
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 02 September 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.967688
 Jieyi Chen1,2,3†
Jieyi Chen1,2,3† Ping Zhang4†Meifang Peng5Bo Liu4
Ping Zhang4†Meifang Peng5Bo Liu4 Xiao Wang4Siyuan Du3Yao Lu6Xiongzheng Mu1
Xiao Wang4Siyuan Du3Yao Lu6Xiongzheng Mu1 Yulan Lu4*
Yulan Lu4* Sijia Wang3*
Sijia Wang3* Yingzhi Wu1*
Yingzhi Wu1*Craniosynostosis (CRS) is a disease with prematurely fused cranial sutures. In the last decade, the whole-exome sequencing (WES) was widely used in Caucasian populations. The WES largely contributed in genetic diagnosis and exploration on new genetic mechanisms of CRS. In this study, we enrolled 264 CRS patients in China. After a 17-gene-panel sequencing designed in the previous study, 139 patients were identified with pathogenic/likely pathogenic (P/LP) variants according to the ACMG guideline as positive genetic diagnosis. WES was then performed on 102 patients with negative genetic diagnosis by panel. Ten P/LP variants were additionally identified in ten patients, increasing the genetic diagnostic yield by 3.8% (10/264). The novel variants in ANKH, H1-4, EIF5A, SOX6, and ARID1B expanded the mutation spectra of CRS. Then we designed a compatible research pipeline (RP) for further exploration. The RP could detect all seven P/LP SNVs and InDels identified above, in addition to 15 candidate variants found in 13 patients with worthy of further study. In sum, the 17-gene panel and WES identified positive genetic diagnosis for 56.4% patients (149/264) in 16 genes. At last, in our estimation, the genetic testing strategy of “Panel-first” saves 24.3% of the cost compared with “WES only”, suggesting the “Panel-first” is an economical strategy.
Craniosynostosis (CRS) is one of the most common congenital craniofacial anomalies in children, with the prevalence of approximately 1/2500 in live births (Lenton et al., 2005). The etiology of CRS is complex. Monogenic, polygenic, chromosomal disorders, and environmental factors might cause CRS in children (Twigg and Wilkie, 2015; Flaherty et al., 2016; Poot, 2019). Previous studies performed different sequencing strategies and focused on genetic diagnosis for CRS patient cohorts (Wilkie et al., 2010; Roscioli et al., 2013; Paumard-Hernandez et al., 2015; Ye et al., 2016; Wilkie et al., 2017; Lee et al., 2018; Topa et al., 2020; Yoon et al., 2020; Tonne et al., 2021; Wu et al., 2021). With the development of the next-generation sequencing (NGS), specific sequencing panels were designed for CRS genetic diagnosis. These panels could achieve 28%–52% diagnostic yield for CRS patients, but the good performance was limited in core CRS genes, such as FGFR2, FGFR3, ERF, TWIST1, TCF12, and EFBN1 (Wilkie et al., 2010; Roscioli et al., 2013; Paumard-Hernandez et al., 2015; Wilkie et al., 2017; Lee et al., 2018; Yoon et al., 2020; Wu et al., 2021). The diagnosis is still complex and ambiguous for a minority of cases. The whole-exome sequencing (WES) is a more general strategy for all exonic genome regions based on NGS. With increasing application of WES in clinical diagnosis and research, many panel-undiagnosed CRS cases have reached positive genetic diagnoses (Miller et al., 2017; Wilkie et al., 2017). Over the last decades, several genes were newly found by WES studies as causal or relevant factors for CRS, like MEGF8, CDC45, SMAD6, BCL11B, TFAP2B, SOX6, and GINS2 (Twigg et al., 2012; Fenwick et al., 2016; Timberlake et al., 2016; Goos et al., 2019; Timberlake et al., 2019; Tolchin et al., 2020; Nabais Sa et al., 2021). The phenotype of CRS is deeply known as an incompletely penetrant phenotype. CRS is also occasional in many syndromes, which could expand the phenotype spectrums of these syndromes. Therefore, WES is recommended for CRS patients who are difficult to be diagnosed by core genes. Recently, we performed the first genetic research in the Chinese CRS cohort, which sequenced 17 genes known to be associated with CRS, including FGFR2, FGFR3, TWIST1, EFNB1, TCF12, SKI, RAB23, FGFR1, TGFBR2, POR, SMAD3, ERF, TGFBR1, MSX2, RECQL4, TGFB2, IFT43 (Wu et al., 2021). About half of these patients were identified with pathogenic/likely pathogenic (P/LP) variants, including single nucleotide variants (SNVs) and small insertions and deletions (InDels). However, the etiology of the rest patients in the previous study was unknown. Thus, WES is necessary to be applied to achieve more genetic diagnoses in Chinese patients. On the other hand, the efficiency and cost of different genetic testing strategies for CRS, such as Sanger sequencing, targeted gene panel sequencing and WES, were still understudied.
This study was performed following the framework described in Figure 1, as an additional study to 17-gene panel sequencing. We first sequenced 102 panel-negative CRS patients by WES, and then identified variants following two different filter pipelines. Both P/LP and candidate variants on candidate genes of CRS were reported in this study. Subsequently, we summarized all P/LP variants detected by panel and WES as the total genetic diagnosis of the Chinese craniosynostosis cohort. Finally, based on the total genetic diagnosis, we retrospectively estimated the cost of two genetic testing strategies, “Panel-first” and “WES-only”.
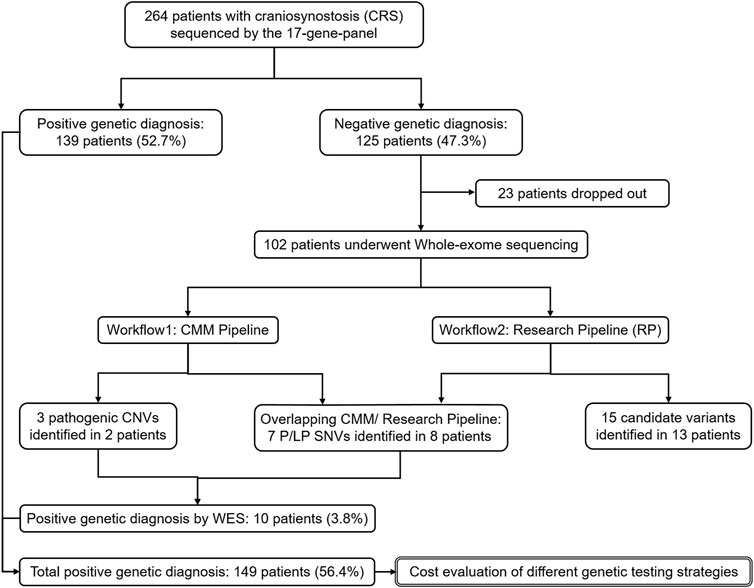
FIGURE 1. The framework of the whole study. Analyses on the genetic diagnosis and variants identification are in single solid line box. Analysis on the economic cost is in double solid line box.
