 Stefania Martone1,2†
Stefania Martone1,2† Autilia Tommasina Buonagura1,2†
Autilia Tommasina Buonagura1,2† Roberta Marra1,2
Roberta Marra1,2 Barbara Eleni Rosato1,2
Barbara Eleni Rosato1,2 Federica Del Giudice2
Federica Del Giudice2 Ferdinando Bonfiglio2,3
Ferdinando Bonfiglio2,3 Mario Capasso1,2
Mario Capasso1,2 Achille Iolascon1,2
Achille Iolascon1,2 Immacolata Andolfo1,2*
Immacolata Andolfo1,2* Roberta Russo1,2*
Roberta Russo1,2*- 1Dipartimento di Medicina Molecolare e Biotecnologie Mediche, Università Degli Studi di Napoli Federico II, Napoli, Italy
- 2CEINGE Biotecnologie Avanzate, Napoli, Italy
- 3Dipartimento di Ingegneria Chimica, Dei Materiali e Della Produzione Industriale, Università Degli Studi di Napoli Federico II, Napoli, Italy
Background: Next-generation sequencing-based genetic testing represents a great opportunity to identify hereditary predispositions to specific pathological conditions and to promptly implement health surveillance or therapeutic protocols in case of disease. The term secondary finding refers to the active search for causative variants in genes associated with medically actionable conditions.
Methods: We evaluated 59 medically actionable ACMG genes using a targeted in silico analysis of clinical exome sequencing performed in 383 consecutive individuals referred to our Medical Genetics Unit. A three-tier classification system of SFs for assessing their clinical impact and supporting a decision-making process for reporting was established.
Results: We identified SFs with high/moderate evidence of pathogenicity in 7.0% (27/383) of analyzed subjects. Among these, 12/27 (44.4%) were carriers of a high-risk recessive disease allele. The most represented disease domains were cancer predisposition (33.3%), cardiac disorders (16.7%), and familial hypercholesterolemia (12.5%).
Conclusion: Although still debated, ensuring during NGS-based genetic testing an opportunistic screening might be valuable for personal and familial early management and surveillance of medically actionable disorders, the individual’s reproductive choices, and the prevalence assessment of underestimated hereditary genetic diseases.
Introduction
Next-generation sequencing (NGS) technologies allow molecular analysis of several genes at the same time (targeted or extended panels), the set of DNA coding sequences (e.g., whole-exome), or the entire genome. These approaches increase the rate of diagnosis of rare diseases; however, they also provide information beyond the clinical question, finding variants in unexpected genes, defined as incidental or secondary findings. (Green et al., 2013) Particularly, the term secondary finding (SF) refers to the active search for causative variants in genes associated with medically actionable conditions. (Green et al., 2013) The use of NGS data could represent a great opportunity to recognize hereditary predispositions to specific pathological conditions and to promptly implement health surveillance or therapeutic protocols in case of the disease. However, the individual and even family psychological impact resulting from the knowledge of a genetic predisposition to a disease is subjective and unpredictable and it should not be overlooked. Therefore, it is debated whether to ensure during a DNA sequencing analysis an opportunistic screening test to deliberately research predispositions to hereditary genetic diseases or to identify the carriership of a recessive disease affecting an individual’s reproductive choices. (Burke et al., 2013; Brothers et al., 2019; Woudstra et al., 2021).
The American College of Medical Genetics (ACMG) recommended that SFs research should be applied as part of a clinical genetic test with diagnostic value to adults and minors, with an opt-out approach. (Green et al., 2013) Since 2013, ACMG periodically updates the list of actionable genes. Originally, a list of 57 actionable genes was proposed, subsequently reduced to 56, and after updated to 59 actionable genes. (Kalia et al., 2017) At the time of writing, this list has been expanded to 73 genes. (Miller et al., 2021a)
According to the concept of opportunistic screening, the European Society of Human Genetics (ESHG) recommended that SFs research should be carried out in the framework of pilot projects; it should only be applied to adults with rare exceptions in minors, with an opt-in approach and the selection of actionable genes should be contextual. Indeed, no actionable gene list was provided by ESHG. (van El et al., 2013; de Wert et al., 2021)
In this study, we aim to assess the frequency of SFs in a case series of 383 consecutive Italian patients referred to our Medical Genetics diagnostic Unit of CEINGE Institute from November 2020 to September 2021. We focused on 59 actionable ACMG genes (Kalia et al., 2017) by a targeted in silico analysis of clinical exome sequencing data. Overall, we identified SFs with high/moderate evidence of pathogenicity in 7.0% of the analyzed subjects. As a second purpose, we herein proposed a three-tier classification system of SFs to support a decision-making process for reporting.
