 Ting Hu1,2,3*†
Ting Hu1,2,3*† Jiamin Wang1,2,3†
Jiamin Wang1,2,3† Qian Zhu1,2,3
Qian Zhu1,2,3 Zhu Zhang1,2,3Rui Hu1,2,3Like Xiao1,2,3Yunyuan Yang1,2,3Na Liao1,2,3Sha Liu1,2,3He Wang1,2,3
Zhu Zhang1,2,3Rui Hu1,2,3Like Xiao1,2,3Yunyuan Yang1,2,3Na Liao1,2,3Sha Liu1,2,3He Wang1,2,3 Xiaoyu Niu2,3*
Xiaoyu Niu2,3* Shanling Liu1,2,3*
Shanling Liu1,2,3*- 1Department of Medical Genetics, West China Second University Hospital, Sichuan University, Chengdu, Sichuan, China
- 2Department of Obstetrics and Gynecology, West China Second University Hospital, Sichuan University, Chengdu, Sichuan, China
- 3Key Laboratory of Birth Defects and Related Diseases of Women and Children (Sichuan University), Ministry of Education, Chengdu, China
Objectives: The study aimed to investigate the clinical use of noninvasive prenatal testing (NIPT) for common fetal aneuploidies as a prenatal screening tool for the detection of rare chromosomal abnormalities (RCAs).
Methods: Gravidas with positive NIPT results for RCAs who subsequently underwent amniocentesis for a single nucleotide polymorphism array (SNP array) were recruited. The degrees of concordance between the NIPT and SNP array were classified into full concordance, partial concordance, and discordance. The positive predictive value (PPV) was used to evaluate the performance of NIPT.
Results: The screen-positivity rate of NIPT for RCAs was 0.5% (842/158,824). Of the 528 gravidas who underwent amniocentesis, 29.2% (154/528) were confirmed to have positive prenatal SNP array results. PPVs for rare autosomal trisomies (RATs) and segmental imbalances were 6.1% (7/115) and 21.1% (87/413), respectively. Regions of homozygosity/uniparental disomy (ROH/UPD) were identified in 9.5% (50/528) of gravidas. The PPV for clinically significant findings was 8.0% (42/528), including 7 cases with mosaic RATs, 30 with pathogenic/likely pathogenic copy number variants, and 5 with imprinting disorders.
Conclusion: NIPT for common fetal aneuploidies yielded low PPVs for RATs, moderate PPVs for segmental imbalances, and incidental findings for ROH/UPD. Due to the low PPV for clinically significant findings, NIPT for common fetal aneuploidies need to be noticed for RCAs.
Introduction
Noninvasive prenatal testing (NIPT), also known as cell-free fetal DNA (cff-DNA) testing, is mainly based on massive parallel sequencing (MPS), and it has been used to screen for common fetal aneuploidies in more than 60 countries since 2011 (Palomaki et al., 2011). NIPT is highly sensitive and specific for the detection of trisomies 13, 18, and 21 (Committee on Genetics Society for Maternal for Maternal-Fetal Medicine, 2015; Taylor-Phillips et al., 2016), which has led to a reduction in invasive diagnostic testing requests by up to 40% to avoid procedure-related miscarriage risks (Wong and Lo, 2016). Recently, rare autosomal trisomies, well-known microdeletion/microduplication syndromes (MMSs), and genome-wide copy number variants (CNVs) have been added by some laboratories as expanded screening items (Wapner et al., 2012; Wapner et al., 2015; Benn and Grati, 2018). Apoptotic placental cells from the cytotrophoblast mixed with maternal cell-free DNA are the primary sources of cff-DNA in maternal circulation (Tjoa et al., 2006; Faas et al., 2012); hence, factors like confined placental mosaicism (CPM) and maternal genomic contributions affect the accuracy of NIPT results (Bianchi and Wilkins-Haug, 2014). Thus, all positive NIPT results should be confirmed by invasive diagnostic testing (Mardy and Wapner, 2016; Cherry et al., 2017).
Chromosomal microarray analysis (CMA), a high-resolution genomic technology used to detect CNVs, has been recommended as a first-tier test for the postnatal evaluation of individuals with unexplained developmental delay, intellectual disability, autism spectrum disorders, or multiple congenital anomalies, including prenatal evaluation of fetuses with structural anomalies observed by ultrasound (Manning et al., 2010; Miller et al., 2010; American College of Obstetricians and Gynecologists Committee on Genetics, 2013). Furthermore, single-nucleotide polymorphism (SNP) arrays can additionally identify haploidy, triploidy, and regions of homozygosity (ROH) (Levy et al., 2014). The pathogenesis of ROHs includes imprinting effects caused by uniparental disomy (UPD) (Robinson, 2000) and increased susceptibility to complex diseases caused by homozygous mutations in autosomal recessive genes (Campbell et al., 2007; Ku et al., 2011).
The utility of NIPT for specific MMSs with moderate to high positive predictive values (PPVs), including DiGeorge syndrome (DGS), Prader–Willi/Angleman syndrome (PWS/AS), cri du chat (CDC), and 1p36 microdeletion (1p36 del) syndrome, has been shown (Dar et al., 2014; Gross et al., 2016; Petersen et al., 2017; Liang et al., 2019). However, there is still a paucity of research focusing on rare chromosomal abnormalities (RCAs) detected by NIPT for common fetal aneuploidies. Therefore, in this retrospective cohort study of 158,919 singleton pregnancies, we evaluated the clinical use of the NIPT as a prenatal screening tool for the detection of RCAs, including aneuploidies and segmental imbalances.
Materials and methods
Patients
For this study, singleton pregnancies at a tertiary-level referral center (West China Second University Hospital, Sichuan University) were included from January 2016 to December 2020. Trained clinical geneticists performed pretest counseling. Before an NIPT or SNP array analysis, we obtained written informed consent from all gravidas who agreed to undergo NIPT or consecutive amniocentesis due to positive NIPT results. The Medical Ethics Committee of the West China Second University Hospital approved the study.
For NIPT, the inclusion criteria were as follows: 1) patients with advanced maternal age ≥35 years who declined invasive procedures; 2) patients with high risk for first- or second-trimester maternal serum screening (T21 ≥ 1/270, T18 ≥ 1/350) who declined invasive procedures; 3) intermediate risk for maternal serum screening (T21:1/270–1/1,000, T18:1/350–1/1,000); 4) fetuses with soft markers detected by ultrasound, including nuchal translucency of 2.5–3.0 mm; 5) positive family history, such as any affected offspring with Down syndrome; and 6) pregnant women who prefer NIPT to maternal serum screening without any clinical indications. The exclusion criteria, according to the current standard practice in China, were as follows: 1) pregnancy gestation period <12 weeks; 2) fetal structural anomalies detected by ultrasound before NIPT; 3) pregnant women with chromosomal abnormalities; 4) multiple pregnancies or co-twin demise after 12 weeks; 5) pregnant women who received stem cell therapy, transplant surgery, allogeneic blood products, or immunotherapy within one year; and 6) pregnant women with malignant tumors. Blood samples (10 ml) from the gravidas were collected in cell-free DNA BCT tubes (Streck, Omaha, United States).