Patients with premature fusion of one or more cranial sutures indicated by either X-ray or computed tomography and without aplasia/hypoplasia of the cerebrum were diagnosed as CRS. CRS could occur in isolation as non-syndromic CRS (nCRS) or be associated with other clinical manifestations as syndromic CRS (sCRS). The nCRS was defined as cranial vault features occur as an isolated defect. The sCRS was defined as patients with accompanying clinical features in addition to craniofacial deformities, such as digital anomalies, cardiac anomalies, and bone defects. CRS patients visiting or being referred to Huashan Hospital Fudan University during 2017–2021 were recruited. We initially enrolled 264 patients and their available parents into the study, including the 201 patients enrolled in the previous study (Wu et al., 2021) (Supplementary Figure S1). Doctors collected their clinical information and a mouth swab sample for genetic analysis for all the enrolled subjects. The parents completed the checklists of clinical features, and then the doctors confirmed them. All samples in this study were collected with appropriate informed consent and approval of the ethics committee of Huashan Hospital Fudan University (HIRB-2018–007).
DNA extraction and 17-gene-panel sequencing were the same as the previous study (Wu et al., 2021). For WES, the sequencing library was constructed and subjected to the exome sequence capture using an AIExome V1-CNV kit (iGeneTech, Beijing, China). The kit consisted of 26,022 genes in 62 Mb of the target region with mean coverage rate of 99.77% and mean depth of 129. The library was then sequenced on an Illumina platform (Illumina, San Diego, CA, United States) to generate 150-bp paired-end reads.
We primarily processed the sequencing data by the following steps: 1) reads trimming by Trimmomatic-0.38; 2) sequence alignment to the GRCh37/hg19 human reference genome by Burrows–Wheeler Aligner-0.7.15; 3) sorting and indexing the alignment into BAM format by SAMTools-1.9; 4) marking and removing PCR duplicates by Picard in Genome Analysis Toolkit (GATK-4.1.8).
The variant calling pipeline for panel was described in the previous study, and that for WES was as following.
For SNVs and InDels, best practice recommended by GATK, including BQSR and VQSR, was employed by GATK-4.1.8. In order to reduce false positive rate, we applied an additional quality control to VCF by removing variants with depth <10. Then we re-calibrated the genotype of the remaining variants by the depth ratio of alternative allele. When the depth ratio of alternative allele (DPRA) was less than 0.3, the variant would be defined as “0/0”, 0.3 ≤ DPRA ≤0.7 as “0/1”, and more than 0.7 as “1/1”. We set this threshold according to our experience of Sanger validation.
For copy number variant (CNV) detection, CANOES and HMZDelFinder were separately applied to BAM files and then merged as the previous work (Qin et al., 2018).
CMM is short for Center for Molecular Medicine of Children’s Hospital of Fudan University, so the diagnosis pipeline is also named as “CMM pipeline” in this study (Yang et al., 2019). The pipeline included annotation, automatic filtrations, and manual mutation curation according to the American College of Medical Genetics and Genomics (ACMG) interpretation guideline. ACMG interpretation guideline could classify variants into five types, including pathogenic (P), likely pathogenic (LP), uncertain significance (VUS), likely benign (LB) and benign (B) (South et al., 2013). All SNVs, InDels and CNVs were processed in the CMM pipeline.
We established a research pipeline (RP) focusing on digging candidate variants in all genes which were potentially associated with CRS in previous studies or databases. RP consists of two parts, and the results are merged at the end (Supplementary Figure S2). Only SNVs and InDels were involved in the RP.
In part one, gene-based and filter-based annotation of variants was performed using ANNOVAR. Loss-of-Function (LoF) variants (including frameshift, nonsense, splicing variants) and deleterious missense variants (deleterious score ≥4 or CADD ≥20) were retained. The deleterious score (DS) was referred to the previous study (Calpena et al., 2020). The allele frequency filter was separately filtered with different inheritance models according to gnomAD (v2.1) and Huabiao Project (Hao et al., 2021). When using database of gnomAD, we especially adjusted the threshold of the autosomal dominant model (AD) to 0.00005, considering the allele count ≤20 in 141,456 samples of gnomAD and the estimation method used by Calpena et al. (2020). When using allele frequency from Huabiao Project, we adjusted the threshold of AD to 0.0005, considering the allele count ≤5 in nearly 5000 samples. It is worth mentioning that the inheritance model in 21 only-probands were assumed due to lack of sequencing result from parents.
In part two, Exomiser (Robinson et al., 2014) utilized by “hiPhive” model with “mouse” & “zebrafish”, with the same allele frequency threshold as in part one. We kept only the top 20 variants referring to previous study (Stark et al., 2017).
In the end, the variants both identified in two parts were retained. Then we selected variants meeting with the candidate gene list for CRS (Supplementary Table S1). We made a literature review for genes associated with or causal to CRS. In order to avoid missing, the candidate gene list also consisted of the following parts: gene associated with “Craniosynostosis HP:0001363” or “Abnormal skull morphology HP:0000929” in the database of HPO (Kohler et al., 2021), and all genes of “Craniosynostosis” and “Skeletal dysplasia” in PanelAPP. There were totally 2,259 genes in the list, including 17 genes involved in the previous study (Wu et al., 2021). Finally, we kept the variants whose inheritance model met with that of the gene in OMIM (Amberger et al., 2019).
The candidate variants identified by both the CMM pipeline and RP were validated by Sanger sequencing or qPCR in individuals with sufficient DNA content. Other remained variants were confirmed by the Integrative Genomics Viewer (IGV-2.4.13), because the amount of DNA extracted from mouth swab sample was limited. The result of Sanger sequencing and IGV were extracted in Supplementary Figure S3 and Supplementary Figure S4.
We performed contingency table analysis and two-tailed Fisher’s exact tests to assess detailed differences between two or more groups. Single-tailed tests were declared. All statistical analysis was done with R (V4.0.4). p ≤ 0.05 was considered as significant.
In total, 264 patients clinically diagnosed with CRS were continually recruited (Table 1). The sCRS patients account for 61.7% in the whole CRS cohort, and the nCRS patients account for 38.3%. The uni/bi-coronal and multiple synostosis were the main types in sCRS patients. Differently, the sagittal and uni/bi-coronal synostosis were the main types in nCRS patients.

TABLE 1. Summary of the CRS patients enrolled.
In the panel stage, 59 distinct P/LP variants were identified in 139 patients as positive genetic diagnoses (Supplementary Table S2). The diagnostic yield of the 17-gene panel was 52.7% (139/264), updating that in the previous study. The 102 patients with negative genetic diagnoses by the panel and informed consent were further sequenced by WES in this study, including 21 only-probands and 81 trio-families (Figure 1 and Supplementary Table S3). With over half of sCRS and only a few nCRS patients solved by the panel, 33 sCRS and 69 nCRS patients were left and involved in the WES stage, including 61 males and 41 females.