Materials and methods
Patients enrolled in this study and genomic DNA preparation
Overall, 383 consecutive subjects referred to our Medical Genetics diagnostic Unit of CEINGE Institute from November 2020 to September 2021 were included in this study. They were analyzed by clinical exome after genetic counseling.
The case series includes 266 unrelated families, 284 subjects were index cases, and 99 were first-degree relatives (parents and/or siblings). One hundred and fifty-six subjects were pediatrics (0–18 years), whereas 227 were adults (19–75 years). One hundred and ninety-three were females (50.4%), and 190 were males (49.6%).
DNA samples were obtained from each adult subject after signed informed consent, and according to the Declaration of Helsinki. Written informed consent was provided by the participants’ legal guardian/next of kin for pediatric subjects. Prior informed consent to explain the advantages and limits of opportunistic genomic screening has been obtained, allowing the communication of secondary findings exclusively to adult enrolled patients, with an opt-in approach as recommended by ESHG guidelines. Subsequently, we reviewed the patient on the delivery of the original report and we investigated the family history (“dynamic” consensus). This approach was used mainly in the case of the identification of SFs with moderate evidence of pathogenicity.
Genomic DNA preparation was performed using the Maxwell RSC Blood DNA Kit for automated extraction (Promega, Milan, Italy). To evaluate the quality of the extracted genomic DNA before fragmentation, samples were quantified using a UV-Vis spectrophotometer (NanoDrop 2000; Thermo Scientific, Waltham, MA, United States). Then, the genomic DNA was run on 0.8% agarose DNA gel electrophoresis.
Sequencing and data analysis
Genetic testing has been performed by clinical exome, i.e., a test that looks at the protein-coding regions of the genome that have a known clinical association with diseases. In particular, we used a commercially available panel composed of >5000 genes associated with hereditary diseases (SureSelect custom Constitutional Panel 17 Mb, Agilent Technologies). Sample preparation was performed using the Illumina platform’s target enrichment (SureSelectQXT, Agilent Technologies) according to the manufacturer’s instructions. High-throughput sequencing was performed using an Illumina NextSeq 500 platform. The alignment of sequencing reads to the genomic reference, quality control, and identification of variants were carried out with Alissa Align and Call software (v1.1.3–4; Agilent Technologies). Reads were aligned to the GRCh37/hg19 version of the human genome using the BWA MEM algorithm, exons and regions with a read depth below 20 were discarded. Variant annotation and analysis were performed using the Alissa Interpret software (v5.2.10; Agilent Technologies).
The opportunistic genomic screening was performed using a targeted in silico panel (OGS panel) composed of 59 medically actionable genes listed in the ACMG guidelines (Supplementary Table S1). (Kalia et al., 2017) The prioritization of each SF was obtained by gathering evidence from various sources: population data, computational, and pathogenicity scores. Data were filtered for variants present in less than 1% gnomAD, ExAC, and 1000Genomes populations. Rare variants with a frequency >1% in the tested cohort (mainly indels located in regions with low mappability or in homopolymeric regions) were further excluded. In agreement with the definition of screening testing, the prioritized SFs were not confirmed by Sanger sequencing. Nevertheless, read depth (>20), alternative allele frequency (>0.25), and visual inspection of the alignment were evaluated for each variant.
Secondary findings interpretation and classification
All prioritized SFs were firstly analyzed by ClinVar, a public repository that reports the correlation between genetic variants and phenotypes, with supporting evidence, to select pathogenic or likely pathogenic (PLP) SFs. Each variant was manually classified according to the current ACMG guidelines for variant interpretation. (Richards et al., 2015)
Based on both ClinVar interpretation and ACMG classification of the identified SFs, we generated a three-tier classification system of prioritized SFs (Table 1). The first and the second tier include SFs with high evidence of pathogenicity, i.e., either PLP ClinVar variants classified as PLP by ACMG or ClinVar variants with conflicting interpretations of pathogenicity (CI) with PLP interpretations ≥80% and classified as PLP by ACMG guidelines. Finally, the third tier includes SFs with moderate evidence of pathogenicity, i.e., either ClinVar variants with CI or variants not reported in ClinVar, but classified as PLP by ACMG.