All gravidas with positive NIPT results for RCAs, including rare autosomal trisomies (RATs) and segmental imbalances, were advised to undergo amniocentesis for SNP analysis after 16 gestational weeks. To assess the clinical use of NIPT for RCAs, the exclusion criteria were as follows: 1) positive NIPT results for common trisomies (T21/T18/T13); 2) positive NIPT results for sex chromosome aneuploidies (SCAs); and 3) pregnant women who declined amniocentesis or who underwent amniocentesis for traditional cytogenetics (e.g., karyotype alone) but declined SNP analysis. Fetal samples (20 ml) were obtained through amniocentesis. Clear amniotic fluid samples were tested directly, while blood-stained samples were cultured before the SNP array experiments. Additionally, peripheral blood samples of the parents were obtained to confirm that fetal CNVs were inherited or de novo, and ROHs were separated from UPDs.
Noninvasive prenatal testing
Plasma from the blood samples was isolated within 24 h by two-step centrifugation. All procedures, including cell-free DNA extraction, purification, library construction, and quantification, were performed using the fetal chromosome aneuploidy (T21/T18/T13) test kit (Berry Genomics, Beijing, China). MPS was performed on the NextSeq CN500 platform (Berry Genomics, Beijing, China) with 36-bp single-end reads, resulting in 5 million total reads, which corresponds to a 0.05× sequencing depth. GC bias was eliminated by bioinformatic methods combined with local weighted polynomial regression. Raw reads were aligned to the human reference genome, GRCh37 (hg19). Each chromosome with an absolute Z-score greater than 3 was marked with chromosome aneuploidies. CNVs of ≥ 2 Mb were detected using the RUPA algorithm developed by Berry Genomics.
Chromosomal microarray analysis
This procedure was described in our previous study (Hu et al., 2021). CNVs >100 kb or those that affected >50 contiguous probes and ROHs >10 Mb were considered. The pathogenicity of the detected CNVs was according to the criteria of the American College of Medical Genetics and Genomics (ACMG) and Clinical Genome Resource (ClinGen) Technical standards (Riggs et al., 2020). When the limit with which CMA can be expected to detect low-level mosaicism was 10%–20% (Cross et al., 2007; Scott et al., 2010; Hall et al., 2014), we simultaneously performed an interphase fluorescence in situ hybridization analysis (FISH) when CMA detected mosaicism (≥10%). When both a gain and loss of more than 5 Mb were detected in one fetal sample, peripheral blood samples of the parents were karyotyped to confirm whether the parents had chromosomal balanced translocations or inversions. The nomenclature of CMA and karyotypes is according to the International System for Human Genomic Nomenclature (ISCN) 2020 (McGowan-Jordan et al., 2020).
Data analysis
The positive results of NIPT for rare RCAs were classified into two groups: 1) RATs and 2) segmental imbalances; however, the positive results of CMA were classified into three groups: 1) RATs (including mosaic aneuploidies), 2) segmental imbalances, and 3) ROH/UPDs. Clinically significant findings included RATs, pathogenic/likely pathogenic (P/LP) CNVs, and UPDs associated with imprinting disorders.
We compared the NIPT results with those of CMA and classified them into four categories: 1) full concordance: those with consistent aneuploidy results, or with consistent cytoband and copy number gain/loss between NIPT and CMA; 2) partial concordance: at least one of the findings was consistent, but additional findings were detected only by one platform (NIPT/CMA) (for example, NIPT was positive for 20p13p12.1 duplication, and CMA detected 20p13p12.1 duplication and 9p24.3p23 deletion); aneuploidies or segmental imbalances were detected by NIPT but ROH/UPD was confirmed by CMA; and 3) discordance: none of the findings detected by NIPT and CMA were consistent (for example, NIPT was positive for trisomy 6, and CMA was negative; or NIPT was positive for trisomy 7, and CMA detected 16p11.2 deletion).
Clinical follow-up assessments
We performed clinical follow-up assessments 6 months to 3 years postpartum on all gravidas who underwent amniocentesis for SNP analysis. In addition, data on circumstances after birth, including the gestational age of delivery, birth weight, postnatal imaging, developmental details diagnosed by pediatricians, and perinatal or infant death, were collected. For fetuses treated with termination of pregnancy (TOP), the indications such as chromosomal aberrations, abnormalities of ultrasound findings, miscarriage following amniocentesis, premature delivery, and still births, were obtained through hospital information systems.
Statistical analysis
A statistical analysis was performed by SPSS Statistics software v24.0 (IBM SPSS, Armonk, NY, United States). Continuous variables were compared by Student’s t-test, and categorical variables were compared by chi-squared or Fisher’s exact analysis, as appropriate. A p-value < 0.05 was considered to indicate statistical significance.
Results
Patient characteristics
A total of 158,919 gravidas were recruited to undergo NIPT in a 5-year retrospective study to evaluate the clinical value of NIPT for rare RCAs, including aneuploidies, segmental imbalances, and ROH/UPD. The test had a failure rate of 0.1% in 95 cases. A total of 508 (0.3%) cases with positive screen results for common trisomies (T21/T18/T13) and 921 (0.6%) cases for SCAs were excluded. In the 842 (0.5%) gravidas with positive screen results for RCAs, including 193 (22.9%) at high risk for RATs and 649 (77.1%) for segmental imbalances, 528 gravidas underwent consecutive amniocentesis for prenatal diagnosis by the SNP array. The flow diagram of the study is shown in Figure 1. The maternal age ranged from 17 to 44 years (29.1 ± 4.7 years), with 13.8% (73/528) being of advanced maternal age. The gestational age for NIPT and amniocentesis ranged from 12 to 27+5 weeks (19.7 ± 5.7 weeks) and 17 to 30+1 weeks (20.5 ± 3.3 weeks), respectively.