Following the CMM pipeline, we identified 10 P/LP variants in 10 patients as positive diagnosis, including four SNVs, three InDels, and three CNVs (Table 2). Four variants were in the CRS-related genes, including ZIC1, SOX6, NFIA, and ARID1B (Twigg et al., 2015; Bayat et al., 2017; Tolchin et al., 2020; Tonne et al., 2021). The other patients with variants in ANKH, H1-4, and EIF5A had partially consistent phenotypes related to corresponding genes or syndromes according to OMIM (Amberger et al., 2019). For example, case W007 was with laryngomalacia and developmental delay meeting with Rahman Syndrome caused by loss-of-function variants in H1-4 (Tatton-Brown et al., 2017). Although these three genes were reported to be related to CRS for the first time, they were already known to be related to skeletal dysplasia and skull abnormalities (Kohler et al., 2021). As WES complemented the limitation of CNV detection by the 17-gene panel, we additionally identified the CNV deletions related to the hot CRS gene TWIST1, meeting with the well-known haploinsufficiency.
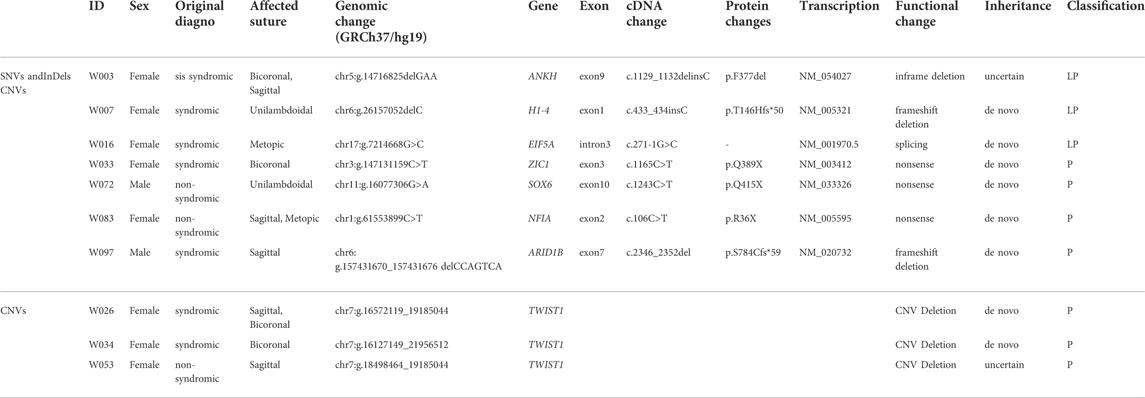
TABLE 2. Pathogenic/likely pathogenic (P/LP) Variants identified by the CMM pipeline.
In addition to the positive diagnoses mentioned above, four variants are noticed in the CMM pipeline but were not classified as P/LP variants (Supplementary Table S3). We reported them as “potentially pathogenic variants” in this study.
A frameshift variant of CDC45 (p.V109fs) in case W030 was defined as a negative diagnosis, because the only one LP variant could not fit to the autosomal recessive (AR) inheritance of CDC45 (Fenwick et al., 2016; Ting et al., 2020). However, many clinical manifestations of W030 matched Meier-gorlin syndrome, a disease caused by CDC45.
A de novo nonsense variant of SNRPB (p.R94X) in case W019 was classified as VUS in the CMM pipeline. The Cerebro-costo-mandibular Syndrome (CCMS) was caused by the mechanism of nonsense-mediated mRNA decay (NMD) of SNRPB (Lynch et al., 2014; Bacrot et al., 2015). This nonsense variant was predicted to efficiently trigger NMD by NMDetective-A, with the score of 0.85 (Lindeboom et al., 2019).
W019 was with cor triatrium, atrial septal defect, atresia of the external auditory canal, and self-report short stature for her age, but without other typical manifestations of CCMS, such as micrognathia or rib abnormalities by X-ray examination. Besides, CRS was never reported as a phenotype of CCMS, which decreasing the classification of the variant.
Although several studies have reported that the variants of IL11RA could cause Crouzon-like craniosynostosis in AR inheritance (Keupp et al., 2013; Miller et al., 2017; Brischoux-Boucher et al., 2018), the two compound heterozygotes of IL11RA (p. [P243R], [L236P]) in case W093 were classified as VUS in the CMM pipeline.
In addition to genetic diagnosis, we performed the research pipeline (RP) to look for candidate variants according to the candidate gene list for CRS (details in Supplementary Table S3). In total, 62 variants were identified in 42 cases by the automatic part of RP (Supplementary Table S3). In order to identify the candidate variants for CRS, we excluded all LB/B variants according to ACMG criteria manually. As a result, all the nine P/LP SNVs and InDels identified in the CMM pipeline and another 16 variants were remained. After reviewing all the genes of remaining variants, we excluded APC, which were mainly associated with cancers. Finally, we listed 15 variants of 11 genes as candidate variants for 13 CRS cases, including the compound heterozygotes of IL11RA (Table 3).
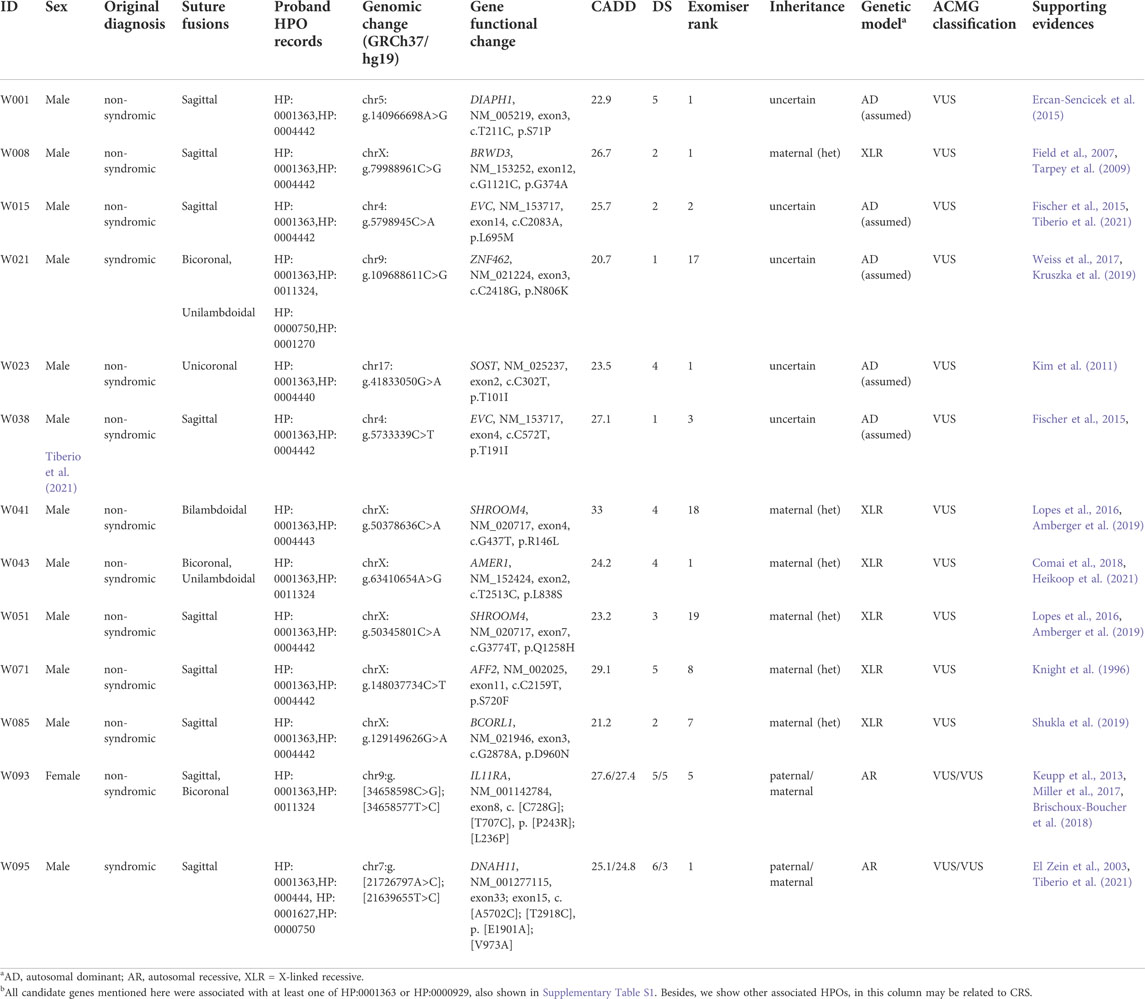
TABLE 3. Candidate variants identified by the research pipeline (RP).
