artery disease. Nat. Genet. 44, 890–894. doi:10.1038/ng.2337
Luckett, L. R., and Gallucci, R. M. (2007). Interleukin-6 (IL-6) modulates migration and matrix metalloproteinase function in dermal fibroblasts from IL-6KO mice. Br. J. Dermatology 156, 1163–1171. doi:10.1111/j.1365-2133.2007.07867.x
Ludwig, J., Viggiano, T. R., Mcgill, D. B., and Oh, B. J. (1980). Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc. 55, 434–438.
Luo, J.-Q., Ren, H., Banh, H. L., Liu, M.-Z., Xu, P., Fang, P.-F., et al. (2017). The associations between apolipoprotein E gene epsilon2/epsilon3/epsilon4 polymorphisms and the risk of coronary artery disease in patients with type 2 diabetes mellitus. Front. Physiology 8. doi:10.3389/fphys.2017.01031
Machado, M. V., and Diehl, A. M. (2016). Pathogenesis of nonalcoholic steatohepatitis. Gastroenterology 150, 1769–1777. doi:10.1053/j.gastro.2016.02.066
Mahajan, A., Wessel, J., Willems, S. M., Zhao, W., Robertson, N. R., Chu, A. Y., et al. (2018). Refining the accuracy of validated target identification through coding variant fine-mapping in type 2 diabetes. Nat. Genet. 50, 559–571. doi:10.1038/s41588-018-0084-1
Mahdessian, H., Taxiarchis, A., Popov, S., Silveira, A., Franco-Cereceda, A., Hamsten, A., et al. (2014). TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc. Natl. Acad. Sci. 111, 8913–8918. doi:10.1073/pnas.1323785111
Mahley, R. (2001). Type III hyperlipoproteinemia (dysbetalipoproteinemia). The metabolic and molecular bases of inherited disease, 2835–2862.
Makishima, S., Boonvisut, S., Ishizuka, Y., Watanabe, K., Nakayama, K., and Iwamoto, S. (2015). Sin3A-associated protein, 18 kDa, a novel binding partner of TRIB1, regulates MTTP expression. J. Lipid Res. 56, 1145–1152. doi:10.1194/jlr.m057802
Manichaikul, A., Palmas, W., Rodriguez, C. J., Peralta, C. A., Divers, J., Guo, X., et al. (2012). Population structure of hispanics in the United States: The multi-ethnic study of atherosclerosis. PLoS Genet. 8, e1002640. doi:10.1371/journal.pgen.1002640
Marais, A. D. (2019). Apolipoprotein E in lipoprotein metabolism, health and cardiovascular disease. Pathology 51, 165–176. doi:10.1016/j.pathol.2018.11.002
Marcus, G. M., Whooley, M. A., Glidden, D. V., Pawlikowska, L., Zaroff, J. G., and Olgin, J. E. (2008). Interleukin-6 and atrial fibrillation in patients with coronary artery disease: Data from the heart and soul study. Am. Heart J. 155, 303–309. doi:10.1016/j.ahj.2007.09.006
Marra, F., and Bertolani, C. (2009). Adipokines in liver diseases. Hepatology 50, 957–969. doi:10.1002/hep.23046
Mcmahon, D., Dinh, A., Kurz, D., Shah, D., Han, G. S., Carman, G. M., et al. (2014). Comparative gene identification 58/α/β hydrolase domain 5 lacks lysophosphatidic acid acyltransferase activity. J. Lipid Res. 55, 1750–1761. doi:10.1194/jlr.m051151
Mcmurray, J., Abdullah, I., Dargie, H. J., and Shapiro, D. (1991). Increased concentrations of tumour necrosis factor in "cachectic" patients with severe chronic heart failure. Br. Heart J. 66, 356–358. doi:10.1136/hrt.66.5.356