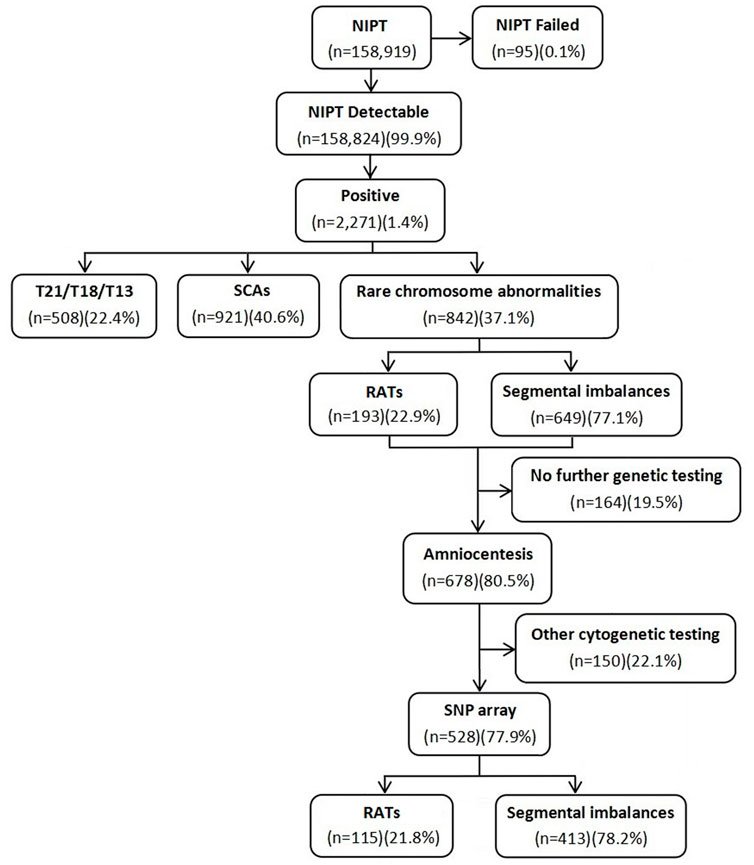
FIGURE 1. Flow diagram of the study.
Of the 528 positive NIPT results, there were 115 fetuses at high risk for RATs, including 10 fetuses with multiple chromosomes, while the remaining 413 fetuses were at high risk for segmental imbalances, including 16 fetuses with multiple chromosomes. The SNP array was successfully performed in all gravidas; 154 (29.2%) cases had positive results, including 7 (4.5%, 7/154) fetuses with mosaic RATs, 97 (63.0%, 97/154) fetuses with segmental imbalances, and 50 (32.5%, 50/154) fetuses with UPD/ROH (Table 1). The concordance between the RCAs detected by NIPT and consecutive CMA results is shown in Table 2. No significant difference in maternal age was observed between the positive and negative SNP array groups (28.8 ± 5.1 years vs. 29.2 ± 4.5; p = 0.350).
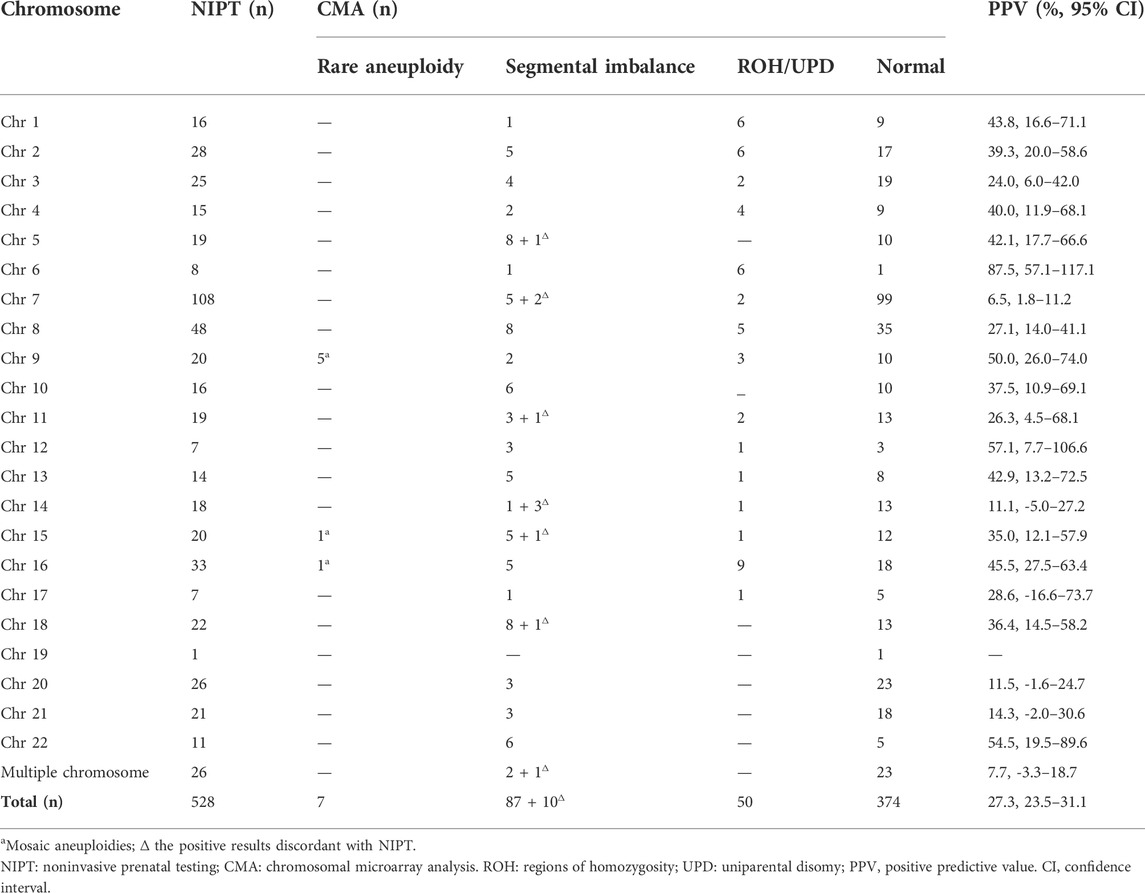
TABLE 1. Summary of the CMA results of 528 fetuses with positive NIPT results.