TABLE 1. Classification criteria of secondary findings.
Results
Description of secondary findings identified in the cohort of patients
Within our case series, the most common indications for genetic testing were neurodevelopmental disorders (61/284 index cases, 21.5%) and malformative syndromes (59/284 index cases, 20.8%) (Figure 1A).
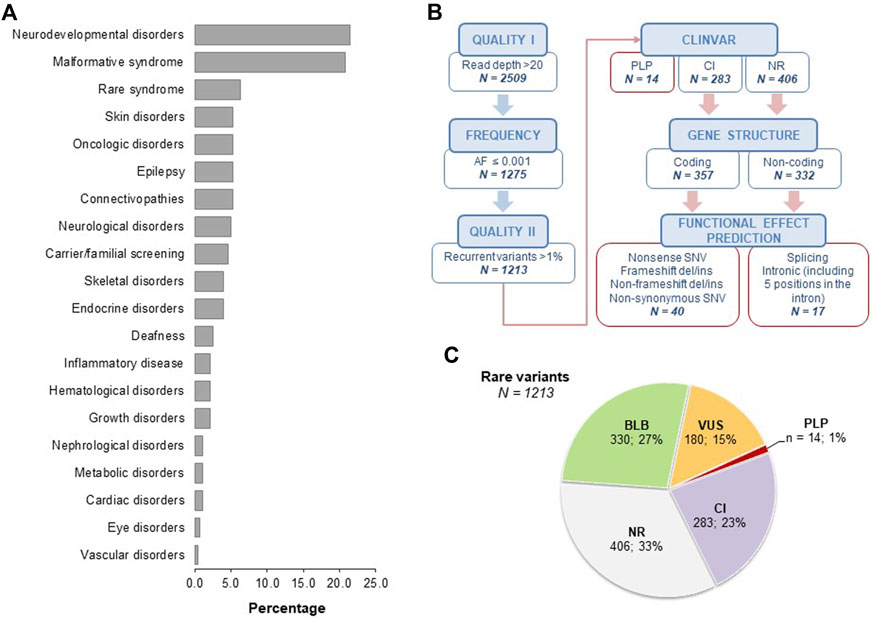
FIGURE 1. Analysis of case series and secondary findings. Panel (A). Clinical indications of the 284 index cases enrolled in the study, subdivided as neurodevelopmental disorders (21.5%), malformative syndromes (20.8%), rare syndrome (6.3%), skin disorders (5.23%), oncologic disorders (5.3%), epilepsy (5.3%), connectivopathies (5.3%), neurological disorders (4.9%), carrier/familial screening (4.6%), skeletal disorders (3.9%), endocrine disorders (3.9%), deafness (2.5%), inflammatory disease (2.1%), hematological disorders (2.1%), growth disorders (2.1%), nephrological disorders (1.1%), metabolic disorders (1.1%), cardiac disorders (1.1%), eye disorders (0.7%), vascular disorders (0.4%). Panel (B). Schematic representation of the filtering strategy for rare SF. The frequency filter was based on data retrieved from GnomAD_exome, GnomAD_genome, ExAC, and 1000Genomes population databases. Recurrent rare variants with frequency >1% in the tested cohort were further excluded. For coding nonsynonymous variants, data were filtered based on the agreement among at least 6/7 prediction tools (CADD Phred >20, LRT: D, MutationAssessor: H-M, MutationTaster: A-D, PolyPhen2 HumDiv: D-P, SIFT: D, FATHMM: D). For noncoding variants, we included essential splice donor/acceptor variants and intronic variants within five positions in the intron. AF, alternative allele frequency; PLP, pathogenic and likely pathogenic variants; CI, conflicting interpretation; NR, not reported; SNV, single nucleotide variant. Panel (C). Pie charts showing the classification of rare SFs as obtained from ClinVar. BLB, benign/likely benign; PLP, pathogenic/likely pathogenic; VUS, variants of uncertain significance; CI, conflicting interpretation; NR, not reported.
