 Lijuan Huang
Lijuan Huang Maosheng Guo
Maosheng Guo Ningdong Li
Ningdong Li- 1Department of Ophthalmology, The Second Affiliated Hospital of Fujian Medical University, Quanzhou, China
- 2Department of Ophthalmology, Beijing Children’s Hospital, Capital Medical University, Beijing, China
- 3Key Laboratory of Major Diseases in Children, Ministry of Education, Beijing, China
- 4Department of Ophthalmology, Children’s Hospital, Capital Institute of Pediatrics, Beijing, China
Background: Waardenburg syndrome (WS) is a rare genetic disorder characterized by congenital sensorineural hearing loss and pigmentary abnormalities of the hair, skin and eyes. However, exotropia is rarely reported. The purpose of this study is to describe the clinical characteristics of three sporadic patients with WS and congenital exotropia and to investigate the disease-causing genes for them.
Methods: Patients underwent detailed physical and ocular examinations. Ocular alignment and binocular status were evaluated. DNA was extracted and whole exome sequencing was performed to detect the pathogenic variations in the disease-causing genes for WS. Cloning sequencing was carried out for those indel variations.
Results: Three unrelated patients were diagnosed with Waardenburg syndrome and congenital exotropia. Four novel variants, including c.136delA (p.I46Sfs*64) and c.668G>T (p.R223L) in PAX3, c.709dupC (p.Q237Pfs*119) in COL11A2, c.426G>A (p.W142X) in SOX10 gene, were detected in this study.
Conclusion: Simultaneous presence of congenital exotropia and WS in our patients is suggested that WS could be involved in malfunction in the multiple nerve systems. Our genetic study will expand the mutation spectrum of PAX3, COL11A2 and SOX10 genes, and is helpful for further study on the molecular pathogenesis of WS.
Introduction
Waardenburg syndrome (WS) is characterized by congenital sensorineural hearing loss and pigmentary abnormalities of the hair, skin and eyes, and can be inherited as an autosomal dominant or an autosomal recessive trait, with an estimated prevalence of 1/42,000 in the global population (Read and Newton, 1997). With the exceptions of sensorineural hearing loss and pigmentary abnormalities, many other clinical features are reported as dystopia canthorum, broad nasal root, bushy eyebrows with synophrys, and abnormalities in the limb, neurologic and visceral organs (Farrer et al., 1992; Gowda and Srinivasan, 2020). Based on the clinical features and molecular diagnosis, WS may be classified into 1–4 four subtypes (Ahmed jan et al., 2022).
WS type 1 (WS1, OMIM193500) is inherited as an autosomal dominant trait and caused by mutations of the PAX3 gene. WS type 2 (WS2) is caused by mutations of MITF (OMIM 193510), SNAI2 (OMIM 608890) and SOX10 (OMIM 611584) genes (Somashekar et al., 2019; Huang et al., 2021). Patients with WS2 do not have dystopia canthorum, which is distinguished from WS1. WS type 3 (WS3, OMIM 148820) is caused by the PAX3 mutations as well, and is distinguished from WS1 by abnormalities of the upper limb. WS type 4 (WS4, OMIM 277580), also known as Waardenburg-Shah syndrome, is usually associated with congenital megacolon and other gastrointestinal malformations. Mutations of EDNRB (OMIM 277580), EDN3 (OMIM 613265) and SOX10 genes have been reported to be responsible for WS4 (Touraine et al., 2000; Verheij and Hofstra, 2016).
Exotropia (XT) is a common form of strabismus with an estimated prevalence of 1% in the population (Govindan et al., 2005). Its etiology is not clear. It is suggested that exotropia could be the interactive results between dynamic and static factors (Von Noorden, 2002). Dynamic factors refer to those innervational factors that tend to maintain ocular alignment through establishing equilibrium between convergence and divergence. Static factors refer to those anatomical factors consisting of orbital structure, anatomy of extraocular muscles and muscle pulleys. XT can be congenital or acquired, determined by the onset of age before or after 6 months.
In the previous reports, exotropia is rarely documented as one of clinical features of Waardenburg syndrome (Dastur et al., 1995). Here, we describe the clinical features of three patients with WS and exotropia, and identify the disease-causing gene variants for these patients.