The recurrence of the same candidate gene in different patients also increased its potential of pathogenicity. Among all pathophysiology of CRS, the genes orchestrating the primary cilium structure and function were also known to play an important role (Tiberio et al., 2021). The EVC encodes a positive regulator downstream the HH signaling, expressed on the ciliary membrane as a single-pass transmembrane protein. We found two sagittal patients, case W015 and W038, carried heterozygous missense variants in EVC. To date, only a single case of sagittal synostosis has been reported in a patient affected by Ellis-van Creveld syndrome, but this disorder is usually caused by homozygous variants in either the EVC or EVC2 genes (Fischer et al., 2015; Tiberio et al., 2021). Although there was no other candidate variants and inheritance validation composing compound heterozygotes in this study, the candidate variants suggest attention on EVC. Another candidate gene identified in more than one patient was SHROOM4. Case W041 and W051 carried different maternally inherited X linked missense variants. The Stocco dos Santos type of X-linked syndromic intellectual developmental disorder and Rett-like syndrome are caused by variants in the SHROOM4, with microcephaly as an occasional phenotype (Lopes et al., 2016; Amberger et al., 2019). Although the parents of two patients did not report intellectual developmental delay for the moment, we will perform a long-term follow-up in the future.
Among other candidate genes with only one case, they are associated with HPO of skeletal dysplasia (Supplementary Table S1). ZNF462 and IL11RA are directly associated with CRS. Besides, we tried to look out the indirect linkages between candidate genes and CRS. The syndromes or intellectual developmental disorders caused by variants in DIAPH1, AFF2, BCORL1 and BRWD3 are occasionally with abnormalities of cranium, such as microcephaly or macrocephaly tall forehead. Furtherly, SOST and AMER1 were causal for cranial sclerosis with craniofacial abnormalities. DNAH11 encodes critical protein for cilia, which might impact development of cranial sutures the primary cilium structure and function (Tiberio et al., 2021). The supporting evidences for above were listed in the last column of Table 3. In additional to literature review, we furtherly analyzed the gene expressions in the cranial neural crest cells (CNCCs), one of the critical cell resources for cranial sutures (Flaherty et al., 2016). Based on the published RNAseq data in GSE70751 (Prescott et al., 2015), we found that the expression levels of SHROOM4, BRWD3, AMER1, EVC, AFF2 and ZNF462 were about or at the top 10% in human CNCCs (Supplementary Figure S5).
In this study, we only defined patients with P/LP patients as positive genetic diagnosis, but potentially pathogenic and candidate variants as negative. The hot gene exonic capture by 17-gene panel in the first stage and the overall exonic capture by WES in the second stage are subset of NGS. We integrated all positive genetic diagnoses in the two stages as the total genetic diagnostic performance of exonic NGS. We totally identified 69 P/LP variants in 16 genes in this study. FGFR2 contributed the most diagnostic yield among all genes (Figure 2). FGFR3, TWIST1, TCF12, and EFNB1 were the following contributor for diagnostic yield. The rest 11 genes only contributed genetic diagnosis for one patient separately.
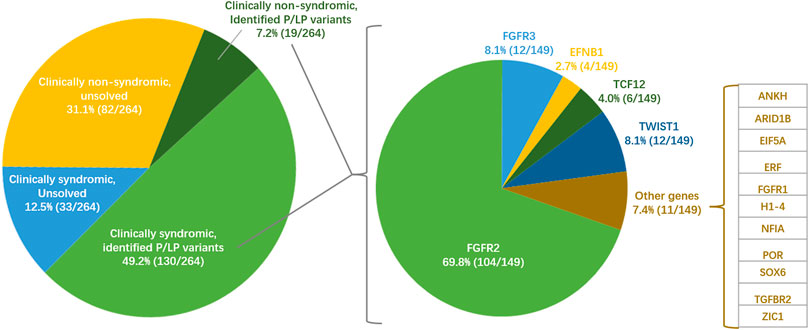
FIGURE 2. Situation of clinical diagnosis, genetic diagnosis and pathogenic genes of craniosynostosis in the Chinese cohort of this study (n = 264). The pie chart on the left shows a broad classification based on clinical diagnosis and genetic diagnosis in this study. The pie chart on the left shows the diagnosis contribution of 16 pathogenic genes in positive genetic diagnosis.
The overall diagnostic yield was 56.4% (149/264). Significantly more positive diagnoses were found in syndromic (79.8%) than that in non-syndromic (18.8%) patients (one-tailed p = 3.78 × 10–23, Figure 2). The diagnostic yields were also different among different suture fusion types (p = 1.30 × 10–15, Supplementary Table S4). To specific suture synostosis, we found that the patients with bi/uni-coronal and multiple synostosis tended to get positive genetic diagnoses than patients with other types (one-tailed, PCoronal = 3.28 × 10–3, PMultiple = 3.50 × 10–6, Supplementary Table S4).
Our study used a genetic testing strategy of “Panel-first and then WES”, which means that we first performed the 17-gene panel genetic testing for CRS patients, and only for panel-negative patients, we would perform the WES. In order to compare the cost between two strategies, we estimated the cost of two situations based on positive genetic diagnoses integrated above. Since the 17-gene panel included all exons of 17 genes, all P/LP variants identified by the 17-gene panel could also be identified by WES. Thus, the diagnostic yield of the a “WES-only” strategy would be same as the “Panel-first” strategy. However, the costs of these two strategies are different. The average fee of a trio-WES was about $945, and 17 gene-panel for a trio was about $315. We retrospectively estimated the cost in all 239 patients who underwent only panel or both panel and WES (Table 4). At the same diagnostic yield, the genetic testing strategy of “Panel-first” ($715 per trio family) could save 24.3% cost ($230) on genetic testing for probands’ families in average, rather than “WES-only” ($945 per trio family).