The average sequencing depth was 142.2 with 98.5% of target regions at 20× coverage and 96.4% at 50×. Among the subjects analyzed by the OGS panel (n = 383), we overall identified 1213 rare variants, on average 3.2 rare variants for each subject (Figure 1B). In particular, 14 out of 1213 variants (1%) were annotated in ClinVar as PLP, 23% (283/1213) as CI, while 33% (406/1213) were not reported (NR) (Figures 1B,C). Based on gene structure and functional effect prediction, we prioritized 40 ClinVar CI variants and 17 ClinVar NR variants (Figure 1B; Supplementary Tables 2–4). Following manual ACMG classification, we identified eight PLP SFs (7 from the ClinVar CI group and one from the ClinVar NR group). Thus, our filtering strategy enabled us to detect 21 unique SFs in 27 subjects (22 unrelated), with a range of 1-2 prioritized SFs for each subject (Table 2). All the identified variants were in the heterozygous state. We detected a single variant for each subject except for one subject who carried two variants.
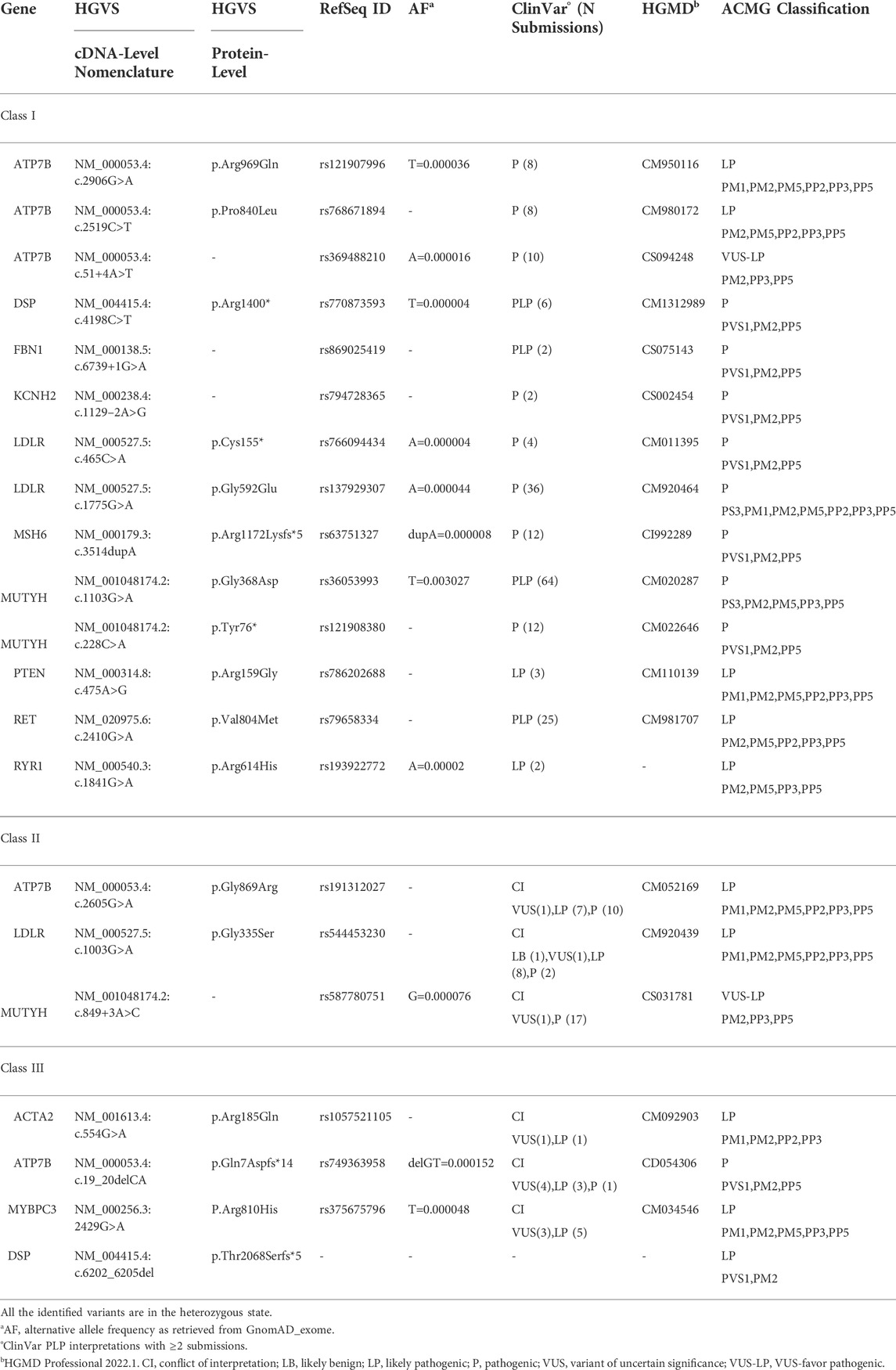
TABLE 2. Clinical exome medically actionable secondary findings identified in 383 participants.