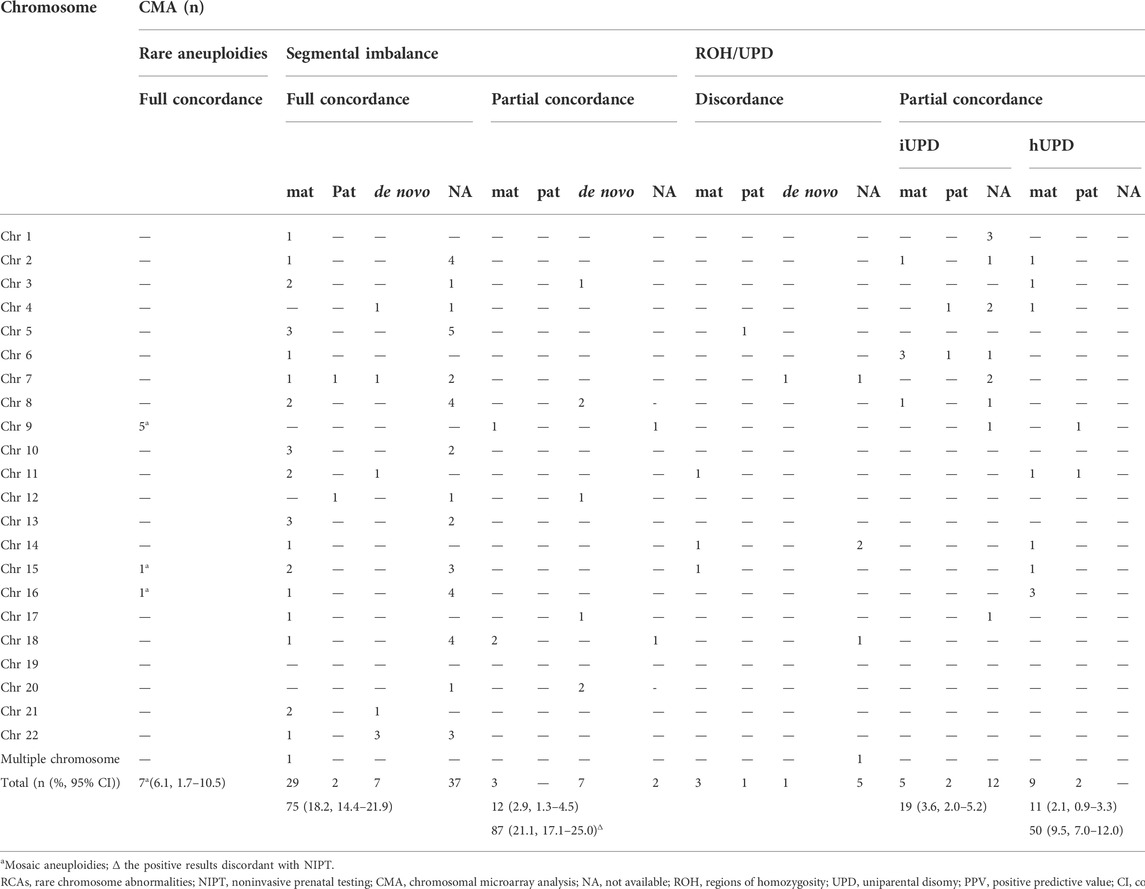
TABLE 2. Concordance between the RCAs detected by NIPT and consecutive CMA results.
Rare autosomal trisomies
The PPV for RATs was 6.1% (7/115, 95% confidence interval (CI), 1.7%–10.5%) (Table 2). The SNP array confirmed that all RATs were as mosaicism. The most common aneuploidy was mosaic trisomy 9, including five cases with mosaic proportions ranging from 15 % to 29%. The other two cases were mosaic trisomy 15 (26%) and mosaic trisomy 16 (13%). All the mosaic aneuploidies were simultaneously confirmed using FISH in amniotic fluids.
Segmental imbalances
The PPV for segmental imbalances was 21.1 % (87/413, 95 % CI, 17.1%–25.0%). A total of 111 segmental imbalances in 97 cases were detected by the SNP array, comprising 75 (77.3%) cases with full concordance, 12 (12.4%) with partial concordance, and 10 (10.3%) with discordance. Approximately, 57.7% (64/111) of the CNVs were <5 Mb, 19.8% (22/111) ranged from 5 to 10 Mb, and 22.5% (25/111) were >10 Mb, with a concordance rate of 75.0% (48/64), 86.4% (19/22), and 88.0% (22/25) between the NIPT and SNP arrays, respectively (p = 0.276).
The PPV for clinically significant CNVs (18 P CNVs and 12 LP CNVs) was 7.3% (30/413; 95 % CI, 4.8%–9.8%). There were 38 clinically significant CNVs including 23 P CNVs and 15 LP CNVs detected by the SNP array in 36 cases, comprising 24 (66.7%) cases with full concordance, six (16.7%) with partial concordance and six (16.7%) with discordance. For the well-known MMSs confirmed by the SNP analysis, 1p36 deletion, 15q11q13 (PWS/AS) duplication, DGS, and 22q11.2 duplication were all detected by NIPT, while 50% (2/4) of the CDC cases were ignored. The details of segmental imbalances detected by the SNP array are shown in Supplementary Table S1.
Parental confirmation by the SNP array was performed in 54.6% (53/97) of cases, while fetuses were detected with segmental imbalances by CMA, including 35, 3, and 15 cases of maternal, paternal inheritance, and de novo inheritance, respectively (Table 2). The proportion of full concordance between NIPT and CMA in cases with maternally inherited CNVs was significantly higher than that in those with paternal-inherited or de novo CNVs (82.9% (29/35) vs. 50.0% (9/18); p = 0.012) (Table 2). In addition, of the 10 cases with subchromosomal unbalanced rearrangements, except for one couple who refused to perform karyotyping (No. 84), six cases had inherited parental balanced translocations (Nos. 78, 79, 81, 83, 86, and 87), one case inherited paternal pericentric inversion (No. 77), and two cases inherited a mother derivative chromosome associated with intellectual disability (Nos. 80 and 82) (Supplementary Table S1).
Regions of homozygosity/uniparental disomy
We incidentally detected 50 (9.5 %) fetuses by the SNP array with ROH larger than 10 Mb restricted to one chromosome, all relatively consistent with the NIPT results. Chromosome 16 was the most frequently involved (nine cases), followed by chromosomes 1, 2, and 6 (six cases). None of the fetuses was from consanguineous couples.
In total, 30 cases were confirmed as UPD: 18 cases were diagnosed with isodisomy as ROHs detected by the SNP analysis involving the whole chromosome (including six cases with a confirmed parental source of ROHs), and 12 iso-heterodisomy cases were verified by parental blood samples. The most frequent UPD was UPD6 (five cases), followed by UPD4 (four cases), and UPD1, UPD2, and UPD16 (three cases).
There were 11 cases of ROHs associated with imprinted chromosomes. Except for three couples who refused to perform parental confirmations, imprinting disorders were confirmed in five of the eight cases, including transient neonatal diabetes mellitus (pUPD6), Silver–Russell syndrome (mUPD11), Beckwith–Wiedemann syndrome (pUPD11), Temple syndrome (mUPD14), and PWS (mUPD15). The details of ROHs detected by the SNP analysis are shown in Supplementary Table S2.
Clinical follow-up assessments
Clinical follow-up results were obtained in 90.2% of cases (476/528). Except for the fetuses lost to follow-up, the rates of normal infants, termination of pregnancy (TOP), and birth with defects (including neonatal demises without physical birth defects) in those without chromosomal aberrations by the SNP array were 94.9% (314/331), 2.4% (8/331), and 2.7% (9/331), respectively, while in those with chromosomal aberrations by the SNP array were 47.5% (69/145), 49.0% (71/145), and 3.4% (5/145), respectively (Table 3).