Mirea, A. M., Tack, C. J., Chavakis, T., Joosten, L. a. B., and Toonen, E. J. M. (2018). IL-1 family cytokine pathways underlying NAFLD: Towards new treatment strategies. Trends Mol. Med. 24, 458–471. doi:10.1016/j.molmed.2018.03.005
Miyaaki, H., Miuma, S., Taura, N., Shibata, H., Soyama, A., Hidaka, M., et al. (2018). PNPLA3 as a liver steatosis risk factor following living-donor liver transplantation for hepatitis C. Hepatol. Res. 48, E335–e339. doi:10.1111/hepr.12920
Nagiec, M. M., Skepner, A. P., Negri, J., Eichhorn, M., Kuperwasser, N., Comer, E., et al. (2015). Modulators of hepatic lipoprotein metabolism identified in a search for small-molecule inducers of tribbles pseudokinase 1 expression. PLoS One 10, e0120295. doi:10.1371/journal.pone.0120295
Naik, A., Košir, R., and Rozman, D. (2013). Genomic aspects of NAFLD pathogenesis. Genomics 102, 84–95. doi:10.1016/j.ygeno.2013.03.007
Namikawa, C., Shu-Ping, Z., Vyselaar, J. R., Nozaki, Y., Nemoto, Y., Ono, M., et al. (2004). Polymorphisms of microsomal triglyceride transfer protein gene and manganese superoxide dismutase gene in non-alcoholic steatohepatitis. J. Hepatology 40, 781–786. doi:10.1016/j.jhep.2004.01.028
Newton-Cheh, C., Johnson, T., Gateva, V., Tobin, M. D., Bochud, M., Coin, L., et al. (2009). Genome-wide association study identifies eight loci associated with blood pressure. Nat. Genet. 41, 666–676. doi:10.1038/ng.361
Ng, C. H., Xiao, J., Lim, W. H., Chin, Y. H., Yong, J. N., Tan, D. J. H., et al. (2022). Placebo effect on progression and regression in NASH: Evidence from a meta-analysis. Hepatology 75 (6), 1647–1661. doi:10.1002/hep.32315
Nobili, V., Bedogni, G., Donati, B., Alisi, A., and Valenti, L. (2013). The I148M variant of PNPLA3 reduces the response to docosahexaenoic acid in children with non-alcoholic fatty liver disease. J. Med. Food 16, 957–960. doi:10.1089/jmf.2013.0043
Noga, A. A., Zhao, Y., and Vance, D. E. (2002). An unexpected requirement for PhosphatidylethanolamineN-methyltransferase in the secretion of very low density lipoproteins. J. Biol. Chem. 277, 42358–42365. doi:10.1074/jbc.m204542200
Ong, J. P., Pitts, A., and Younossi, Z. M. (2008). Increased overall mortality and liver-related mortality in non-alcoholic fatty liver disease. J. Hepatol. 49, 608–612. doi:10.1016/j.jhep.2008.06.018
Oscarsson, J., Önnerhag, K., Risérus, U., Sundén, M., Johansson, L., Jansson, P. A., et al. (2018). Effects of free omega-3 carboxylic acids and fenofibrate on liver fat content in patients with hypertriglyceridemia and non-alcoholic fatty liver disease: A double-blind, randomized, placebo-controlled study. J. Clin. Lipidol. 12, 1390–1403. e1394. doi:10.1016/j.jacl.2018.08.003
Palmer, C. N., Maglio, C., Pirazzi, C., Burza, M. A., Adiels, M., Burch, L., et al. (2012). Paradoxical lower serum triglyceride levels and higher type 2 diabetes mellitus susceptibility in obese individuals with the PNPLA3 148M variant. PLoS One 7, e39362. doi:10.1371/journal.pone.0039362
Pan, J. X. (2017). LncRNA H19 promotes atherosclerosis by regulating MAPK and NF-kB signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 21, 322–328.