Case presentation
Clinical features
Patient 01 from Family 01 was a 22-year old male. Medical history was negative for family history, brain injury, prematurity or hypoxia at his delivery. He had developed a constant exotropia at the age of 4 months but had not undergone surgical treatment for his strabismus. He showed a large angle exotropia of more than 45° in primary gaze position (Figure 1A). He was diagnosed as Waardenburg syndrome type 3 based on his dystopia canthorum, broad nasal root, synophrys, heterochromia irides, hypopigmented fundus appearance, normal fovea and malformed fingers, and hearing loss (Figures 1A, D, G; Figures 2A–C). His best-corrected visual acuity was 20/20 in the right eye and 16/20 in the left eye. He did not have binocular vision assessed through the Bagolini striated glasses and fixated alternatively with each of his eyes. Neurological and mental disorders were excluded by a neurologist.
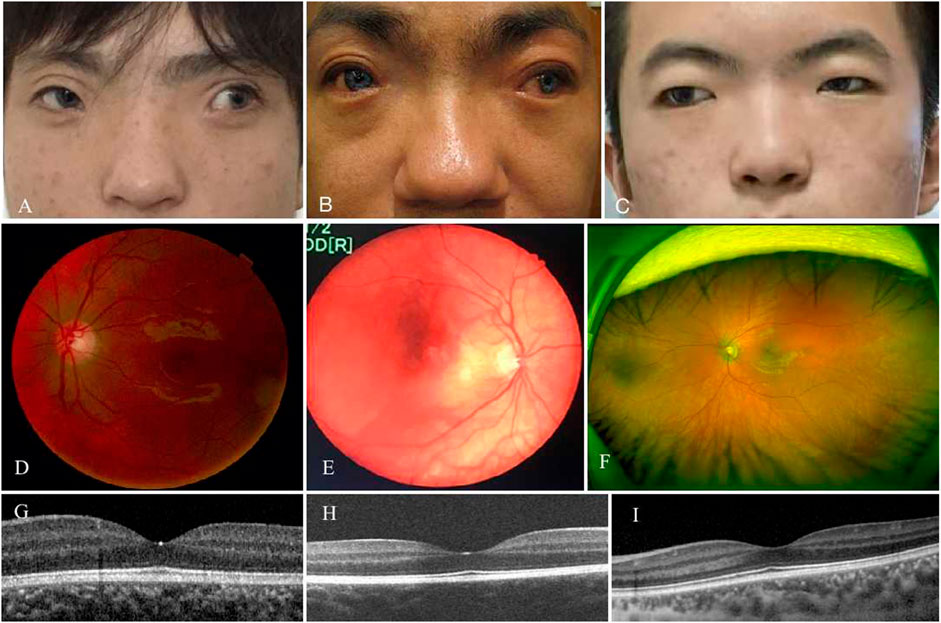
FIGURE 1. Phenotypes of three patients with WS and exotropia. All patients had an large angle exotropia (A–C). They were diagnosed as WS by dystopia canthorum, broad nasal root, synophrys (A–C), and hypopigmented fundus (D–F). Their fovea structure were normal (G–I).
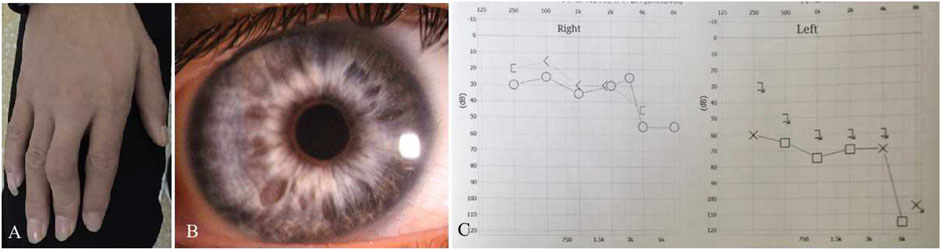
FIGURE 2. Clinical features of Patient 01. He had deformity of the fingers (A) and heterochromia irides (B). The audiograms showed that his hearing was severely reduced on the left more than the right side (C). X-axis, frequency in hertz (Hz); Y-axis, hearing level in decibels (dB).