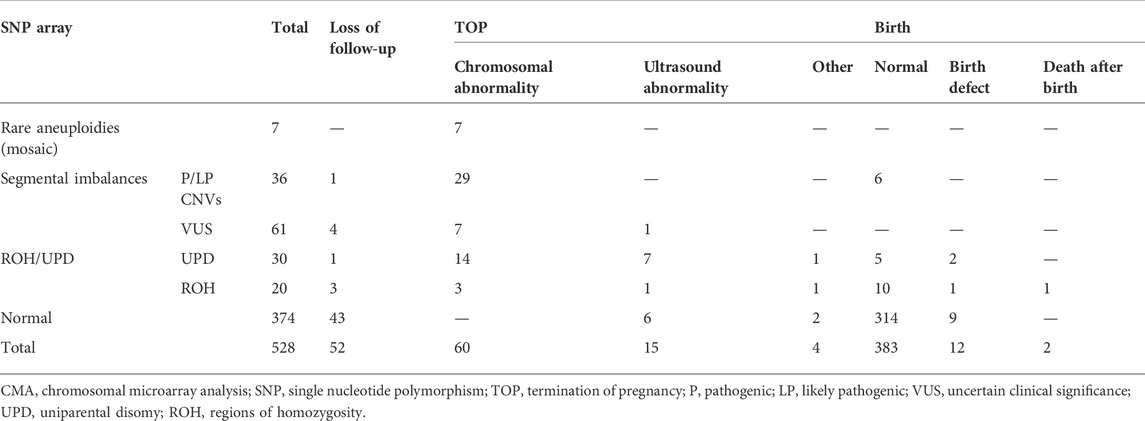
TABLE 3. Clinical follow-up assessment of the 528 fetuses detected by CMA.
For the 97 cases with segmental imbalances, except for five (5.2 %) cases that were lost during follow-up, the rate of elective TOP in fetuses with clinically significant CNVs (82.9 %, 29/35) was significantly higher than that in those with variants of uncertain clinical significance (VUS) (14.0%, 8/57) (p < 0.001). The proportion of TOP for fetuses with VUS of de novo or refused parental confirmation (28.6 %, 8/30) was significantly higher than that for those with inherited VUS (0.0%, 0/33) (p = 0.002) (Supplementary Table S1). For the 50 cases with ROHs, except for four (8.0 %) cases that were lost to follow-up, the rate of elective TOP in fetuses with UPD (75.9 %, 22/29) was significantly higher than that in those with ROHs (29.4%, 5/17) (p < 0.001). There was no significant difference in the rate of elective TOP between fetuses with UPD-related imprinting disorders (100.0 %, 5/5) and those with UPD-unrelated imprinting disorders (70.8%, 17/24) (p = 0.222) (Supplementary Table S2). The most common reason for elective TOP in fetuses with UPD-unrelated imprinting disorders was fetal growth restriction (FGR) (29.4%, 5/17).
Discussion
Currently, NIPT has been widely used for the detection of common fetal aneuploidies and SCAs; however, expanding the clinical applications of rare RCAs remains controversial (Rose et al., 2016; Chitty et al., 2018). According to the current guidelines (Gregg et al., 2016; American College of Obstetricians and Gynecologists’ Committee on Practice Bulletins—Obstetrics; Committee on Genetics; Society for Maternal-Fetal Medicine, 2020), NIPT is not recommended for screening RATs and genome-wide CNVs because the screening accuracy for detection and false-positive rates has not been established. The PPVs for these disorders are much lower than those for common trisomies, which may lead to unnecessary invasive procedures.
Our study showed that the PPV for RATs was low (6.1%), which is consistent with the results of previous studies (Chen et al., 2019; Zhu et al., 2021). A possible explanation for the high false-positive rate is that these RATs are less prevalent, while many of them have high rates of CPM, whereby a chromosomal abnormality occurs only in the placenta but not in the fetus, with an incidence of approximately 1%–2% in typical CVS (Grati et al., 2014; Mardy and Wapner, 2016; Grati et al., 2017; Chen et al., 2019). Notably, all the aneuploidies were confirmed to be low-level mosaicisms (13%–29%) by the SNP array of the amniotic fluid, arising from mitotic rescue of a meiotic error or a very early mitotic error (Taylor et al., 2014), which is consistent with previous studies (Liang et al., 2019; Zhu et al., 2021). The explanation for this is that almost all patients with RATs experienced pregnancy loss before amniocentesis and were excluded from our study. In addition, all parents decided to terminate the pregnancies even though no significant ultrasound abnormality was detected; this may have induced bias in comprehensively evaluating the clinical value of NIPT for RATs, especially for low-level mosaicisms without postnatal clinical features.
The PPV for segmental imbalances was moderate (21.1 %), which is consistent with the studies of Zhu et al. (2021) (28.9%) and Chen et al. (2019) (29.0%) but much lower than those reported by Liang et al. (2019) (40.8 %). The depth of sequencing may be attributable to this difference as Liang et al. (2019) performed NIPT-Plus with 20 Mb reads per sample, which was approximately four times our data. Additionally, NIPT-Plus uses combinatorial data analysis algorithms to identify genome-wide CNVs associated with MMSs (Liang et al., 2019). Comparing the results of NIPT with the SNP array, there was no significant difference among the concordance rates for subgroups of CNVs <5 Mb (75.0%), ranging from 5 to 10 Mb (86.4%), and >10 Mb (88.0%). The results oppose the empirical hypothesis that NIPT yields a higher positive rate for larger segmental imbalances than for smaller ones. This could be attributed to the optimization and validation of the regions of well-known MMSs. It is exemplified that all the four detected CNVs involving the 22q11.2 recurrent (DGS/VCFS) region were less than 3.5 Mb, which was consistent with previous studies (Ravi et al., 2018; Liang et al., 2019; Zhu et al., 2021).
For cases with clinically significant CNVs, the PPV was only 7.3% (30/413). Except for nine cases with parental chromosome rearrangements, the PPVs for clinically significant CNVs were extremely low (5.1%, 21/413). VUS accounted for 62.9 % (61/97) of the imbalanced segments; thus, the detection of VUS following positive NIPT is associated with increased family economic burden, maternal anxiety, or even panic, and the potential risk of terminating pregnancy. For the fully concordant segmental imbalances between NIPT and the SNP array, parental confirmations showed that the rate of maternal-inherited CNVs (85.3 %, 29/34) was significantly higher than that those with paternal inheritance or in a de novo manner (47.4 %, 9/19). Thus, it is reasonable to suspect that for gravidas with positive NIPT but negative CMA results in segmental imbalances, maternal CNVs may be detected. This could also reduce the PPVs of NIPT, as reported by Kaseniit et al. (2018) in a large-scale study. It is necessary to determine whether NIPT is an effective way to screen for clinically significant CNVs.
