 Yi-Han Chang
Yi-Han Chang Pei Lin4
Pei Lin4 Jia-Ling Lin
Jia-Ling Lin Chao-Kai Hsu
Chao-Kai Hsu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet. , 23 September 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.954931
Around one-third of patients diagnosed with idiopathic dilated cardiomyopathy (DCM) turn out to be familial cases, in only a few of which the identification of a pathogenic/likely pathogenic variant could be achieved. Cardiomyopathy caused by desmoplakin gene mutations represents a distinct form with a high prevalence of left ventricle involvement. We report a novel desmoplakin mutation carried by two individuals in a Taiwanese family, in which the proband recovered well after heart transplantation and under medical control, while her son had received an implantable cardioverter defibrillator and has been under guideline-directed medical therapy. The present study broadens the genetic spectrum of this disease entity and strengthens the notion that a detailed family history with genetic study contributes to the early detection and treatment of inherited diseases.
Dilated cardiomyopathy (DCM) is one of the most common causes of heart failure and is a leading diagnosis among heart transplant recipients (Cuenca et al., 2016). DCM is characterized by left ventricular or biventricular dilation and impaired systolic function that is not explained by abnormal loading conditions (e.g., hypertension or valvular heart disease) nor by coronary artery disease (Schultheiss et al., 2019). The underlying etiologies of DCM include genetic mutations, infection, autoimmune disease, toxin exposure, metabolic or endocrine dysfunction, neuromuscular disease, and pregnancy/peripartum cardiomyopathy. In some cases, however, there is no recognizable cause even after thorough assessments. These cases are given the clinical diagnosis of “idiopathic” DCM. Previous studies have demonstrated that familial DCM accounts for 20%–35% of patients who are initially classified as idiopathic (Petretta et al., 2011; Hershberger et al., 2013). Therefore, the guidelines of the Heart Failure Society of America/American College of Medical Genetics and Genomics recommend genetic testing for all patients with idiopathic DCM, regardless of family history (Hershberger et al., 2018).
Desmoplakin (DSP) is a cytoskeletal linker protein member of the plakin family. Its main function is to connect intermediate filament proteins with other desmosomal components, playing an important role in cell–cell adhesion. Structurally intact desmosomes provide mechanical strength and resilience, which are crucial to combat stress in the epidermis and the heart (Garrod & Chidgey, 2008). According to previous studies, DSP (6p24.3) mutations are mostly associated with arrhythmogenic right ventricular cardiomyopathy (ARVC; OMIM #607450), which primarily affects the structure and function of right ventricle (RV), with clinical manifestations of right and/or left ventricular ectopy and sudden cardiac death, and with pathological features of fibro-fatty replacement (Rampazzo et al., 2002; Azaouagh et al., 2011). Recent studies suggest that DSP cardiomyopathy represents an entity distinct from typical forms of ARVC or DCM. Instead, DSP cardiomyopathy is characterized by early and predominant left ventricle (LV) involvement, frequent ventricular arrhythmia, and a specific fibrosis pattern seen in magnetic resonance imaging (Castelletti et al., 2017; Helio et al., 2020; Smith et al., 2020).
In the present study, we report a female patient with familial DCM who underwent heart transplantation. Genetic study identified a novel DSP mutation, c.6384delG, p.(Glu2128SerfsTer18), in the proband and her son. Consistent with the current concept of DSP cardiomyopathy, these two cases exhibited impairments in LV structure and function.