Patient 02 from Family 02 was a 31-year old male. He had a notable appearance of a bald head, exotropia, broad nasal root, blue color irides, and bushy eyebrows with synophrys on his face (Figure 1B). Medical history showed that he was normally delivered without brain injury, hypoxia or prematurity at birth. He had exotropia at the age of 4∼5 months. His exotropia was intermittent before the age of 5 years, and progressively developed to a constant exotropia with growth. He never underwent strabismus surgery before visiting our clinic. He complained that his “blue eyes” came from his father. Ocular examination showed that his best-corrected visual acuity was 20/20 in the right eye and 10/20 in the left eye. He had hypopigmented fundus appearance as well (Figure 1E) and normal fovea pit (Figure 1H). Examinations of ocular alignment revealed that he had a constant exotropia with a large deviation angle of 100 prism diopter (PD) at near and distance. Prior to genetic testing, diagnosis of Waardenburg syndrome type 1 was considered according to his synophrys, broad nasal root, pigmentary disturbance of iris, hypopigmented fundus, and hearing loss in his left ear. Neurologic and visceral abnormalities were excluded after physical examinations.
Patient 03 from Family 03 was a 19-year-old male and was not found to have neurological or mental disorders or family history in his medical records. He developed a constant exotropia at the age of 5 months, and presented as a large angle exotropia of more than 35° in primary gaze position, without binocular vision and fixating alternatively with each of his eyes. He was diagnosed as Waardenburg syndrome type 1 in terms of his dystopia canthorum, broad nasal root, synophrys, depigmented irides, and hypopigmented fundus (Figures 1C,F,I). His best-corrected visual acuity was 18/20 in both eyes.
Genetic analysis
Genetic testing revealed that a double gene variation was detected in Patient 01 who carried the pathogenic variations of c.136delA (p.Ile46Serfs*64) in exon2 in PAX3 and c.709dupC (p.Gln237Profs*119) in exon5 in COL11A2. Both of these variations were novel identified in this study, and predicted to produce an abnormal mRNA with a premature termination codon (PTC), which would be probably