ROHs, termed copy number neutral segments showing continuous homozygosity with no intervening heterozygosity (Broman and Weber, 1999), were incidental findings (9.5 %) whereas the NIPT results involved aneuploidies or segmental imbalances related to the chromosomes. UPD is defined as both homologous chromosomes inherited from one parent with no contribution (for that chromosome) from the other parent (Engel, 1980). The common mechanisms resulting in UPD involve trisomy rescue, monosomy rescue, and somatic mitotic crossover (Del Gaudio et al., 2020). CPM is a well-known biologic phenomenon that is likely to result from mitotic or meiotic non-disjunction errors and trisomy rescue (Grati et al., 2014). One hypothesis is that mosaicism may be a marker for UPD (Mardy and Wapner, 2016). Although the nature process is that abnormal cell lines are encountered more frequently in placental tissues than in the fetus itself (Simoni and Sirchia, 1994), we speculated that CPM could induce abnormal NIPT results which may also be associated with UPD. The SNP array can detect isodisomy directly; however, up to one-third of UPD (heterodisomy) cases may be undetectable (Hoppman et al., 2018). Heterodisomy (exactly combined iso- and heterodisomy (mixtures of both subtypes)) may be detected by 1 or more ROHs on a single chromosome that does not include the pericentromeric region (Gonzales et al., 2022). Notably, 12 cases with combined iso- and heterodisomy were detected in our study. Thus, we recommend the prenatal SNP array for gravidas with positive NIPT results of RCAs, especially for those involved in imprinted chromosomes.
Consistent with previous studies (Wen et al., 2019; Liu et al., 2021), ROHs most frequently involved chromosomes 16 and 2, followed by chromosomes 1 and 6. After further parental confirmation, 60.0% of ROH cases were diagnosed as UPD, with a maternal to paternal ratio of 14:4, which is consistent with previous studies due to the higher propensity for maternal non-disjunction (Liehr, 2010; Yamazawa et al., 2010). In addition, five fetuses with imprinting disorders were detected. As the results of the prenatal diagnosis were obtained before detailed second-trimester fetal anomaly scans, these families opted for TOP prior to the typical ultrasound presentation of these disorders. When imprinting disorders were excluded, UPD had almost no clinical consequences (Gonzales et al., 2022). However, ROH/UPD fetuses with ultrasound abnormalities showed worse prognoses than those without abnormalities (Del Gaudio et al., 2020). In our study, 12.0% (6/50) of the cases showed FGR, one of the common ultrasound abnormalities in fetuses with ROH/UPD, which indicated adverse perinatal outcomes, and those families opted for TOP. Notably, the fetus (No. 123) with mUPD15 and fetus (No. 126) with mUPD16 were subsequently confirmed to have placental trisomy 15 and trisomy 16, respectively, which further verified the mechanism of CPM.
Our study had several limitations. First, we detected several maternal-inherited VUSs, reflecting the limitations to the analytical performance. Better algorithms to differentiate between fetal and maternal CNVs and improve clinically significant CNV calling should be performed in the future. Second, we expanded the clinical utility of NIPT for RCAs, which is recommended worldwide for traditionally screened aneuploidies. The low depth of sequencing influences the PPVs for RCAs compared with NIPT-Plus. Third, our study only included gravidas with positive NIPT results of RCAs who subsequently underwent amniocentesis for SNP analysis. We did not follow up the gravidas with negative NIPT results or refused invasive procedures. Thus, we failed to obtain a negative predictive value for comprehensively assessing the clinical use of NIPT for RCAs. Fourth, for cases with negative CMA results, we did not obtain maternal or placental results to assess the potential proportion to induce unnecessary invasive procedures. Fifth, for those fetuses with UPD/ROHs, although parental consanguinity was excluded, autosomal recessive disorders associated with ROHs were not detected regularly.
In summary, this retrospective study demonstrated that NIPT for common fetal aneuploidies yielded a low PPV for RATs (6.1 %), moderate PPV for segmental imbalances (21.1 %), and incidental findings (9.5 %) for ROH/UPD. This study provides valuable information for genetic counseling and management of gravidas with positive NIPT results for RCAs. Due to the low PPV (8.0 %) for clinically significant findings, NIPT for common fetal aneuploidies need to be noticed for rare RCAs. The prenatal SNP array should be regarded as the first-tier test for positive NIPT, particularly when imprinted chromosomes are involved.
Data availability statement
All datasets generated for this study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving human participants were reviewed and approved by the Medical Ethics Committee of West China Second University Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
The study was conceived and designed by TH, HW, XN, and SL. Patient recruitment and sample collection were undertaken by TH and JW. Experiments and data collection were performed by TH, JW, RH, LX, NL, and SL. Data analyses and interpretation were performed by TH, JW, QZ, and ZZ. All figures and tables were generated by TH and JW. The manuscript was written by TH and JW. All authors critically reviewed the manuscript and approved the final manuscript for publication.
Funding
The study was supported by the National Key Research and Development Program of China (2021YFC1005300); and the Sichuan Province Science and Technology Support Program, China (2021YFS0078 and 2022YFS0078).
Acknowledgments
The authors would like to thank all the doctors who helped with patient recruitment. We are grateful to all the team members for their contributions to data collection and integrity.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.955694/full#supplementary-material
References
American College of Obstetricians and Gynecologists Committee on Genetics (2013). Committee opinion no. 581: The use of chromosomal microarray analysis in prenatal diagnosis. Obstet. Gynecol. 122 (6), 1374–1377. doi:10.1097/01.AOG.0000438962.16108.d1
American College of Obstetricians and Gynecologists’ Committee on Practice Bulletins—Obstetrics; Committee on Genetics; Society for Maternal-Fetal Medicine (2020). Screening for fetal chromosomal abnormalities: ACOG practice bulletin, number 226. Obstet. Gynecol. 136 (4), e48–e69. doi:10.1097/AOG.0000000000003070
Committee on Genetics Society for Maternal-Fetal Medicine (2015). Committee opinion No. 640: Cell-Free DNA screening for fetal aneuploidy. Obstet. Gynecol. 126(3), e31–e37. doi:10.1097/AOG.0000000000001051
Benn, P., and Grati, F. R. (2018). Genome-wide non-invasive prenatal screening for all cytogenetically visible imbalances. Ultrasound Obstet. Gynecol. 51 (4), 429–433. doi:10.1002/uog.19014