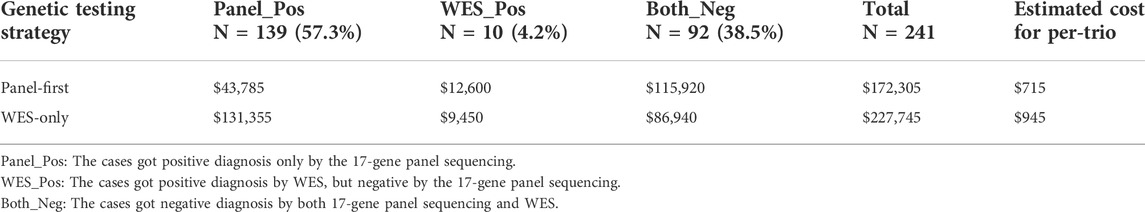
TABLE 4. Cost estimation for two genetic testing strategies.
Among all 264 patients involved in our genetic testing study, the diagnostic yield of the 17-gene panel was 52.7%, and the additional diagnostic yield by WES was 3.8% (10/264, Table 2), which might mainly due to the two reasons in this study. The first one is the additional detection of CNVs. The panel used in the first stage could only detect SNPs and InDels, so the three deletions of TWIST1 were missed in the panel stage. The other reason is the expand for more gene detection by WES than the 17-gene panel. However, the actual performance of WES might be better than the current observation in this study. The exclusion of 23 patients lost to follow-up and lacking of functional experiments for VUS might decrease the diagnostic yield of WES. Another reason for underestimation of diagnostic yield is the detection of CNVs might be missed. The CNV calling pipeline used in this study depend on at least 20 samples from the same batch (Qin et al., 2018), but we sequenced samples in many different batches in practice. Thus, the sample size of many batches is fewer than ten, resulting failure of CNV detection.
Among all seven P/LP SNVs and InDels, only two of ZIC1 and NFIA were reported, while the other diagnosed variants are novel. They exactly gave the genetic diagnosis to patients’ families and doctors, and expanded the variant spectra of CRS. The phenotype spectra of several gene-related diseases were also expanded. For example, we were not aware of previous descriptions of CRS associated with variants in ANKH, H1-4, and EIF5A, but the clinical features and the functional of these variants identified were considered sufficient to assign positive diagnosis. To the four candidate variants identified in W019, W030 and W093, although they could not lead to positive diagnosis temporarily, they pointed out the direction of experimental verification indicating their physiology for CRS.
Clinical interpretation of genomic variants requires the standard classification guidelines and workflows, as well as considering the consistency between a variant and a disease phenotype by enough solid evidence. Research-based analysis performed by the Clinical Genetics Group, Oxford (CGG) identified additionally over one-fold P/LP variants than the GE/GMC pipeline for rare diseases, which demonstrated the value of research analysis and the importance of continually improving algorithms to maximize the potential of clinical genome sequencing (Hyder et al., 2021). Our study combined the CMM pipeline following the ACMG guideline and the RP as a supplement. These ACMG germline variant curation guidelines have been broadly adopted by clinical genetic testing laboratories globally to report genetic diagnoses of genetic diseases (Niehaus et al., 2019). However, the limitation of clinical practice for genetic diagnosis is the strictness and rigorousness. It is difficult to explore more novel genes for CRS by the CMM pipeline only. Thus, we referred to several studies and established the RP to identify candidate variants for genetic diagnosis to support further research.
All the P/LP variants and one potentially pathogenic variant identified by the CMM pipeline could be covered by the RP. Moreover, the RP could identify more candidate variants classified as VUS by the ACMG guideline. Although these specific RP variants were not validated by experiments in this study, they could serve as evidence to look for any other patients with variants in the same genes or a potential direction of functional experiments. For example, the gene of EVC and SHROOM4 were reported in more than one case among candidate variants identified by RP, which should be considered for functional validation first. The indirect linkages between the rest candidate genes and CRS could be inferred by literature review. The high expression of SHROOM4, BRWD3, AMER1, EVC, AFF2, and ZNF462 in CNCCs suggested the probable pathophysiology of CRS from another perspective.
In sum, the CMM pipeline is reliable for genetic diagnosis, while the RP is a loose analysis process to identify both P/LP variants and candidate variants. The shortage of both pipelines is the requirement of manual work in ACMG classification, which is also a worldwide challenge. On the other hand, since the RP is specific to the research for CRS, the current list of candidate genes only considered skeletal abnormalities as the cause of CRS, but ignored other possibilities of CRS as a secondary phenotype of other disorders.
With the comparison between sCRS and nCRS patients, the NGS based on exonic regions significantly contributed more to the genetic diagnosis in sCRS, regardless of panel or WES in this study. Such difference between sCRS and nCRS was also reported in previous studies (Roscioli et al., 2013; Wilkie et al., 2017; Lee et al., 2018; Yoon et al., 2020). Meanwhile, the significantly higher genetic diagnostic yield was observed in the patients with bi/uni-coronal or multiple synostosis. Therefore, the genetic testing, especially in exonic genome regions, is recommended for sCRS patients and patients with bi/uni-coronal or multiple synostosis for genetic diagnosis.
There are many genetic testing strategies for CRS in previous studies conducted in different countries (Kutkowska-Kazmierczak et al., 2018). In general, the doctor should give a primary clinical diagnosis and recommend a genetic testing, seemed like a “Step-by-step” strategy, including Sanger sequencing of specific exons of FGFR2, FGFR3, and TWIST1, and structural variants detection around TWIST1. However, the match of primary clinical diagnosis and proper genetic testing depends on the experience of doctors, so the “Step-by-step” strategy often lasts a long time and cause financial burden, also known as a long diagnosis odyssey. With the increasing knowledge about CRS genes and the advances of NGS, several sequencing panels were designed for CRS (Lee et al., 2018; Yoon et al., 2020; Tonne et al., 2021; Wu et al., 2021). The reported panels could cover all the exons in the “Step-by-step” strategy or core genes for CRS in only one step. Therefore, we proposed “a CRS sequencing panel first and then WES for panel-negative patients” strategy (Panel-first strategy) as a new “Step-by-step” strategy for genetic testing of CRS patients, of which the efficiency is not limited by the experience of doctors.
As WES is recommended as the first-tier for genetic testing in comprehensive hospitals in China (Yang et al., 2013; Hu et al., 2017), we compared the price of “Panel-first” and “WES-only”. We found the cost of former is 24.3% lower compared with the latter in average. Since over 50% patients could get genetic diagnoses only by the 17-gene panel, it is obvious that the “Panel-first” strategy has a lower cost without losing the genetic diagnostic yield, which is more suitable for price-sensitive patients and their families. On the other hand, only several hospitals and doctors are famous for diagnosing and treating CRS, causing resource imbalance. Thus, we especially recommend a “Panel-first” strategy to the hospitals with a large number of CRS patients.