degraded due to the nonsense mediated decay (NMD) mechanism (Figure 3A). A novel heterozygous variation of c.426G>A (p.Trp142X) in the SOX10 gene was detected in Patient 02 (Figure 3B). This variation would lead to a wild-type amino acid of Tryptophan at Codon 142 replaced by a premature termination codon, which would be degraded due to NMD mechanism. The novel identified pathogenic variation of c.668G>T (p.Arg223Leu) in PAX3 (Figure 3C) was detected in Patient 03. A positive polar charged Argnine at codon 223 replaced by a nonpolar Leucine would be predicted to damage the stability of the protein structure and function, which was proven in the 3-D model construction using the PyMOL program (Figure 4). All above four variations were not detected in 100 ethnically matched control individuals through the Sanger sequencing. The pathogenicity assessment was listed in Table 1, based on the ACMG guidelines.
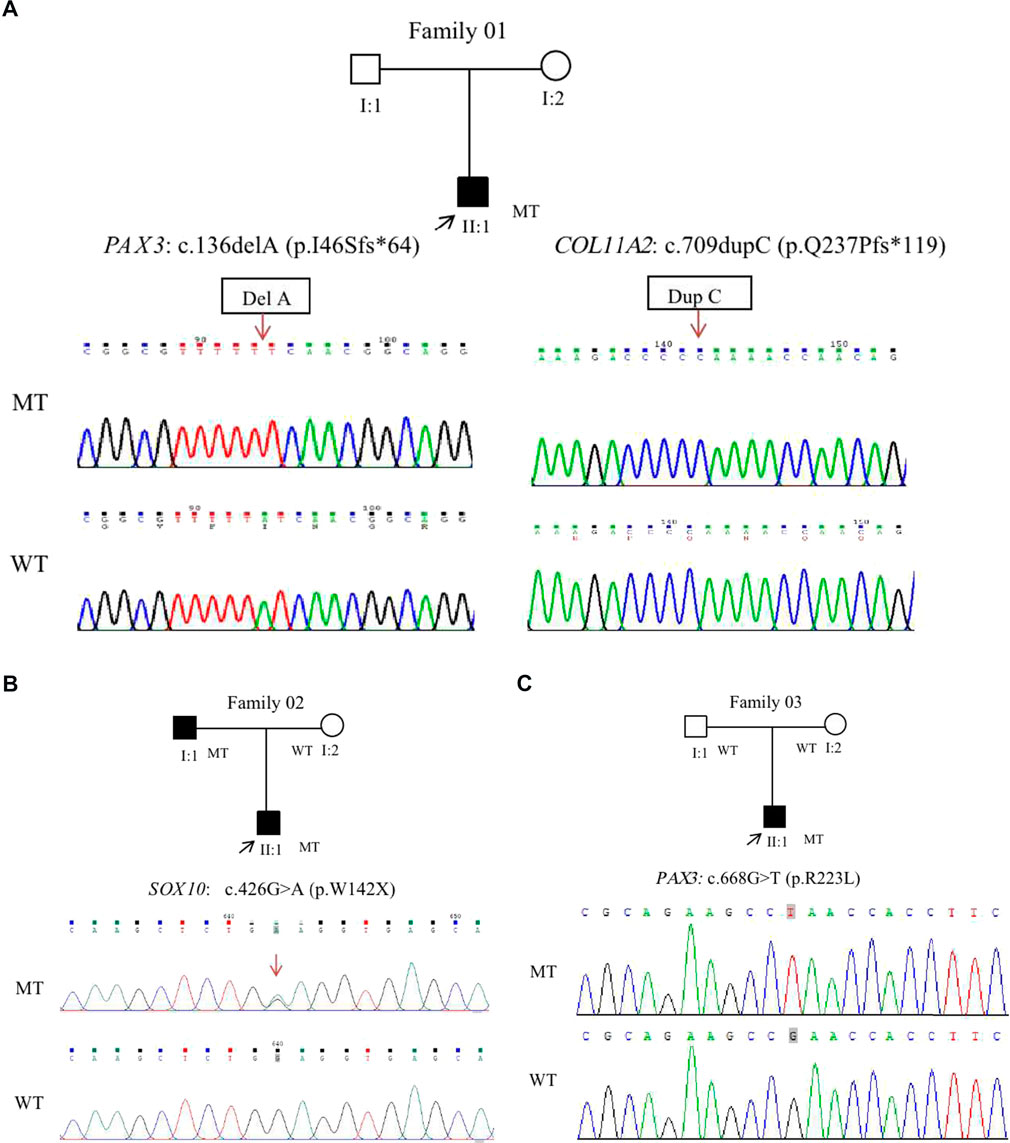
FIGURE 3. Four novel variants were identified from three WS patients in this study. A double gene variant in the PAX3 and COL11A2 was detected in Patient 01. Cloning sequencing showed that a single nucleotide deletion of c.136del A was detected in the PAX3, and a single nucleotide duplication of c.709dupC was found in the COL11A2 gene (A). A heterozygote missense variation of c.426G>A in SOX10 was detected in the Patient 02 (B). This variant resulted in an amino acid of Tryptophan (W) at Codon 142 replaced by a premature terminate codon. A novel heterozygote missense variation of c.668G>T in PAX3 was detected in Patient 03 (C). The variant led to an amino acid of Arginine at Codon 223 replaced by Leucine. Mutant type (upper row), wide type (lower row); the arrows denote the locations of the indel mutations.
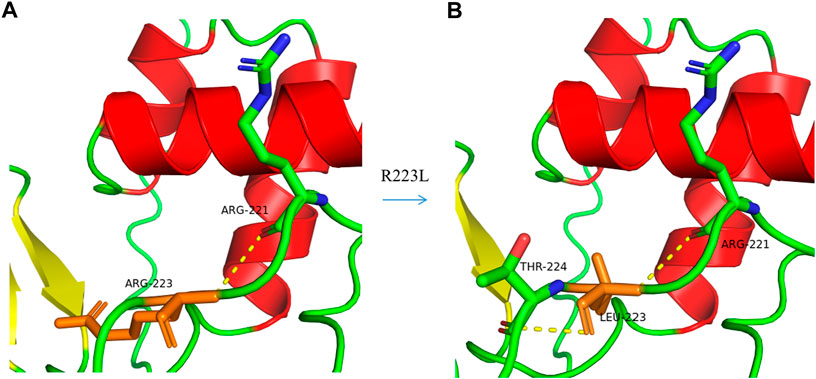
FIGURE 4. A 3-D model construction showed that the novel missense variation of c .668G>T in PAX3 resulted in a large and positively charged amino acid of Arginine (R) at codon 223 being replaced by small and uncharged nonpolar amino acid of Leucine (L). The wild-type Arginine formed a hydrogen bond with R221 (A), while Leucine could form hydrogen bonds with R221 and T224 (B), which would damage the stability of the protein structure and function.