To select SFs highly likely to be causative, we applied a three-tier classification system to prioritize SFs according to different evidence levels of pathogenicity (Table 1). Based on this classification, we observed SFs with high/moderate evidence of pathogenicity in 7.0% of analyzed subjects (27/383) and 8.3% of unrelated families (22/266). In particular, SFs with high evidence of pathogenicity (tiers I-II) were found in 6.0% of analyzed subjects (23/383) and 7.1% of unrelated families (19/266). Whereas SFs with moderate evidence of pathogenicity (tier III) were identified in 1.0% of analyzed subjects (4/383) and 1.5% of unrelated families (4/266).
Molecular and clinical classification of secondary findings
Most of the subjects presented dominant medically actionable variants (15/27, 55.5%). The most recurrent genes harboring SFs with high/moderate levels of pathogenic evidence were LDLR and DSP (Figure 2A). Besides, 12 subjects carried at least one high-risk recessive allele (12/27, 44.4%) either in ATP7B or in MUTYH gene, causative of Wilson disease and MUTYH-associated polyposis, respectively (Figure 2A).
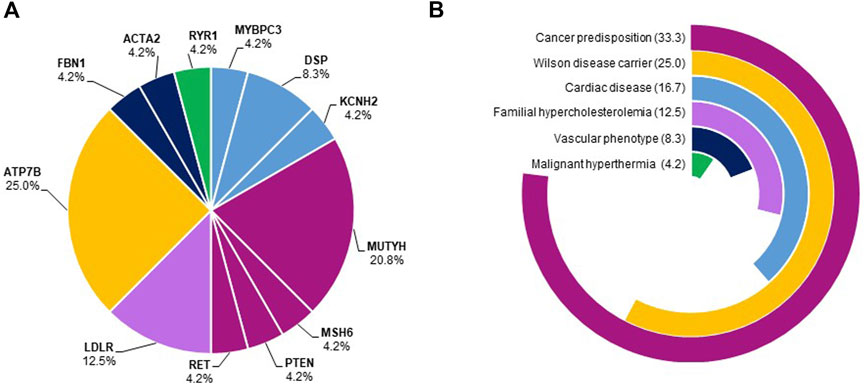
FIGURE 2. Frequency of genes harboring secondary findings and disease domains. Panel (A). The pie chart shows the frequency of genes harboring SFs. Genes belonging to the same disease domain were shown using the color code reported in panel B. Panel (B). The pie chart shows the proportions of disease domains of identified SFs. The most frequent disease domains were cancer predisposition (33.3%), cardiac diseases (16.7%), and familial hypercholesterolemia (12.5%).
Of note, 33.3% of SFs with high/moderate evidence of pathogenicity were identified in loci related to cancer predisposition (MUTYH-associated polyposis, Lynch syndrome, multiple endocrine neoplasia type 2, Cowden syndrome), 16.7% in genes causative of cardiac disorders (arrhythmogenic right ventricular dysplasia, dilated cardiomyopathy, long QT syndrome), and 12.5% in genes associated with familial hypercholesterolemia (Figure 2B).