There are three limitations to the estimation in our study. The first one is about the price. We used the present prices in only a few three-A hospitals in Shanghai as reference, regardless of price fluctuation and regional difference. The second is about the gene selection of panel. The total genetic diagnosis covered 16 genes, while only five genes, including FGFR2, FGFR3, TWIST1, TCF12, and EFNB1 contributed most. Thus, “which genes should be selected in the panel with the highest efficiency”, should be considered in the future study. Due to the limited budget and sample size, we only performed the “Panel-first” strategy in the study, and assumed all positive cases could be detected in the group of “WES-only”. Independent groups of different strategies and their real cost should be compared to make more precise estimation. Despite the limitations mentioned above, estimating the cost of different genetic testing strategies could be useful for clinical practice.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Huashan Hospital Fudan University. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
YZ, SW and YL designed and supervised the study. YW, MP, JC and ZM acquired patient information and samples. JC, PZ, BL, XW and YL performed the genetic and bioinformatic analysis and the variants interpretation. PZ and YaoL performed validation of variants by Sanger sequencing and qPCR. JC and PZ developed the manuscript with critical review input from YL and YW. All authors approved the final manuscript.
This work was supported by the foundation of the Shanghai Municipal Science and Technology Major Project (No. 2017SHZDZX01). We also thank the support from the National Key Research and Development Program (No. 2020YFC2006402) and the National Natural Science Foundation of China (No. 82102268).
We are very grateful to the families who participated in this study. We thank all suggestions and comments from clinicians and researchers. We thank the shared data from “The HuaBiao Project: Whole-Exome Database of Han Chinese”.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.967688/full#supplementary-material
Amberger, J. S., Bocchini, C. A., Scott, A. F., and Hamosh, A. (2019). OMIM.org: Leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res. 47, D1038–D1043. doi:10.1093/nar/gky1151
Bacrot, S., Doyard, M., Huber, C., Alibeu, O., Feldhahn, N., Lehalle, D., et al. (2015). Mutations in SNRPB, encoding components of the core splicing machinery, cause cerebro-costo-mandibular syndrome. Hum. Mutat. 36, 187–190. doi:10.1002/humu.22729
Bayat, A., Kirchhoff, M., Madsen, C. G., Roos, L., and Kreiborg, S. (2017). Familial craniofacial abnormality and polymicrogyria associated with a microdeletion affecting the NFIA gene. Clin. Dysmorphol. 26, 148–153. doi:10.1097/MCD.0000000000000182
Brischoux-Boucher, E., Trimouille, A., Baujat, G., Goldenberg, A., Schaefer, E., Guichard, B., et al. (2018). IL11RA-related Crouzon-like autosomal recessive craniosynostosis in 10 new patients: Resemblances and differences. Clin. Genet. 94, 373–380. doi:10.1111/cge.13409
Calpena, E., Cuellar, A., Bala, K., Swagemakers, S. M. A., Koelling, N., Mcgowan, S. J., et al. (2020). SMAD6 variants in craniosynostosis: Genotype and phenotype evaluation. Genet. Med. 22, 1498–1506. doi:10.1038/s41436-020-0817-2
Comai, G., Boutet, A., Tanneberger, K., Massa, F., Rocha, A. S., Charlet, A., et al. (2018). Genetic and molecular insights into genotype-phenotype relationships in osteopathia striata with cranial sclerosis (OSCS) through the analysis of novel mouse wtx mutant alleles. J. Bone Min. Res. 33, 875–887. doi:10.1002/jbmr.3387
El Zein, L., Omran, H., and Bouvagnet, P. (2003). Lateralization defects and ciliary dyskinesia: Lessons from algae. Trends Genet. 19, 162–167. doi:10.1016/S0168-9525(03)00026-X
Ercan-Sencicek, A. G., Jambi, S., Franjic, D., Nishimura, S., Li, M., El-Fishawy, P., et al. (2015). Homozygous loss of DIAPH1 is a novel cause of microcephaly in humans. Eur. J. Hum. Genet. 23, 165–172. doi:10.1038/ejhg.2014.82
Fenwick, A. L., Kliszczak, M., Cooper, F., Murray, J., Sanchez-Pulido, L., Twigg, S. R., et al. (2016). Mutations in CDC45, encoding an essential component of the pre-initiation complex, cause meier-gorlin syndrome and craniosynostosis. Am. J. Hum. Genet. 99, 125–138. doi:10.1016/j.ajhg.2016.05.019
Field, M., Tarpey, P. S., Smith, R., Edkins, S., O'Meara, S., Stevens, C., et al. (2007). Mutations in the BRWD3 gene cause X-linked mental retardation associated with macrocephaly. Am. J. Hum. Genet. 81, 367–374. doi:10.1086/520677
Fischer, A. S., Weathers, W. M., Wolfswinkel, E. M., Bollo, R. J., Hollier, L. H., and Buchanan, E. P. (2015). Ellis-van Creveld syndrome with sagittal craniosynostosis. Craniomaxillofac. Trauma Reconstr. 8, 132–135. doi:10.1055/s-0034-1393733
Flaherty, K., Singh, N., and Richtsmeier, J. T. (2016). Understanding craniosynostosis as a growth disorder. Wiley Interdiscip. Rev. Dev. Biol. 5, 429–459. doi:10.1002/wdev.227
Goos, J. A. C., Vogel, W. K., Mlcochova, H., Millard, C. J., Esfandiari, E., Selman, W. H., et al. (2019). A de novo substitution in BCL11B leads to loss of interaction with transcriptional complexes and craniosynostosis. Hum. Mol. Genet. 28, 2501–2513. doi:10.1093/hmg/ddz072
Hao, M., Pu, W., Li, Y., Wen, S., Sun, C., Ma, Y., et al. (2021). The HuaBiao project: Whole-exome sequencing of 5000 han Chinese individuals. J. Genet. Genomics 48, 1032–1035. doi:10.1016/j.jgg.2021.07.013
Heikoop, D., Brick, L., Chitayat, D., Colaiacovo, S., Dupuis, L., Faghfoury, H., et al. (2021). The phenotypic spectrum of AMER1-related osteopathia striata with cranial sclerosis: The first Canadian cohort. Am. J. Med. Genet. A 185, 3793–3803. doi:10.1002/ajmg.a.62452
Hu, X., Li, N., Xu, Y., Li, G., Yu, T., Yao, R. E., et al. (2017). Proband-only medical exome sequencing as a cost-effective first-tier genetic diagnostic test for patients without prior molecular tests and clinical diagnosis in a developing country: The China experience. Genet. Med. 20, 1045–1053. doi:10.1038/gim.2017.195
Hyder, Z., Calpena, E., Pei, Y., Tooze, R. S., Brittain, H., Twigg, S. R. F., et al. (2021). Evaluating the performance of a clinical genome sequencing program for diagnosis of rare genetic disease, seen through the lens of craniosynostosis. Genet. Med. 23, 2360–2368. doi:10.1038/s41436-021-01297-5
Keupp, K., Li, Y., Vargel, I., Hoischen, A., Richardson, R., Neveling, K., et al. (2013). Mutations in the interleukin receptor IL11RA cause autosomal recessive Crouzon-like craniosynostosis. Mol. Genet. Genomic Med. 1, 223–237. doi:10.1002/mgg3.28
Kim, S. J., Bieganski, T., Sohn, Y. B., Kozlowski, K., Semenov, M., Okamoto, N., et al. (2011). Identification of signal peptide domain SOST mutations in autosomal dominant craniodiaphyseal dysplasia. Hum. Genet. 129, 497–502. doi:10.1007/s00439-011-0947-3
Knight, S., Ritchie, R. J., Chakrabarti, L., Cross, G., Davies, K. E., Mueller, R. F., et al. (1996). A study of FRAXE in mentally retarded individuals referred for fragile X syndrome (FRAXA) testing in the United Kingdom. Am. J. Hum. Genet. 58, 906–913.