Petersen, K. F., Dufour, S., Hariri, A., Nelson-Williams, C., Foo, J. N., Zhang, X.-M., et al. (2010). Apolipoprotein C3 gene variants in nonalcoholic fatty liver disease. N. Engl. J. Med. 362, 1082–1089. doi:10.1056/nejmoa0907295
Pfeffer, K. (2003). Biological functions of tumor necrosis factor cytokines and their receptors. Cytokine & Growth Factor Rev. 14, 185–191. doi:10.1016/s1359-6101(03)00022-4
Pingitore, P., Dongiovanni, P., Motta, B. M., Meroni, M., Lepore, S. M., Mancina, R. M., et al. (2016). PNPLA3 overexpression results in reduction of proteins predisposing to fibrosis. Hum. Mol. Genet. 25, 5212–5222. doi:10.1093/hmg/ddw341
Pingitore, P., Pirazzi, C., Mancina, R. M., Motta, B. M., Indiveri, C., Pujia, A., et al. (2014). Recombinant PNPLA3 protein shows triglyceride hydrolase activity and its I148M mutation results in loss of function. Biochimica Biophysica Acta (BBA) - Mol. Cell Biol. Lipids 1841, 574–580. doi:10.1016/j.bbalip.2013.12.006
Pirazzi, C., Valenti, L., Motta, B. M., Pingitore, P., Hedfalk, K., Mancina, R. M., et al. (2014). PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells. Hum. Mol. Genet. 23, 4077–4085. doi:10.1093/hmg/ddu121
Pollin, T. I., Damcott, C. M., Shen, H., Ott, S. H., Shelton, J., Horenstein, R. B., et al. (2008). A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science 322, 1702–1705. doi:10.1126/science.1161524
Psychari, S. N., Apostolou, T. S., Sinos, L., Hamodraka, E., Liakos, G., and Kremastinos, D. T. (2005). Relation of elevated C-reactive protein and interleukin-6 levels to left atrial size and duration of episodes in patients with atrial fibrillation. Am. J. Cardiol. 95, 764–767. doi:10.1016/j.amjcard.2004.11.032
Qidwai, T., and Khan, F. (2011). Tumour necrosis factor gene polymorphism and disease prevalence. Scand. J. Immunol. 74, 522–547. doi:10.1111/j.1365-3083.2011.02602.x
Rees, M. G., Wincovitch, S., Schultz, J., Waterstradt, R., Beer, N. L., Baltrusch, S., et al. (2012). Cellular characterisation of the GCKR P446L variant associated with type 2 diabetes risk. Diabetologia 55, 114–122. doi:10.1007/s00125-011-2348-5
Reustle, A., and Torzewski, M. (2018). Role of p38 MAPK in atherosclerosis and aortic valve sclerosis. Int. J. Mol. Sci. 19. doi:10.3390/ijms19123761
Riddell, D. R., Graham, A., and Owen, J. S. (1997). Apolipoprotein E inhibits platelet aggregation through the L-arginine:nitric oxide pathway: Implications for vascular disease. J. Biol. Chem. 272, 89–95. doi:10.1074/jbc.272.1.89
Ridker, P. M., Everett, B. M., Thuren, T., Macfadyen, J. G., Chang, W. H., Ballantyne, C., et al. (2017). Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 377, 1119–1131. doi:10.1056/nejmoa1707914
Rinella, M. E. (2015). Nonalcoholic fatty liver disease: A systematic review. JAMA 313, 2263–2273. doi:10.1001/jama.2015.5370
Romeo, S., Kozlitina, J., Xing, C., Pertsemlidis, A., Cox, D., Pennacchio, L. A., et al. (2008). Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 40, 1461–1465. doi:10.1038/ng.257
Romeo, S., Sentinelli, F., Cambuli, V. M., Incani, M., Congiu, T., Matta, V., et al. (2010a). The 148M allele of the PNPLA3 gene is associated with indices of liver damage early in life. J. Hepatol. 53, 335–338. doi:10.1016/j.jhep.2010.02.034
Romeo, S., Sentinelli, F., Dash, S., Yeo, G. S., Savage, D. B., Leonetti, F., et al. (2010b). Morbid obesity exposes the association between PNPLA3 I148M (rs738409) and indices of hepatic injury in individuals of European descent. Int. J. Obes. (Lond) 34, 190–194. doi:10.1038/ijo.2009.216
Rotman, Y., Koh, C., Zmuda, J. M., Kleiner, D. E., and Liang, T. J. (2010). The association of genetic variability in patatin-like phospholipase domain-containing protein 3 (PNPLA3) with histological severity of nonalcoholic fatty liver disease. Hepatology 52, 894–903. doi:10.1002/hep.23759