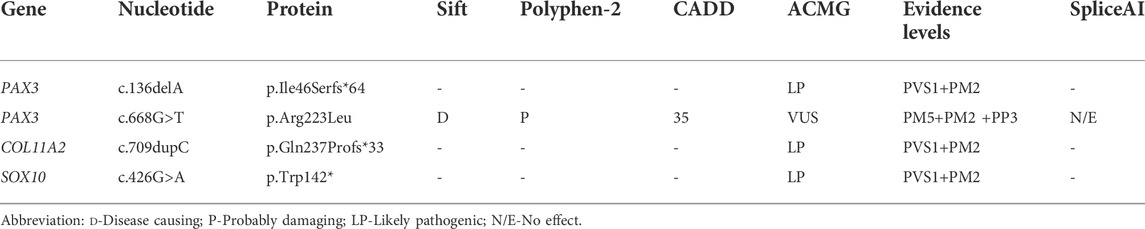
TABLE 1. Summary of Detected WS-related Novel Variants in this Study.
Discussion
In this study, we have described the clinical features of three patients with WS and large exotropia. The most prominent characteristic of our patients was their large angle congenital exotropia. Previous studies showed that few patients with WS had strabismus, and of those patients esotropia was common but exotropia was rare (Delleman and Hageman, 1978; Tang et al., 2020). In one of the reports, five of 26 patients (19%) with WS had esotropia (Delleman and Hageman, 1978). However, only two patients were described with mild exotropia in two separated reports (Dastur et al., 1995). To our knowledge, congenital exotropia with a large angle deviation is reported for the first time in our patients with WS.
Waardenburg syndrome (WS) is believed to be caused by anomalies in neural crest cell development and migration, which results in abnormal distribution of the NSC-derived melanocytes and produces an abnormal color in the hair and iris (Huang et al., 2021). In addition, another significant feature of WS is hearing loss caused by acoustic nerve injury (Boudewyns et al., 2020). As a company symptom, exotropia occurs in all of our WS patients. However, we are not sure the exact cause of their exotropia. We assume that it could be caused by abnormal nerve development due to anomalies in neural crest cell development and migration, based on the fact that congenital exotropia is usually associated with neurological disorders, craniofacial anomalies, and cerebral palsy (Choi and Kim, 2013; Lueder and Galli, 2018). In addition, recent studies demonstrate that melanin is involved in nerve development. Melanin metabolic disorders such as oculocutaneous albinism (OCA) and ocular albinism (OA) usually present as reduced visual acuity, strabismus and nystagmus (Federico and Krishnamurthy, 2022).
With the exception of exotropia, our patients present clinical and genetic heterogeneity with clinical manifestations and genotypes that are not completely consistent. Patient 01 is presumed to have WS3 because of the deformity of his fingers. He carries a double mutation of PAX3 and COL11A2. Mutations of the PAX3 gene have been recognized to be the underlying molecular pathogenesis of WS1 and WS3. As a member of the paired box family of transcription factors, PAX3 plays an important role in neural development and myogenesis during the fetal period (Boudjadi et al., 2018). Mutations of PAX3 have been proven to be associated with hearing loss (Kim et al., 2014). COL11A2 gene is a protein-coding gene, playing an important role in fibrillogenesis (Vázquez-Villa et al., 2015). Mutations of COL11A2 result in deafness in patients with DFNA13 (non-syndromic sensorineural deafness type 13) (Kunst et al., 2000). To our knowledge, hearing loss caused by a double gene mutation of both PAX3 and COL11A2 has not been reported in previous studies. However, we are not sure whether or not these two mutations would have a superimposed effect on hearing damage. We also are not able to compare the severity of hearing loss caused by a single gene mutation with a double gene mutation due to the small sample size.