Discussion
Secondary findings arise from the active search for causative variants in genes associated with medically actionable conditions. To date, it is debated whether to ensure, during NGS-based genetic testing, an opportunistic screening to research deliberately predispositions to hereditary genetic diseases or identify a state of carrier for recessive disorders affecting the individual’s reproductive choices. (Woudstra et al., 2021)
Using an in silico targeting of 59 medically actionable genes, we investigated SFs in a monocentric case series of 383 consecutive subjects undergoing a clinical exome in suspicion of different genetic conditions, mainly neurodevelopmental disorders, and malformative syndromes. Our study allowed us to detect overall 21 unique actionable SFs with high/moderate evidence of pathogenicity in 7.0% of analyzed subjects, 17 of them classified as SFs with high evidence of pathogenicity in 6.0% of individuals, in line with reported SFs frequencies ranging between 1.2% and 11%. (Xue et al., 2012; Dorschner et al., 2013; Cassa et al., 2013; Olfson et al., 2015; Jang et al., 2015; eMERGE Clinical Annotation Working Group, 2020; Jalkh et al., 2020; Aloraini et al., 2022) Different factors including study design, sequencing technology, tested population, the knowledge of variants in literature, and data interpretation explain the different rates of SFs reported in previous studies. Data interpretation is a complicated phase of the study of a variant, and the choice of a specific methodology may underestimate or overestimate the finding. We herein established a three-class ranking to prioritize secondary events based on their annotation/prediction using the ClinVar database and ACMG guidelines. (Richards et al., 2015) Through this approach, we tried to provide a clinically-driven method for assessing the impact of medically actionable genetic variations with direct implications on the choice to report or not a specific SF. In particular, we suggested that SFs with high evidence of pathogenicity (i.e., those included in the first two tiers) should be recommended for reporting in adult subjects after signing their opt-in informed consent regardless of information about their family history. Conversely, those variants with moderate evidence of pathogenicity (i.e., those included in the third tier) should be carefully re-evaluated considering the family history before inclusion in the genetic testing report. In these specific cases, the collection of family history through a “dynamic” consensus system as proposed by the SFMPP could be the right approach to reanalyze SFs with uncertain pathogenicity, in order not to miss SFs. (Pujol et al., 2018) The three-tier classification highlighted the difficulty of the molecular geneticists to report those SFs with moderate evidence of pathogenicity (tier III). Only the knowledge of positive family history for that specific disease can overcome the problem of reporting those SFs included in tier III. Of note, the current recommendations for reporting of SFs in clinical exome establish that all variants that are classified as P or LP according to ACMG/AMP standards and guidelines should be reported as SFs. (Miller et al., 2021b)
In agreement with previous similar studies, we identified SFs in genes mostly associated with cardiac disorders (16.7%), cancer predisposition (33.3%), and familial hypercholesterolemia (12.5%). (eMERGE Clinical Annotation Working Group, 2020; Aloraini et al., 2022; Ramensky et al., 2021) These findings underline the importance of establishing rapid and early screening methods to avoid the underestimation of medically actionable disorders.
In Southern Italy, the prevalence of hypertrophic cardiomyopathy, Lynch syndrome, or familial hypercholesterolemia is not well-established since prevalence studies are mainly based on screening in young adults and therefore biased by age-related penetrance. Similarly, incomplete penetrance may account for underestimated prevalence, as in the case of some mismatch repair genes causative of Lynch syndrome.
Based on the identified SF, the clinicians might suggest extending the communication to first-degree family members and eventually performing a single-variant diagnostic test on the relatives. Indeed, according to a multicenter study, the return of SFs in apparently healthy family members increases the diagnosis of hereditary conditions. (Hart et al., 2019) In parallel, clinicians should start monitoring the condition with adequate clinical and/or biochemical testing. Our data confirm the importance of detecting underdiagnosed disorders, such as familial hypercholesterolemia, especially in those countries where clinical scores are lacking, such as Italy, in contrast to those that routinely use clinical scores for diagnosis, such as the DUTCH score. (Nordestgaard et al., 2013; Bertolini et al., 2017; Reeskamp et al., 2020) Additionally, they also underline the relevance of identifying susceptibility to disorders for which the risk-to-benefit ratio of the recommended interventions favors such interventions since they are not overly burdensome or costly. This is the case of malignant hyperthermia susceptibility whose clinical diagnostic test is invasive and expensive. Nevertheless, most surgical procedures can be performed by avoiding known triggering agents, and effective measures can be taken if knowledge of the risk is communicated to the anesthesiologist. (Katz et al., 2020)
We identified 12 individuals with a recessive pathogenic disease allele in our case series. Reporting carrier status (e.g., in ATP7B gene) might be highly beneficial, especially in a population characterized by a high rate of consanguineous marriages for the recurrence risk assessment and valuable for the carrier itself. Indeed, it is well-known that heterozygous carriers of MUTYH alleles have an increased risk of colorectal cancer and MUTYH carriership is associated with younger age of diagnosis, and a higher prevalence of polyps, right-sided and synchronous cancers.
The return of genomic SFs in NGS testing represents a point of discussion among molecular and clinical geneticists. Our study estimated the frequency of clinically actionable SFs in a cohort of Italian patients. We herein provided a classification system of SFs to assess their clinical impact and guide the reporting, focusing on a manual clinically-driven approach to study each variant, and adopting a dynamic consensus.