Rung, J., Cauchi, S., Albrechtsen, A., Shen, L., Rocheleau, G., Cavalcanti-Proença, C., et al. (2009). Genetic variant near IRS1 is associated with type 2 diabetes, insulin resistance and hyperinsulinemia. Nat. Genet. 41, 1110–1115. doi:10.1038/ng.443
Said, M. A., Verweij, N., and Van Der Harst, P. (2018). Associations of combined genetic and lifestyle risks with incident cardiovascular disease and diabetes in the UK Biobank study. JAMA Cardiol. 3, 693–702. doi:10.1001/jamacardio.2018.1717
Samani, N. J., Erdmann, J., Hall, A. S., Hengstenberg, C., Mangino, M., Mayer, B., et al. (2007). Genomewide association analysis of coronary artery disease. N. Engl. J. Med. 357, 443–453. doi:10.1056/nejmoa072366
Samuel, V. T., Liu, Z.-X., Qu, X., Elder, B. D., Bilz, S., Befroy, D., et al. (2004). Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem. 279, 32345–32353. doi:10.1074/jbc.m313478200
Santoro, N., Caprio, S., Pierpont, B., Van Name, M., Savoye, M., and Parks, E. J. (2015). Hepatic de novo lipogenesis in obese youth is modulated by a common variant in the GCKR gene. J. Clin. Endocrinol. Metabolism 100, E1125–E1132. doi:10.1210/jc.2015-1587
Santoro, N., Zhang, C. K., Zhao, H., Pakstis, A. J., Kim, G., Kursawe, R., et al. (2012). Variant in the glucokinase regulatory protein (GCKR) gene is associated with fatty liver in obese children and adolescents. Hepatology 55, 781–789. doi:10.1002/hep.24806
Santos, R. D., Valenti, L., and Romeo, S. (2019). Does nonalcoholic fatty liver disease cause cardiovascular disease? Current knowledge and gaps. Atherosclerosis 282, 110–120. doi:10.1016/j.atherosclerosis.2019.01.029
Sanyal, A. J., Friedman, S. L., Mccullough, A. J., and Dimick-Santos, L. (2015). Challenges and opportunities in drug and biomarker development for nonalcoholic steatohepatitis: Findings and recommendations from an American association for the study of liver diseases-U.S. Food and drug administration joint workshop. Hepatology 61, 1392–1405. doi:10.1002/hep.27678
Satoh, T., Kidoya, H., Naito, H., Yamamoto, M., Takemura, N., Nakagawa, K., et al. (2013). Critical role of Trib1 in differentiation of tissue-resident M2-like macrophages. Nature 495, 524–528. doi:10.1038/nature11930
Sazci, A., Akpinar, G., Aygun, C., Ergul, E., Senturk, O., and Hulagu, S. (2008). Association of apolipoprotein E polymorphisms in patients with non-alcoholic steatohepatitis. Dig. Dis. Sci. 53, 3218–3224. doi:10.1007/s10620-008-0271-5
Scorletti, E., West, A. L., Bhatia, L., Hoile, S. P., Mccormick, K. G., Burdge, G. C., et al. (2015). Treating liver fat and serum triglyceride levels in NAFLD, effects of PNPLA3 and TM6SF2 genotypes: Results from the WELCOME trial. J. Hepatol. 63, 1476–1483. doi:10.1016/j.jhep.2015.07.036
Sentinelli, F., Romeo, S., Maglio, C., Incani, M., Burza, M. A., Scano, F., et al. (2011). Lack of effect of apolipoprotein C3 polymorphisms on indices of liver steatosis, lipid profile and insulin resistance in obese Southern Europeans. Lipids Health Dis. 10, 93. doi:10.1186/1476-511x-10-93
Shen, J., Wong, G. L., Chan, H. L., Chan, R. S., Chan, H. Y., Chu, W. C., et al. (2015). PNPLA3 gene polymorphism and response to lifestyle modification in patients with nonalcoholic fatty liver disease. J. Gastroenterol. Hepatol. 30, 139–146. doi:10.1111/jgh.12656
Simons, N., Dekker, J. M., Van Greevenbroek, M. M. J., Nijpels, G., ’T Hart, L. M., Van Der Kallen, C. J. H., et al. (2016). A common gene variant in glucokinase regulatory protein interacts with glucose metabolism on diabetic dyslipidemia: The combined CODAM and hoorn studies. Diabetes Care 39, 1811–1817. doi:10.2337/dc16-0153