Bianchi, D. W., and Wilkins-Haug, L. (2014). Integration of noninvasive DNA testing for aneuploidy into prenatal care: What has happened since the rubber met the road? Clin. Chem. 60 (1), 78–87. doi:10.1373/clinchem.2013.202663
Broman, K. W., and Weber, J. L. (1999). Long homozygous chromosomal segments in reference families from the centre d'Etude du polymorphisme humain. Am. J. Hum. Genet. 65 (6), 1493–1500. doi:10.1086/302661
Campbell, H., Carothers, A. D., Rudan, I., Hayward, C., Biloglav, Z., Barac, L., et al. (2007). Effects of genome-wide heterozygosity on a range of biomedically relevant human quantitative traits. Hum. Mol. Genet. 16 (2), 233–241. doi:10.1093/hmg/ddl473
Chen, Y., Yu, Q., Mao, X., Lei, W., He, M., and Lu, W. (2019). Noninvasive prenatal testing for chromosome aneuploidies and subchromosomal microdeletions/microduplications in a cohort of 42, 910 single pregnancies with different clinical Features. Hum. Genomics 13 (1), 60. doi:10.1186/s40246-019-0250-2
Cherry, A. M., Akkari, Y. M., Barr, K. M., Kearney, H. M., Rose, N. C., South, S. T., et al. (2017). Diagnostic cytogenetic testing following positive noninvasive prenatal screening results: A clinical laboratory practice resource of the American College of medical genetics and genomics (ACMG). Genet. Med. 19 (8), 845–850. doi:10.1038/gim.2017.91
Chitty, L. S., Hudgins, L., and Norton, M. E. (2018). Current controversies in prenatal diagnosis 2: Cell-free DNA prenatal screening should be used to identify all chromosome abnormalities. Prenat. Diagn. 38 (3), 160–165. doi:10.1002/pd.5216
Cross, J., Peters, G., Wu, Z., Brohede, J., and Hannan, G. N. (2007). Resolution of trisomic mosaicism in prenatal diagnosis: Estimated performance of a 50K SNP microarray. Prenat. Diagn. 27 (13), 1197–1204. doi:10.1002/pd.1884
Dar, P., Curnow, K. J., Gross, S. J., Hall, M. P., Stosic, M., Demko, Z., et al. (2014). Clinical experience and follow-up with large scale single-nucleotide polymorphism-based noninvasive prenatal aneuploidy testing. Am. J. Obstet. Gynecol. 211 (5), 527.e1–527. doi:10.1016/j.ajog.2014.08.006
Del Gaudio, D., Shinawi, M., Astbury, C., Tayeh, M. K., Deak, K. L., Raca, G., et al. (2020). Diagnostic testing for uniparental disomy: A points to consider statement from the American College of medical genetics and genomics (ACMG). Genet. Med. 22 (7), 1133–1141. doi:10.1038/s41436-020-0782-9
Engel, E. (1980). A new genetic concept: Uniparental disomy and its potential effect, isodisomy. Am. J. Med. Genet. 6 (2), 137–143. doi:10.1002/ajmg.1320060207
Faas, B. H., de Ligt, J., Janssen, I., Eggink, A. J., Wijnberger, L. D., van Vugt, J. M., et al. (2012). Non-invasive prenatal diagnosis of fetal aneuploidies using massively parallel sequencing-by-ligation and evidence that cell-free fetal DNA in the maternal plasma originates from cytotrophoblastic cells. Expert Opin. Biol. Ther. 12, S19–S26. doi:10.1517/14712598.2012.670632
Gonzales, P. R., Andersen, E. F., Brown, T. R., Horner, V. L., Horwitz, J., Rehder, C. W., et al. (2022). Interpretation and reporting of large regions of homozygosity and suspected consanguinity/uniparental disomy, 2021 revision: A technical standard of the American College of medical genetics and genomics (ACMG). Genet. Med. 24 (2), 255–261. doi:10.1016/j.gim.2021.10.004
Grati, F. R., Malvestiti, F., Branca, L., Agrati, C., Maggi, F., and Simoni, G. (2017). Chromosomal mosaicism in the fetoplacental unit. Best. Pract. Res. Clin. Obstet. Gynaecol. 42, 39–52. doi:10.1016/j.bpobgyn.2017.02.004
Grati, F. R., Malvestiti, F., Ferreira, J. C., Bajaj, K., Gaetani, E., Agrati, C., et al. (2014). Fetoplacental mosaicism: Potential implications for false-positive and false-negative noninvasive prenatal screening results. Genet. Med. 16 (8), 620–624. doi:10.1038/gim.2014.3
Gregg, A. R., Skotko, B. G., Benkendorf, J. L., Monaghan, K. G., Bajaj, K., Best, R. G., et al. (2016). Noninvasive prenatal screening for fetal aneuploidy, 2016 update: A position statement of the American College of medical genetics and genomics. Genet. Med. 18 (10), 1056–1065. doi:10.1038/gim.2016.97
Gross, S. J., Stosic, M., McDonald-McGinn, D. M., Bassett, A. S., Norvez, A., Dhamankar, R., et al. (2016). Clinical experience with single-nucleotide polymorphism-based non-invasive prenatal screening for 22q11.2 deletion syndrome. Ultrasound Obstet. Gynecol. 47 (2), 177–183. doi:10.1002/uog.15754
Hall, G. K., Mackie, F. L., Hamilton, S., Evans, A., McMullan, D. J., Williams, D., et al. (2014). Chromosomal microarray analysis allows prenatal detection of low level mosaic autosomal aneuploidy. Prenat. Diagn. 34 (5), 505–507. doi:10.1002/pd.4333
Hoppman, N., Rumilla, K., Lauer, E., Kearney, H., and Thorland, E. (2018). Patterns of homozygosity in patients with uniparental disomy: Detection rate and suggested reporting thresholds for SNP microarrays. Genet. Med. 20 (12), 1522–1527. doi:10.1038/gim.2018.24
Hu, T., Tian, T., Zhang, Z., Wang, J., Hu, R., Xiao, L., et al. (2021). Prenatal chromosomal microarray analysis in 2466 fetuses with ultrasonographic soft markers: a prospective cohort study. Am. J. Obstet. Gynecol. 224 (5), 516.e1–516.e16. e16. doi:10.1016/j.ajog.2020.10.039
Kaseniit, K. E., Hogan, G. J., D'Auria, K. M., Haverty, C., and Muzzey, D. (2018). Strategies to minimize false positives and interpret novel microdeletions based on maternal copy-number variants in 87, 000 noninvasive prenatal screens. BMC Med. Genomics 11 (1), 90. doi:10.1186/s12920-018-0410-6
Ku, C. S., Naidoo, N., Teo, S. M., and Pawitan, Y. (2011). Regions of homozygosity and their impact on complex diseases and traits. Hum. Genet. 129 (1), 1–15. doi:10.1007/s00439-010-0920-6
Levy, B., Sigurjonsson, S., Pettersen, B., Maisenbacher, M. K., Hall, M. P., Demko, Z., et al. (2014). Genomic imbalance in products of conception: Single-nucleotide polymorphism chromosomal microarray analysis. Obstet. Gynecol. 124, 202–209. doi:10.1097/AOG.0000000000000325
Liang, D., Cram, D. S., Tan, H., Linpeng, S., Liu, Y., Sun, H., et al. (2019). Clinical utility of noninvasive prenatal screening for expanded chromosome disease syndromes. Genet. Med. 21 (9), 1998–2006. doi:10.1038/s41436-019-0467-4
Liehr, T. (2010). Cytogenetic contribution to uniparental disomy (UPD). Mol. Cytogenet. 3, 8. doi:10.1186/1755-8166-3-8
Liu, J., He, Z., Lin, S., Wang, Y., Huang, L., Huang, X., et al. (2021). Absence of heterozygosity detected by single-nucleotide polymorphism array in prenatal diagnosis. Ultrasound Obstet. Gynecol. 57 (2), 314–323. doi:10.1002/uog.21951
Manning, M., Hudgins, L., Practice, P., and Committee, G. (2010). Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities. Genet. Med. 12 (11), 742–745. doi:10.1097/GIM.0b013e3181f8baad
Mardy, A., and Wapner, R. J. (2016). Confined placental mosaicism and its impact on confirmation of NIPT results. Am. J. Med. Genet. C Semin. Med. Genet. 172 (2), 118–122. doi:10.1002/ajmg.c.31505
J. McGowan-Jordan, R. J. Hastings, and S. Moore (Editors) (2020). An international system for human cytogenomic nomenclature (Basel, Switzerland: Karger).