The establishment of a three-tier ranking of prioritized SFs might facilitate the reporting of these variants thus significantly impacting patients’ follow-up and management. Our data showed that most medically actionable variants were in genes associated with cardiac disorders, oncological predisposition, and familial hypercholesterolemia, conditions that are probably underestimated. With the report of these findings, the patients will benefit from a multi-disciplinary approach, including genetic counseling, involving a detailed personal and family history, surveillance of the disease, and family plan.
Data availability statement
Aggregated variant frequency information can be requested to the corresponding author. Individual genotype information cannot be made available in order to protect participant privacy.
Ethics statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
SM, ATB, IA, and RR designed and conducted the study and prepared the manuscript; BER, RM, and FDG prepared the library enrichment and the sequencing analysis; RR and FB performed the bioinformatic analysis; SM and ATB performed in silico analysis of secondary findings; SM and ATB cared for the patient; MC and AI carried out a critical revision of the manuscript.
Acknowledgments
The authors kindly thank the Bioinformatic Service for NGS and the NGS service of CEINGE Advanced Biotechnologies institute.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.956723/full#supplementary-material
References
Aloraini, T., Alsubaie, L., Alasker, S., Al Muitiri, A., Alswaid, A., Eyiad, W., et al. (2022). The rate of secondary genomic findings in the Saudi population. Am. J. Med. Genet. A 188, 83–88. doi:10.1002/ajmg.a.62491
Bertolini, S., Pisciotta, L., Fasano, T., Rabacchi, C., and Calandra, S. (2017). The study of familial hypercholesterolemia in Italy: A narrative review. Atheroscler. Suppl 29, 1–10. doi:10.1016/j.atherosclerosissup.2017.07.003
Brothers, K. B., Vassy, J. L., and Green, R. C. (2019). Reconciling opportunistic and population screening in clinical genomics. Mayo Clin. Proc. 94, 103–109. doi:10.1016/j.mayocp.2018.08.028
Burke, W., Antommaria, A. H., Bennett, R., Botkin, J., Clayton, E. W., Henderson, G. E., et al. (2013). Recommendations for returning genomic incidental findings? We need to talk. Genet. Med 15, 854–859. doi:10.1038/gim.2013.113
Cassa, C. A., Tong, M. Y., and Jordan, D. M. (2013). Large numbers of genetic variants considered to be pathogenic are common in asymptomatic individuals. Hum. Mutat. 34, 1216–1220. doi:10.1002/humu.22375
de Wert, G., Dondorp, W., Clarke, A., Dequeker, E. M. C., Cordier, C., Deans, Z., et al. (2021). Opportunistic genomic screening. Recommendations of the European society of human genetics. Eur. J. Hum. Genet 29, 365–377. doi:10.1038/s41431-020-00758-w
Dorschner, M. O., Amendola, L. M., Turner, E. H., Robertson, P. D., Shirts, B. H., Gallego, C. J., et al. (2013). Actionable, pathogenic incidental findings in 1, 000 participants' exomes. Am. J. Hum. Gene 93, 631–640. doi:10.1016/j.ajhg.2013.08.006
eMERGE Clinical Annotation Working Group, (2020). Frequency of genomic secondary findings among 21, 915 eMERGE network participants. Genet. Med 22, 1470–1477. doi:10.1038/s41436-020-0810-9
Green, R. C., Berg, J. S., Grody, W. W., Kalia, S. S., Korf, B. R., Martin, C. L., et al. (2013). ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet. Med 15, 565–574. doi:10.1038/gim.2013.73
Hart, M. R., Biesecker, B. B., Blout, C. L., Christensen, K. D., Amendola, L. M., Bergstrom, K. L., et al. (2019). Secondary findings from clinical genomic sequencing: Prevalence, patient perspectives, family history assessment, and health-care costs from a multisite study. Genet. Med 21, 1100–1110. doi:10.1038/s41436-018-0308-x
Jalkh, N., Mehawej, C., and Chouery, E. (2020). Actionable exomic secondary findings in 280 Lebanese participants. Front. Genet 11, 208. doi:10.3389/fgene.2020.00208
Jang, M. A., Lee, S. H., Kim, N., and Ki, C. S. (2015). Frequency and spectrum of actionable pathogenic secondary findings in 196 Korean exomes. Genet. Med 17, 1007–1011. doi:10.1038/gim.2015.26