Simons, N., Isaacs, A., Koek, G. H., Kuč, S., Schaper, N. C., and Brouwers, M. (2017). PNPLA3, TM6SF2, and MBOAT7 genotypes and coronary artery disease. Gastroenterology 152, 912–913. doi:10.1053/j.gastro.2016.12.020
Simons, P., Simons, N., Stehouwer, C. D. A., Schalkwijk, C. G., Schaper, N. C., and Brouwers, M. (2018). Association of common gene variants in glucokinase regulatory protein with cardiorenal disease: A systematic review and meta-analysis. PLoS One 13, e0206174. doi:10.1371/journal.pone.0206174
Sliz, E., Sebert, S., Würtz, P., Kangas, A. J., Soininen, P., Lehtimäki, T., et al. (2018). NAFLD risk alleles in PNPLA3, TM6SF2, GCKR and LYPLAL1 show divergent metabolic effects. Hum. Mol. Genet. 27, 2214–2223. doi:10.1093/hmg/ddy124
Softic, S., Gupta, M. K., Wang, G. X., Fujisaka, S., O'neill, B. T., Rao, T. N., et al. (2017). Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J. Clin. Invest 127, 4059–4074. doi:10.1172/jci94585
Song, C., Burgess, S., Eicher, J. D., O'donnell, C. J., Johnson, A. D., Huang, J., et al. (2017). Causal effect of plasminogen activator inhibitor type 1 on coronary heart disease. J. Am. Heart Assoc. 6, e004918. doi:10.1161/JAHA.116.004918
Song, J., Da Costa, K. A., Fischer, L. M., Kohlmeier, M., Kwock, L., Wang, S., et al. (2005). Polymorphism of the PEMT gene and susceptibility to nonalcoholic fatty liver disease (NAFLD). FASEB J. 19, 1266–1271. doi:10.1096/fj.04-3580com
Sookoian, S., Castaño, G. O., Scian, R., Mallardi, P., Fernández Gianotti, T., Burgueño, A. L., et al. (2015). Genetic variation in transmembrane 6 superfamily member 2 and the risk of nonalcoholic fatty liver disease and histological disease severity. Hepatology 61, 515–525. doi:10.1002/hep.27556
Sookoian, S., and Pirola, C. J. (2011). Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology 53, 1883–1894. doi:10.1002/hep.24283
Soubeyrand, S., Martinuk, A., Naing, T., Lau, P., and Mcpherson, R. (2016). Role of Tribbles Pseudokinase 1 (TRIB1) in human hepatocyte metabolism. Biochim. Biophys. Acta 1862, 223–232. doi:10.1016/j.bbadis.2015.12.003
Speliotes, E. K., Butler, J. L., Palmer, C. D., Voight, B. F., and Hirschhorn, J. N. (2010). PNPLA3 variants specifically confer increased risk for histologic nonalcoholic fatty liver disease but not metabolic disease. Hepatology 52, 904–912. doi:10.1002/hep.23768
Speliotes, E. K., Yerges-Armstrong, L. M., Wu, J., Hernaez, R., Kim, L. J., Palmer, C. D., et al. (2011). Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 7, e1001324. doi:10.1371/journal.pgen.1001324
Stefan, N., Fritsche, A., Schick, F., and Häring, H. U. (2016). Phenotypes of prediabetes and stratification of cardiometabolic risk. Lancet Diabetes Endocrinol. 4, 789–798. doi:10.1016/s2213-8587(16)00082-6
Stender, S., Kozlitina, J., Nordestgaard, B. G., Tybjærg-Hansen, A., Hobbs, H. H., and Cohen, J. C. (2017). Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nat. Genet. 49, 842–847. doi:10.1038/ng.3855
Stender, S., and Loomba, R. (2020). PNPLA3 genotype and risk of liver and all-cause mortality. Hepatology 71, 777–779. doi:10.1002/hep.31113
Stumvoll, M., and Jacob, S. (1999). Multiple sites of insulin resistance: Muscle, liver and adipose tissue. Exp. Clin. Endocrinol. diabetes 107, 107–110. doi:10.1055/s-0029-1212083
Tabas, I., García-Cardeña, G., and Owens, G. K. (2015). Recent insights into the cellular biology of atherosclerosis. J. Cell Biol. 209, 13–22. doi:10.1083/jcb.201412052
Tada, H., Melander, O., Louie, J. Z., Catanese, J. J., Rowland, C. M., Devlin, J. J., et al. (2015). Risk prediction by genetic risk scores for coronary heart disease is independent of self-reported family history. Eur. Heart J. 37, 561–567. doi:10.1093/eurheartj/ehv462