As a member of the SRY-related HMG-box family, SOX10 plays an important role in embryonic development, especially in the nervous system, determining the cell fate (Røge et al., 2017). Mutations of SOX10 may result in either the WS2 or the WS4 (Ahmed jan et al., 2022). Patient 02 was detected to have a nonsense variant of c.426G>A (p.W142X) in SOX10 gene. Because congenital megacolon and other gastrointestinal disorders were excluded, he was regarded as WS2. However, with clinical findings alone, he would be diagnosed as WS1 due to his dystopia canthorum, which is traditionally noted to be absent from WS2. This conflict illustrates the clinical and genetic heterogeneities that exist in Waardenburg syndrome. Only Patient 03 was diagnosed as WS1 based on his typical characteristics of dystopia canthorum, broad nasal root, synophrys, depigmented iris and fundus. His molecular diagnosis further supported he is WS1 because he carries with a pathogenic variation of c.668G>T (p.R223L) in PAX3.
In summary, we present three separate cases of WS with congenital exotropia and find four novel variants in PAX3, COL11A2 and SOX10 genes. Our finding would expand the mutation spectrum for these three genes. In addition, we find that there is not a widespread association between WS and congenital exotropia, based on our review of the literature. Through genetic experiments, we find that a rare double gene mutation of PAX3 and COL11A2 could cause hearing loss in our patient with WS. All mutations detected in PAX3, COL11A2 and SOX10 are heterozygous variations. As an autosomal dominant disorder, haplotype insufficiency could be the underlying molecular pathogenesis caused by these mutations because these variants are predicted to produce abnormal mRNA and to be degraded by NMD mechanism.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by the Beijing Children's Hospital. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author contributions
LH did the clinical investigation of patients and drafted the manuscript. MG did the clinical investigation of patients. NL did clinical advice and critical revision of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (81670883), and Key Clinical Specialty Discipline Construction Program of Fujian, P. R. C (Fujian Health Medicine and Politics [2022]884).
Acknowledgments
The authors would like to thank the patients and their families.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a shared affiliation with the author(s) (NL) at the time of review.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ahmed jan, N., Mui, R. K., and Masood, S. (2022). Waardenburg syndrome. Tampa, Florida, United States: Treasure Island: StatPearls. Internet.
Boudewyns, A., van den Ende, J., Declau, F., Wuyts, W., Peeters, N., Brandt, A. H. D., et al. (2020). Etiological work-up in referrals from neonatal hearing screening: 20 Years of experience. Otol. Neurotol. 41 (9), 1240–1248. doi:10.1097/MAO.0000000000002758
Boudjadi, S., Chatterjee, B., Sun, W., Vemu, P., and Barr, F. G. (2018). The expression and function of PAX3 in development and disease. Gene 666, 145–157. doi:10.1016/j.gene.2018.04.087
Choi, Y. M., and Kim, S. H. (2013). Comparison of clinical features between two different types of exotropia before 12 Months of age based on stereopsis outcome. Ophthalmology 120 (1), 3–7. doi:10.1016/j.ophtha.2012.07.062
Dastur, Y. K., Chitale, A., Dasgupta, S., and DudhAni, A. (1995). Waardenburg syndrome with anisocoria and exotropia. J. Postgrad. Med. 41 (4), 111–112.
Delleman, J. W., and Hageman, M. J. (1978). Ophthalmological findings in 34 patients with Waardenburg syndrome. J. Pediatr. Ophthalmol. Strabismus 15 (6), 341–345. doi:10.3928/0191-3913-19781101-03
Farrer, L. A., Grudfast, K. M., Amos, J., Arnos, K. S., Asher, J. H., Beighton, P., et al. (1992). Waardenburg syndrome (WS) type I is caused by defects at multiple loci, one of which is near ALPP on chromosome 2: First report of the WS consortium. Am. J. Hum. Genet. 50 (5), 902–913.
Federico, J. R., and Krishnamurthy, K. (2022). Albinism. Tampa, Florida, United States: Treasure Island (FL): StatPearls.