Miller, D. T., Adam, M. P., Aradhya, S., Biesecker, L. G., Brothman, A. R., Carter, N. P., et al. (2010). Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 86 (5), 749–764. doi:10.1016/j.ajhg.2010.04.006
Palomaki, G. E., Kloza, E. M., Lambert-Messerlian, G. M., Haddow, J. E., Neveux, L. M., Ehrich, M., et al. (2011). DNA sequencing of maternal plasma to detect Down syndrome: An international clinical validation study. Genet. Med. 13 (11), 913–920. doi:10.1097/GIM.0b013e3182368a0e
Petersen, A. K., Cheung, S. W., Smith, J. L., Bi, W., Ward, P. A., Peacock, S., et al. (2017). Positive predictive value estimates for cell-free noninvasive prenatal screening from data of a large referral genetic diagnostic laboratory. Am. J. Obstet. Gynecol. 217 (6), 691.e1–691. doi:10.1016/j.ajog.2017.10.005
Ravi, H., McNeill, G., Goel, S., Meltzer, S. D., Hunkapiller, N., Ryan, A., et al. (2018). Validation of a SNP-based non-invasive prenatal test to detect the fetal 22q11.2 deletion in maternal plasma samples. PLoS One 13 (2), e0193476. doi:10.1371/journal.pone.0193476
Riggs, E. R., Andersen, E. F., Cherry, A. M., Kantarci, S., Kearney, H., Patel, A., et al. (2020). Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of medical genetics and genomics (ACMG) and the clinical genome resource (ClinGen). Genet. Med. 22 (2), 245–257. doi:10.1038/s41436-019-0686-8
Robinson, W. P. (2000). Mechanisms leading to uniparental disomy and their clinical consequences. Bioessays 22 (5), 452–459. doi:10.1002/(SICI)1521-1878(200005)22:5<452:AID-BIES7>3.0.CO;2-K
Rose, N. C., Benn, P., and Milunsky, A. (2016). Current controversies in prenatal diagnosis 1: Should NIPT routinely include microdeletions/microduplications? Prenat. Diagn. 36 (1), 10–14. doi:10.1002/pd.4710
Scott, S. A., Cohen, N., Brandt, T., Toruner, G., Desnick, R. J., and Edelmann, L. (2010). Detection of low-level mosaicism and placental mosaicism by oligonucleotide array comparative genomic hybridization. Genet. Med. 12 (2), 85–92. doi:10.1097/GIM.0b013e3181cc75d0
Simoni, G., and Sirchia, S. M. (1994). Confined placental mosaicism. Prenat. Diagn. 14 (13), 1185–1189. doi:10.1002/pd.1970141304
Taylor, T. H., Gitlin, S. A., Patrick, J. L., Crain, J. L., Wilson, J. M., and Griffin, D. K. (2014). The origin, mechanisms, incidence and clinical consequences of chromosomal mosaicism in humans. Hum. Reprod. Update 20 (4), 571–581. doi:10.1093/humupd/dmu016
Taylor-Phillips, S., Freeman, K., Geppert, J., Agbebiyi, A., Uthman, O. A., Madan, J., et al. (2016). Accuracy of non-invasive prenatal testing using cell-free DNA for detection of Down, edwards and patau syndromes: A systematic review and meta-analysis. BMJ Open 6 (1), e010002. doi:10.1136/bmjopen-2015-010002
Tjoa, M. L., Cindrova-Davies, T., Spasic-Boskovic, O., Bianchi, D. W., and Burton, G. J. (2006). Trophoblastic oxidative stress and the release of cell-free feto-placental DNA. Am. J. Pathol. 169 (2), 400–404. doi:10.2353/ajpath.2006.060161
Wapner, R. J., Babiarz, J. E., Levy, B., Stosic, M., Zimmermann, B., Sigurjonsson, S., et al. (2015). Expanding the scope of noninvasive prenatal testing: Detection of fetal microdeletion syndromes. Am. J. Obstet. Gynecol. 212 (3), 332.e1–e9. doi:10.1016/j.ajog.2014.11.041
Wapner, R. J., Martin, C. L., Levy, B., Ballif, B. C., Eng, C. M., Zachary, J. M., et al. (2012). Chromosomal microarray versus karyotyping for prenatal diagnosis. N. Engl. J. Med. 367 (23), 2175–2184. doi:10.1056/NEJMoa1203382
Wen, J., Comerford, K., Xu, Z., Wu, W., Amato, K., Grommisch, B., et al. (2019). Analytical validation and chromosomal distribution of regions of homozygosity by oligonucleotide array comparative genomic hybridization from normal prenatal and postnatal case series. Mol. Cytogenet. 12, 12. doi:10.1186/s13039-019-0424-6
Wong, F. C., and Lo, Y. M. (2016). Prenatal diagnosis innovation: Genome sequencing of maternal plasma. Annu. Rev. Med. 67, 419–432. doi:10.1146/annurev-med-091014-115715
Yamazawa, K., Ogata, T., and Ferguson-Smith, A. C. (2010). Uniparental disomy and human disease: An overview. Am. J. Med. Genet. C Semin. Med. Genet. 154C (3), 329–334. doi:10.1002/ajmg.c.30270
Keywords: NIPS for rare chromosome abnormalities noninvasive prenatal screening, rare chromosomal abnormality, positive predictive value, chromosomal microarray analysis, prenatal diagnosis
Citation: Hu T, Wang J, Zhu Q, Zhang Z, Hu R, Xiao L, Yang Y, Liao N, Liu S, Wang H, Niu X and Liu S (2022) Clinical experience of noninvasive prenatal testing for rare chromosome abnormalities in singleton pregnancies. Front. Genet. 13:955694. doi: 10.3389/fgene.2022.955694
Received: 29 May 2022; Accepted: 26 August 2022;
Published: 26 September 2022.
Edited by:
Fan Jin, Zhejiang University, ChinaReviewed by:
Thomas Liehr, Friedrich Schiller University Jena, GermanyHui Xi, Hunan Provincial Maternal and Child Health Care Hospital, China
Copyright © 2022 Hu, Wang, Zhu, Zhang, Hu, Xiao, Yang, Liao, Liu, Wang, Niu and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ting Hu, aHV0aW5nNDEyM0AxNjMuY29t; Xiaoyu Niu, bml1eHlAc2N1LmVkdS5jbg==; Shanling Liu, c3Vubnk2MzBAMTI2LmNvbQ==
†These authors have contributed equally to this work