Takeuchi, F., Yokota, M., Yamamoto, K., Nakashima, E., Katsuya, T., Asano, H., et al. (2012). Genome-wide association study of coronary artery disease in the Japanese. Eur. J. Hum. Genet. 20, 333–340. doi:10.1038/ejhg.2011.184
Tang, C. S., Zhang, H., Cheung, C. Y. Y., Xu, M., Ho, J. C. Y., Zhou, W., et al. (2015). Exome-wide association analysis reveals novel coding sequence variants associated with lipid traits in Chinese. Nat. Commun. 6, 10206. doi:10.1038/ncomms10206
Targher, G., Byrne, C. D., Lonardo, A., Zoppini, G., and Barbui, C. (2016). Non-alcoholic fatty liver disease and risk of incident cardiovascular disease: A meta-analysis. J. Hepatol. 65, 589–600. doi:10.1016/j.jhep.2016.05.013
Targher, G., Day, C. P., and Bonora, E. (2010). Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N. Engl. J. Med. 363, 1341–1350. doi:10.1056/nejmra0912063
Taskinen, M. R. (2003). Diabetic dyslipidaemia: From basic research to clinical practice. Diabetologia 46, 733–749. doi:10.1007/s00125-003-1111-y
Teslovich, T. M., Musunuru, K., Smith, A. V., Edmondson, A. C., Stylianou, I. M., Koseki, M., et al. (2010). Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466, 707–713. doi:10.1038/nature09270
Tiret, L., Mallet, C., Poirier, O., Nicaud, V., Millaire, A., Bouhour, J.-B., et al. (2000). Lack of association between polymorphisms of eight candidate genes and idiopathic dilated cardiomyopathy. J. Am. Coll. Cardiol. 35, 29–35. doi:10.1016/s0735-1097(99)00522-7
Tokushige, K., Takakura, M., Tsuchiya-Matsushita, N., Taniai, M., Hashimoto, E., and Shiratori, K. (2007). Influence of TNF gene polymorphisms in Japanese patients with NASH and simple steatosis. J. Hepatology 46, 1104–1110. doi:10.1016/j.jhep.2007.01.028
Trépo, E., Romeo, S., Zucman-Rossi, J., and Nahon, P. (2016). PNPLA3 gene in liver diseases. J. Hepatol. 65, 399–412. doi:10.1016/j.jhep.2016.03.011
Trujillo-Murillo, K., Bosques-Padilla, F. J., Calderón-Lozano, I., Navar-Vizcarra, S., Garza-González, E., Niderhauser-García, A., et al. (2011). Association of tumor necrosis factor α and manganese superoxide dismutase polymorphisms in patients with non-alcoholic steatohepatitis from northeast Mexico. Open Hepatology J. 3. doi:10.2174/1876517301103010001
Unalp-Arida, A., and Ruhl, C. E. (2020). Patatin-like phospholipase domain-containing protein 3 I148M and liver fat and fibrosis scores predict liver disease mortality in the U.S. Population. Hepatology 71, 820–834. doi:10.1002/hep.31032
Valenti, L., Al-Serri, A., Daly, A. K., Galmozzi, E., Rametta, R., Dongiovanni, P., et al. (2010a). Homozygosity for the patatin-like phospholipase-3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 51, 1209–1217. doi:10.1002/hep.23622
Valenti, L., Alisi, A., Galmozzi, E., Bartuli, A., Del Menico, B., Alterio, A., et al. (2010b). I148M patatin-like phospholipase domain-containing 3 gene variant and severity of pediatric nonalcoholic fatty liver disease. Hepatology 52, 1274–1280. doi:10.1002/hep.23823
Valenti, L., Fracanzani, A. L., Dongiovanni, P., Santorelli, G., Branchi, A., Taioli, E., et al. (2002). Tumor necrosis factor α promoter polymorphisms and insulin resistance in nonalcoholic fatty liver disease. Gastroenterology 122, 274–280. doi:10.1053/gast.2002.31065
Vance, D. E. (2014). Phospholipid methylation in mammals: From biochemistry to physiological function. Biochim. Biophys. Acta 1838, 1477–1487. doi:10.1016/j.bbamem.2013.10.018
Vance, D. E., and Ridgway, N. D. (1988). The methylation of phosphatidylethanolamine. Prog. Lipid Res. 27, 61–79. doi:10.1016/0163-7827(88)90005-7
Vance, D. E., Walkey, C. J., and Cui, Z. (1997). Phosphatidylethanolamine N-methyltransferase from liver. Biochimica Biophysica Acta (BBA) - Lipids Lipid Metabolism 1348, 142–150. doi:10.1016/s0005-2760(97)00108-2