Govindan, M., Mohney, B. G., Diehl, N., and Burke, J. (2005). Incidence and types of childhood exotropia: A population-based study. Ophthalmology 112, 104–108. doi:10.1016/j.ophtha.2004.07.033
Gowda, V. K., and Srinivasan, V. M. (2020). Waardenburg syndrome type I. Indian J. Pediatr. 87 (3), 244. doi:10.1007/s12098-019-03170-5
Huang, S., Song, J., He, C., Cai, X., Yuan, K., Mei, L., et al. (2021). Genetic insights, disease mechanisms, and biological therapeutics for Waardenburg syndrome. Gene Ther. (25), 1–19. doi:10.1038/s41434-021-00240-2
Kim, H. K., Ankamreddy, H., Lee, D. J., Kong, K. A., Ko, H. W., Kim, M. H., et al. (2014). Pax3 function is required specifically for inner ear structures with melanogenic fates. Biochem. Biophys. Res. Commun. 445 (3), 608–614. doi:10.1016/j.bbrc.2014.02.047
Kunst, H., Huybrechts, C., Marres, H., Huygen, P., Cremers, C., and Van Camp, G. (2000). The phenotype of DFNA13/col11a2: Nonsyndromic autosomal dominant mid-frequency and high-frequency sensorineural hearing impairment. Am. J. Otol. 21 (2), 181–187. doi:10.1016/s0196-0709(00)80006-x
Lueder, G. T., and Galli, M. (2018). Infantile exotropia and developmental delay. J. Pediatr. Ophthalmol. Strabismus 55 (4), 225–228. doi:10.3928/01913913-20180213-05
Read, A. P., and Newton, V. E. (1997). Waardenburg syndrome. J. Med. Genet. 34 (8), 656–665. doi:10.1136/jmg.34.8.656
Røge, R., Nielsen, S., Bzorek, M., and Vyberg, M. (2017). NordiQC assessments of SOX10 immunoassays. Appl. Immunohistochem. Mol. Morphol. 25 (6), 377–380. doi:10.1097/PAI.0000000000000536
Somashekar, P. H., Girisha, K. M., Nampoothiri, S., Gowrishankar, K., Devi, R. R., Gupta, N., et al. (2019). Locus and allelic heterogeneity and phenotypic variability in Waardenburg syndrome. Clin. Genet. 95 (3), 398–402. doi:10.1111/cge.13468
Tang, X. J., Ping, X. Y., Luo, C. Q., Yu, X. N., Tang, Y. L., and Shentu, X. C. (2020). Dystrophia canthorum in Waardenburg syndrome with a novel MITF mutation. Int. J. Ophthalmol. 13 (7), 1054–1059. doi:10.18240/ijo.2020.07.06
Touraine, R. L., Attie-Bitach, A., Manceau, E., Korsch, E., Sarda, P., Pingault, V., et al. (2000). Neurological phenotype in Waardenburg syndrome type 4 correlates with novel SOX10 truncating mutations and expression in developing brain. Am. J. Hum. Genet. 66 (6), 1496–1503. doi:10.1086/302895
Vázquez-Villa, F., Garcia-Ocana, M., Galván, J. A., García-Martínez, J., García-Pravia, C., Menéndez-Rodríguez, P., et al. (2015). COL11A1/(pro)collagen 11A1 expression is a remarkable biomarker of human invasive carcinoma-associated stromal cells and carcinoma progression. Tumour Biol. 36 (4), 2213–2222. doi:10.1007/s13277-015-3295-4
Verheij, J. B. G. M., and Hofstra, R. M. W. (2016). “EDNRB, EDN3, SOX10, and the shah-waardenburg syndrome (WS4): The molecular basis of clinical disorders of morphogenesis: Epstein's inborn errors of development,” in Epstein's inborn errors of development, 531–535.
Keywords: waardenburg syndrome, exotropia, PAX3, SOX10, COL11A2
Citation: Huang L, Guo M and Li N (2022) Case report: Exotropia in waardenburg syndrome with novel variations. Front. Genet. 13:969680. doi: 10.3389/fgene.2022.969680
Received: 15 June 2022; Accepted: 10 August 2022;
Published: 02 September 2022.
Edited by:
Zi-Bing Jin, Capital Medical University, ChinaReviewed by:
Wenmin Sun, Sun Yat-sen University, ChinaTahir Khan, National University of Medical Sciences (NUMS), Pakistan
Copyright © 2022 Huang, Guo and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ningdong Li, bG5kMzBAMTYzLmNvbQ==
†These authors have contributed equally to this work