Kohler, S., Gargano, M., Matentzoglu, N., Carmody, L. C., Lewis-Smith, D., Vasilevsky, N. A., et al. (2021). The human phenotype ontology in 2021. Nucleic Acids Res. 49, D1207–D1217. doi:10.1093/nar/gkaa1043
Kruszka, P., Hu, T., Hong, S., Signer, R., Cogne, B., Isidor, B., et al. (2019). Phenotype delineation of ZNF462 related syndrome. Am. J. Med. Genet. A 179, 2075–2082. doi:10.1002/ajmg.a.61306
Kutkowska-Kazmierczak, A., Gos, M., and Obersztyn, E. (2018). Craniosynostosis as a clinical and diagnostic problem: molecular pathology and genetic counseling. J. Appl. Genet. 59, 133–147. doi:10.1007/s13353-017-0423-4
Lee, E., Le, T., Zhu, Y., Elakis, G., Turner, A., Lo, W., et al. (2018). A craniosynostosis massively parallel sequencing panel study in 309 Australian and New Zealand patients: findings and recommendations. Genet. Med. 20, 1061–1068. doi:10.1038/gim.2017.214
Lenton, K. A., Nacamuli, R. P., Wan, D. C., Helms, J. A., and Longaker, M. T. (2005). Cranial suture biology. Curr. Top. Dev. Biol. 66, 287–328. doi:10.1016/s0070-2153(05)66009-7
Lindeboom, R. G., Vermeulen, M., Lehner, B., and Supek, F. (2019). The impact of nonsense-mediated mRNA decay on genetic disease, gene editing and cancer immunotherapy. Nat. Genet. 51, 1645–1651. doi:10.1038/s41588-019-0517-5
Lopes, F., Barbosa, M., Ameur, A., Soares, G., De Sá, J., Dias, A. I., et al. (2016). Identification of novel genetic causes of Rett syndrome-like phenotypes. J. Med. Genet. 53, 190–199. doi:10.1136/jmedgenet-2015-103568
Lynch, D. C., Revil, T., Schwartzentruber, J., Bhoj, E. J., Innes, A. M., Lamont, R. E., et al. (2014). Disrupted auto-regulation of the spliceosomal gene SNRPB causes cerebro-costo-mandibular syndrome. Nat. Commun. 5, 4483. doi:10.1038/ncomms5483
Miller, K. A., Twigg, S. R., Mcgowan, S. J., Phipps, J. M., Fenwick, A. L., Johnson, D., et al. (2017). Diagnostic value of exome and whole genome sequencing in craniosynostosis. J. Med. Genet. 54, 260–268. doi:10.1136/jmedgenet-2016-104215
Nabais Sa, M. J., Miller, K. A., Mcquaid, M., Koelling, N., Wilkie, A. O. M., Wurtele, H., et al. (2021). Biallelic GINS2 variant p.(Arg114Leu) causes Meier-Gorlin syndrome with craniosynostosis. J. Med. Genet. 59, 776–780. doi:10.1136/jmedgenet-2020-107572
Niehaus, A., Azzariti, D. R., Harrison, S. M., Distefano, M. T., Hemphill, S. E., Senol-Cosar, O., et al. (2019). A survey assessing adoption of the ACMG-AMP guidelines for interpreting sequence variants and identification of areas for continued improvement. Genet. Med. 21, 1699–1701. doi:10.1038/s41436-018-0432-7
Paumard-Hernandez, B., Berges-Soria, J., Barroso, E., Rivera-Pedroza, C. I., Perez-Carrizosa, V., Benito-Sanz, S., et al. (2015). Expanding the mutation spectrum in 182 Spanish probands with craniosynostosis: identification and characterization of novel TCF12 variants. Eur. J. Hum. Genet. 23, 907–914. doi:10.1038/ejhg.2014.205
Poot, M. (2019). Structural genome variations related to craniosynostosis. Mol. Syndromol. 10, 24–39. doi:10.1159/000490480
Prescott, S. L., Srinivasan, R., Marchetto, M. C., Grishina, I., Narvaiza, I., Selleri, L., et al. (2015). Enhancer divergence and cis-regulatory evolution in the human and chimp neural crest. Cell 163, 68–83. doi:10.1016/j.cell.2015.08.036
Qin, Q., Liu, B., Yang, L., Wu, B., Wang, H., Dong, X., et al. (2018). Application of copy number variation screening analysis process based on high-throughputsequencing technology. Chin. J. Evid. Based Pediatr. 13, 5.