Vance, J. E., and Vance, D. E. (1985). The role of phosphatidylcholine biosynthesis in the secretion of lipoproteins from hepatocytes. Can. J. Biochem. Cell Biol. 63, 870–881. doi:10.1139/o85-108
Vanni, E., Bugianesi, E., Kotronen, A., De Minicis, S., Yki-Järvinen, H., and Svegliati-Baroni, G. (2010). From the metabolic syndrome to NAFLD or vice versa? Dig. Liver Dis. 42, 320–330. doi:10.1016/j.dld.2010.01.016
Vermeulen, P. S., Lingrell, S., Yao, Z., and Vance, D. E. (1997). Phosphatidylcholine biosynthesis is required for secretion of truncated apolipoprotein Bs from McArdle RH7777 cells only when a neutral lipid core is formed. J. Lipid Res. 38, 447–458. doi:10.1016/s0022-2275(20)37253-9
Walker, A. K., Jacobs, R. L., Watts, J. L., Rottiers, V., Jiang, K., Finnegan, D. M., et al. (2011). A conserved SREBP-1/phosphatidylcholine feedback circuit regulates lipogenesis in metazoans. Cell 147, 840–852. doi:10.1016/j.cell.2011.09.045
Wang, J.-K., Feng, Z.-W., Li, Y.-C., Li, Q.-Y., and Tao, X.-Y. (2012). Association of tumor necrosis factor-α gene promoter polymorphism at sites -308 and -238 with non-alcoholic fatty liver disease: A meta-analysis. J. Gastroenterology Hepatology 27, 670–676. doi:10.1111/j.1440-1746.2011.06978.x
Wang, L., Jing, J., Fu, Q., Tang, X., Su, L., Wu, S., et al. (2015). Association study of genetic variants at newly identified lipid gene TRIB1 with coronary heart disease in Chinese Han population. Lipids Health Dis. 14, 46. doi:10.1186/s12944-015-0043-0
Wang, M., Menon, R., Mishra, S., Patel, A. P., Chaffin, M., Tanneeru, D., et al. (2020). Validation of a genome-wide polygenic score for coronary artery disease in south asians. J. Am. Coll. Cardiol. 76, 703–714. doi:10.1016/j.jacc.2020.06.024
Wattacheril, J., Issa, D., and Sanyal, A. (2018). Nonalcoholic steatohepatitis (NASH) and hepatic fibrosis: Emerging therapies. Annu. Rev. Pharmacol. Toxicol. 58, 649–662. doi:10.1146/annurev-pharmtox-010617-052545
Weintraub, M. S., Eisenberg, S., and Breslow, J. L. (1987). Different patterns of postprandial lipoprotein metabolism in normal, type IIa, type III, and type IV hyperlipoproteinemic individuals. Effects of treatment with cholestyramine and gemfibrozil. J. Clin. Investigation 79, 1110–1119. doi:10.1172/jci112926
Willer, C. J., Sanna, S., Jackson, A. U., Scuteri, A., Bonnycastle, L. L., Clarke, R., et al. (2008). Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat. Genet. 40, 161–169. doi:10.1038/ng.76
Wobst, J., Schunkert, H., and Kessler, T. (2018). Genetic alterations in the NO-cGMP pathway and cardiovascular risk. Nitric Oxide 76, 105–112. doi:10.1016/j.niox.2018.03.019
Wong, V. W.-S., Wong, G. L.-H., Tsang, S. W.-C., Hui, A. Y., Chan, A. W.-H., Choi, P. C.-L., et al. (2008). Genetic polymorphisms of adiponectin and tumor necrosis factor-alpha and nonalcoholic fatty liver disease in Chinese People. J. Gastroenterology Hepatology 23, 914–921. doi:10.1111/j.1440-1746.2008.05344.x
Xi, G., Shen, X., Wai, C., White, M. F., and Clemmons, D. R. (2019). Hyperglycemia induces vascular smooth muscle cell dedifferentiation by suppressing insulin receptor substrate-1–mediated p53/KLF4 complex stabilization. J. Biol. Chem. 294, 2407–2421. doi:10.1074/jbc.RA118.005398
Xu, J., Cui, G., Esmailian, F., Plunkett, M., Marelli, D., Ardehali, A., et al. (2004). Atrial extracellular matrix remodeling and the maintenance of atrial fibrillation. Circulation 109, 363–368. doi:10.1161/01.cir.0000109495.02213.52
Xu, Y., Liu, H., Liu, M., Li, F., Liu, L., Du, F., et al. (2016). A human apolipoprotein E mimetic peptide reduces atherosclerosis in aged apolipoprotein E null mice. Am. J. Transl. Res. 8, 3482–3492.