Kalia, S. S., Adelman, K., Bale, S. J., Chung, W. K., Eng, C., Evans, J. P., et al. (2017). Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the American College of medical genetics and genomics. Genet. Med 19, 249–255. doi:10.1038/gim.2016.190
Katz, A. E., Nussbaum, R. L., Solomon, B. D., Rehm, H. L., Williams, M. S., and Biesecker, L. G. (2020). Management of secondary genomic findings. Am. J. Hum. Genet 107, 3–14. doi:10.1016/j.ajhg.2020.05.002
Miller, D. T., Lee, K., Chung, W. K., Gordon, A. S., Herman, G. E., Klein, T. E., et al. (2021). ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of medical genetics and genomics (ACMG). Genet. Med. 23, 1381–1390. doi:10.1038/s41436-021-01172-3
Miller, D. T., Lee, K., Gordon, A. S., Amendola, L. M., Adelman, K., Bale, S. J., et al. (2021). Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2021 update: A policy statement of the American College of medical genetics and genomics (ACMG). Genet. Med 23, 1391–1398. doi:10.1038/s41436-021-01171-4
Nordestgaard, B. G., Chapman, M. J., Humphries, S. E., Ginsberg, H. N., Masana, L., Descamps, O. S., et al. (2013). Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: Guidance for clinicians to prevent coronary heart disease: Consensus statement of the European atherosclerosis society. Eur. Heart J 34, 3478–90a. doi:10.1093/eurheartj/eht273
Olfson, E., Cottrell, C. E., Davidson, N. O., Gurnett, C. A., Heusel, J. W., Stitziel, N. O., et al. (2015). Identification of medically actionable secondary findings in the 1000 genomes. PLoS One 10, e0135193. doi:10.1371/journal.pone.0135193
Pujol, P., Vande Perre, P., Faivre, L., Sanlaville, D., Corsini, C., Baertschi, B., et al. (2018). Guidelines for reporting secondary findings of genome sequencing in cancer genes: The SFMPP recommendations. Eur. J. Hum. Genet 26, 1732–1742. doi:10.1038/s41431-018-0224-1
Ramensky, V. E., Ershova, A. I., Zaicenoka, M., Kiseleva, A. V., Zharikova, A. A., Vyatkin, Y. V., et al. (2021). Targeted sequencing of 242 clinically important genes in the Russian population from the ivanovo region. Front. Genet. 12, 709419. doi:10.3389/fgene.2021.709419
Reeskamp, L. F., Tromp, T. R., Defesche, J. C., Grefhorst, A., Stroes, E. S., Hovingh, G. K., et al. (2020). Next-generation sequencing to confirm clinical familial hypercholesterolemia. Eur. J. Prev. Cardiol, 204748732094299. doi:10.1177/2047487320942996
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. ,(2015). ACMG Laboratory Quality Assurance Committee Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med 17, 405–424. doi:10.1038/gim.2015.30
van El, C. G., Cornel, M. C., Borry, P., Hastings, R. J., Fellmann, F., Hodgson, S. V., et al. (2013). Whole-genome sequencing in health care: Recommendations of the European society of human genetics. Eur. J. Hum. Genet 21, 580–584. doi:10.1038/ejhg.2013.46
Woudstra, A., Dondorp, W., and de Wert, G. (2021). Stakeholder views on opportunistic genomic screening in The Netherlands: A qualitative study. Eur. J. Hum. Genet 29, 949–956. doi:10.1038/s41431-021-00828-7
Keywords: secondary findings, clinical exome, genetic testing, ACMG actionable genes, NGS testing
Citation: Martone S, Buonagura AT, Marra R, Rosato BE, Del Giudice F, Bonfiglio F, Capasso M, Iolascon A, Andolfo I and Russo R (2022) Clinical exome-based panel testing for medically actionable secondary findings in a cohort of 383 Italian participants. Front. Genet. 13:956723. doi: 10.3389/fgene.2022.956723
Received: 30 May 2022; Accepted: 19 October 2022;
Published: 10 November 2022.
Edited by:
Antonio Percesepe, University of Parma, ItalyReviewed by:
Moien Kanaan, Bethlehem University, PalestinePraseetha Kizhakkedath, United Arab Emirates University, United Arab Emirates
Nadia Akawi, United Arab Emirates University, United Arab Emirates
Copyright © 2022 Martone, Buonagura, Marra, Rosato, Del Giudice, Bonfiglio, Capasso, Iolascon, Andolfo and Russo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Immacolata Andolfo, aW1tYWNvbGF0YS5hbmRvbGZvQHVuaW5hLml0; Roberta Russo, cm9iZXJ0YS5ydXNzb0B1bmluYS5pdA==
†These authors have contributed equally to this work