Robinson, P. N., Kohler, S., Oellrich, A., Sanger Mouse Genetics, P., Wang, K., Mungall, C. J., et al. (2014). Improved exome prioritization of disease genes through cross-species phenotype comparison. Genome Res. 24, 340–348. doi:10.1101/gr.160325.113
Roscioli, T., Elakis, G., Cox, T. C., Moon, D. J., Venselaar, H., Turner, A. M., et al. (2013). Genotype and clinical care correlations in craniosynostosis: findings from a cohort of 630 Australian and New Zealand patients. Am. J. Med. Genet. C Semin. Med. Genet. 163C, 259–270. doi:10.1002/ajmg.c.31378
Shukla, A., Girisha, K. M., Somashekar, P. H., Nampoothiri, S., Mcclellan, R., and Vernon, H. J. (2019). Variants in the transcriptional corepressor BCORL1 are associated with an X-linked disorder of intellectual disability, dysmorphic features, and behavioral abnormalities. Am. J. Med. Genet. A 179, 870–874. doi:10.1002/ajmg.a.61118
South, S. T., Lee, C., Lamb, A. N., Higgins, A. W., Kearney, H. M., Working Group For The American College Of Medical, G., et al. (2013). ACMG standards and guidelines for constitutional cytogenomic microarray analysis, including postnatal and prenatal applications: revision 2013. Genet. Med. 15, 901–909. doi:10.1038/gim.2013.129
Stark, Z., Dashnow, H., Lunke, S., Tan, T. Y., Yeung, A., Sadedin, S., et al. (2017). A clinically driven variant prioritization framework outperforms purely computational approaches for the diagnostic analysis of singleton WES data. Eur. J. Hum. Genet. 25, 1268–1272. doi:10.1038/ejhg.2017.123
Tarpey, P. S., Smith, R., Pleasance, E., Whibley, A., Edkins, S., Hardy, C., et al. (2009). A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat. Genet. 41, 535–543. doi:10.1038/ng.367
Tatton-Brown, K., Loveday, C., Yost, S., Clarke, M., Ramsay, E., Zachariou, A., et al. (2017). Mutations in epigenetic regulation genes are a major cause of overgrowth with intellectual disability. Am. J. Hum. Genet. 100, 725–736. doi:10.1016/j.ajhg.2017.03.010
Tiberio, F., Parolini, O., and Lattanzi, W. (2021). Ciliary signalling and mechanotransduction in the pathophysiology of craniosynostosis. Genes 12, 1073. doi:10.3390/genes12071073
Timberlake, A. T., Choi, J., Zaidi, S., Lu, Q., Nelson-Williams, C., Brooks, E. D., et al. (2016). Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and common BMP2 alleles. Elife 5, e20125. doi:10.7554/eLife.20125
Timberlake, A. T., Jin, S. C., Nelson-Williams, C., Wu, R., Furey, C. G., Islam, B., et al. (2019). Mutations in TFAP2B and previously unimplicated genes of the BMP, Wnt, and Hedgehog pathways in syndromic craniosynostosis. Proc. Natl. Acad. Sci. U. S. A. 116, 15116–15121. doi:10.1073/pnas.1902041116
Ting, C. Y., Bhatia, N. S., Lim, J. Y., Goh, C. J., Vasanwala, R. F., Ong, C. C., et al. (2020). Further delineation of CDC45-related Meier-Gorlin syndrome with craniosynostosis and review of literature. Eur. J. Med. Genet. 63, 103652. doi:10.1016/j.ejmg.2019.04.009
Tolchin, D., Yeager, J. P., Prasad, P., Dorrani, N., Russi, A. S., Martinez-Agosto, J. A., et al. (2020). De novo SOX6 variants cause a neurodevelopmental syndrome associated with ADHD, craniosynostosis, and osteochondromas. Am. J. Hum. Genet. 106, 830–845. doi:10.1016/j.ajhg.2020.04.015
Tonne, E., Due-Tonnessen, B. J., Mero, I. L., Wiig, U. S., Kulseth, M. A., Vigeland, M. D., et al. (2021). Benefits of clinical criteria and high-throughput sequencing for diagnosing children with syndromic craniosynostosis. Eur. J. Hum. Genet. 29, 920–929. doi:10.1038/s41431-020-00788-4
Topa, A., Rohlin, A., Andersson, M. K., Fehr, A., Lovmar, L., Stenman, G., et al. (2020). NGS targeted screening of 100 Scandinavian patients with coronal synostosis. Am. J. Med. Genet. A 182, 348–356. doi:10.1002/ajmg.a.61427
Twigg, S. R., Forecki, J., Goos, J. A., Richardson, I. C., Hoogeboom, A. J. M., Van Den Ouweland, A. M., et al. (2015). Gain-of-function mutations in ZIC1 are associated with coronal craniosynostosis and learning disability. Am. J. Hum. Genet. 97, 378–388. doi:10.1016/j.ajhg.2015.07.007
Twigg, S. R., Lloyd, D., Jenkins, D., Elcioglu, N. E., Cooper, C. D., Al-Sannaa, N., et al. (2012). Mutations in multidomain protein MEGF8 identify a Carpenter syndrome subtype associated with defective lateralization. Am. J. Hum. Genet. 91, 897–905. doi:10.1016/j.ajhg.2012.08.027
Twigg, S. R., and Wilkie, A. O. (2015). A genetic-pathophysiological framework for craniosynostosis. Am. J. Hum. Genet. 97, 359–377. doi:10.1016/j.ajhg.2015.07.006
Weiss, K., Wigby, K., Fannemel, M., Henderson, L. B., Beck, N., Ghali, N., et al. (2017). Haploinsufficiency of ZNF462 is associated with craniofacial anomalies, corpus callosum dysgenesis, ptosis, and developmental delay. Eur. J. Hum. Genet. 25, 946–951. doi:10.1038/ejhg.2017.86
Wilkie, A. O., Byren, J. C., Hurst, J. A., Jayamohan, J., Johnson, D., Knight, S. J., et al. (2010). Prevalence and complications of single-gene and chromosomal disorders in craniosynostosis. Pediatrics 126, e391–400. doi:10.1542/peds.2009-3491
Wilkie, A. O. M., Johnson, D., and Wall, S. A. (2017). Clinical genetics of craniosynostosis. Curr. Opin. Pediatr. 29, 622–628. doi:10.1097/mop.0000000000000542
Wu, Y., Peng, M., Chen, J., Suo, J., Zou, S., Xu, Y., et al. (2021). A custom-designed panel sequencing study in 201 Chinese patients with craniosynostosis revealed novel variants and distinct mutation spectra. J. Genet. Genomics 48, 167–171. doi:10.1016/j.jgg.2020.11.004
Yang, L., Kong, Y., Dong, X., Hu, L., Lin, Y., Chen, X., et al. (2019). Clinical and genetic spectrum of a large cohort of children with epilepsy in China. Genet. Med. 21, 564–571. doi:10.1038/s41436-018-0091-8
Yang, Y., Muzny, D. M., Reid, J. G., Bainbridge, M. N., Willis, A., Ward, P. A., et al. (2013). Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 369, 1502–1511. doi:10.1056/NEJMoa1306555
Ye, X., Guilmatre, A., Reva, B., Peter, I., Heuze, Y., Richtsmeier, J. T., et al. (2016). Mutation screening of candidate genes in patients with nonsyndromic sagittal craniosynostosis. Plast. Reconstr. Surg. 137, 952–961. doi:10.1097/01.prs.0000479978.75545.ee
Keywords: craniosynostosis, whole-exome sequencing, genetic diagnosis, research pipeline, candidate variants, cost estimation
Citation: Chen J, Zhang P, Peng M, Liu B, Wang X, Du S, Lu Y, Mu X, Lu Y, Wang S and Wu Y (2022) An additional whole-exome sequencing study in 102 panel-undiagnosed patients: A retrospective study in a Chinese craniosynostosis cohort. Front. Genet. 13:967688. doi: 10.3389/fgene.2022.967688
Received: 13 June 2022; Accepted: 01 August 2022;
Published: 02 September 2022.
Edited by:
Long Guo, RIKEN Center for Integrative Medical Sciences, JapanReviewed by:
Michael Cunningham, University of Washington, United StatesCopyright © 2022 Chen, Zhang, Peng, Liu, Wang, Du, Lu, Mu, Lu, Wang and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yingzhi Wu, d3V5aW5nemhpODE1QDE2My5jb20=; Sijia Wang, d2FuZ3NpamlhQHBpY2IuYWMuY24=; Yulan Lu, eXVsYW5sdUBmdWRhbi5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.