Yao, Z. M., and Vance, D. E. (1989). Head group specificity in the requirement of phosphatidylcholine biosynthesis for very low density lipoprotein secretion from cultured hepatocytes. J. Biol. Chem. 264, 11373–11380. doi:10.1016/s0021-9258(18)60474-0
Yao, Z. M., and Vance, D. E. (1988). The active synthesis of phosphatidylcholine is required for very low density lipoprotein secretion from rat hepatocytes. J. Biol. Chem. 263, 2998–3004. doi:10.1016/s0021-9258(18)69166-5
Younossi, Z. M., Koenig, A. B., Abdelatif, D., Fazel, Y., Henry, L., and Wymer, M. (2016). Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64, 73–84. doi:10.1002/hep.28431
Zain, S. M., Mohamed, R., Mahadeva, S., Cheah, P. L., Rampal, S., Basu, R. C., et al. (2012). A multi-ethnic study of a PNPLA3 gene variant and its association with disease severity in non-alcoholic fatty liver disease. Hum. Genet. 131, 1145–1152. doi:10.1007/s00439-012-1141-y
Zeggini, E., Scott, L. J., Saxena, R., Voight, B. F., Marchini, J. L., Hu, T., et al. (2008). Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat. Genet. 40, 638–645. doi:10.1038/ng.120
Zhang, D., Liu, Z.-X., Choi, C. S., Tian, L., Kibbey, R., Dong, J., et al. (2007). Mitochondrial dysfunction due to long-chain Acyl-CoA dehydrogenase deficiency causes hepatic steatosis and hepatic insulin resistance. Proc. Natl. Acad. Sci. 104, 17075–17080. doi:10.1073/pnas.0707060104
Zhang, H., Sun, X., and Liu, Y. (2011). Food safety and technological implications of food traceability systems. IFIP Adv. Inf. Commun. Technol. 345, 1–10. doi:10.1007/978-3-642-18336-2_1
Zhang, X., Zhang, B., Zhang, C., Sun, G., and Sun, X. (2021). Current progress in delineating the roles of pseudokinase TRIB1 in controlling human diseases. J. Cancer 12, 6012–6020. doi:10.7150/jca.51627
Zhao, Q. R., Lei, Y. Y., Li, J., Jiang, N., and Shi, J. P. (2017). Association between apolipoprotein E polymorphisms and premature coronary artery disease: A meta-analysis. Clin. Chem. Laboratory Med. (CCLM) 55, 284–298. doi:10.1515/cclm-2016-0145
Zhao, W., Du, F., Zhang, M., Sun, S., Yu, H., and Fan, D. (2011). A new recombinant human apolipoprotein E mimetic peptide with high-density lipoprotein binding and function enhancing activity. Exp. Biol. Med. 236, 1468–1476. doi:10.1258/ebm.2011.011169
Zhou, Y.-J., Li, Y.-Y., Nie, Y.-Q., Yang, H., Zhan, Q., Huang, J., et al. (2010). Influence of polygenetic polymorphisms on the susceptibility to non-alcoholic fatty liver disease of Chinese People. J. Gastroenterology Hepatology 25, 772–777. doi:10.1111/j.1440-1746.2009.06144.x
Keywords: genetics, cardiovascular diseases, non-alcoholic fatty liver disease, coronary artery disease, cardiomyopathy, atrial fibrillation
Citation: Chew NWS, Chong B, Ng CH, Kong G, Chin YH, Xiao W, Lee M, Dan YY, Muthiah MD and Foo R (2022) The genetic interactions between non-alcoholic fatty liver disease and cardiovascular diseases. Front. Genet. 13:971484. doi: 10.3389/fgene.2022.971484
Received: 17 June 2022; Accepted: 19 July 2022;
Published: 10 August 2022.
Edited by:
Wanqing Liu, Wayne State University, United StatesReviewed by:
Zheyun Peng, Wayne State University, United StatesCopyright © 2022 Chew, Chong, Ng, Kong, Chin, Xiao, Lee, Dan, Muthiah and Foo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicholas W.S. Chew, bmljaG9sYXNfd3NfY2hld0BudWhzLmVkdS5zZw==; Roger Foo, cm9nZXIuZm9vQG51cy5lZHUuc2c=
†ORCID: Nicholas Chew, orcid.org/0000-0002-0640-0430
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.