Germline DNA extracted from the proband and her family members (the proband’s two sons and her younger sister) was used for paired-end library preparation using the SureSelect All Exon 50 Mb Version 4.0 kit (Agilent, Santa Clara, CA, United States) according to the manufacturer’s recommendations. Sequencing as carried out by massively parallel sequencing with 100-bp paired-end reads using the HiSeq-2000 platform (Illumina, CA, United States). The Novoalign software package (Novocraft Technologies Sdn Bhd) was used to align reads generated to the reference human genome. Reads mapping to multiple locations on the reference human genome were excluded from downstream analysis. The BedTools package was used to calculate the depth and breadth of sequence coverage (Quinlan & Hall, 2010). Single-nucleotide substitutions and small indels were detected with the SamTools package (Li et al., 2009). Sequence variants were annotated with the Annovar tool (Wang et al., 2010). To assess the pathogenicity of the candidate variants, an in-house variant-filtering pipeline was used. Nonsense variants or indels resulting in frameshift mutations with minor allele frequencies (MAF) of less than 0.5% in the 1,000 Genomes Project (Siva, 2008) and Exome Aggregation Consortium (ExAC) were included. The damage prediction criteria for filtering the candidate variants included a Combined Annotation Dependent Depletion (CADD) score of above 15, a Deleterious Annotation of Genetic variants using Neural Networks (DANN) score of above 0.95, and a Polymorphism Phenotyping v2 (PolyPhen-2) score of above 0.95. Variants with MAF exceeding 0.5% or with damage prediction scores not fulfilling our criteria were excluded as non-pathogenic. BAM files of WES were visualized via Integrative Genomics Viewer (IGV) (Robinson et al., 2011).
Confirmative polymerase chain reaction (PCR) and Sanger sequencing tests were performed on the DNA from the proband and her family members (the proband’s two sons and her younger sister) to validate the filtered variants detected by WES and for segregation analysis. Primers were designed using the Ensembl database (Howe et al., 2021) and Primer3 (Untergasser et al., 2012) online software.
A 49-year-old female presented to our hospital with dyspnea on exertion and chest tightness for 2 months (Figure 1A). Physical examination found a 2/6 pansystolic murmur over the left lower sternal border and apex. Electrocardiography showed sinus rhythm with left anterior hemiblock and left ventricular hypertrophy. Chest X-ray showed cardiomegaly. In echocardiography, her left ventricular end-diastolic diameter (LVEDD) was 78 mm and the ejection fraction by Simpson’s biplane method was 32% with moderate mitral regurgitation (Figure 1B). Coronary angiography showed normal coronary arteries. Radionuclide ventriculography showed impaired systolic and diastolic function of the LV with global ejection fraction 25% but normal systolic function of the RV. She was diagnosed with DCM. Due to frequent ventricular extrasystoles and non-sustained ventricular tachycardia, she received an implantable cardioverter defibrillator. She was treated with guideline-directed medical therapy for heart failure, including a beta blocker, an angiotensin-converting enzyme inhibitor (ACEi), a mineralocorticoid receptor antagonist, and ivabradine. Later, the ACEi was replaced with an angiotensin receptor–neprilysin inhibitor. Her heart function and daily performance did not improve, but deteriorated rapidly in the following 2 years. The highest N-terminal pro-brain natriuretic peptide (NT-pro-BNP) concentration was 7,000 ng/L. At the age of 59 years, she received a heart transplant. Currently, she is in healthy condition under immunosuppressants with mycophenolate, tacrolimus, and prednisolone.

FIGURE 1. The pedigree and cardiac examinations of the proband (II-2) and her son (III-1). (A) The pedigree of the proband (arrow) and her family. (B) Echocardiography of the proband (II-2) shows dilated LV and LA with LVEDD 78 mm and poor LV function, with EF 32% and ICD lead in situ. (C) Echocardiography of the proband’s son (III-1) shows dilated LV and LA with LVEDD 78 mm and poor LV function with EF 22.8%, with the ICD lead in situ. (D) Genetic study of the proband’s family. Sanger sequencing reveals a deletion mutation in the proband (II-2) and her son (III-1), suggesting a dominant inheritance pattern for this variant. EF: ejection fraction; ICD: implantable cardioverter defibrillator; LA: left atrium; LV: left ventricle; LVEDD: left ventricular end-diastolic diameter.
Notably, the proband’s son was diagnosed with DCM at the age of 24 years with ejection fraction 22.8% and LVEDD 78.2 mm in the initial echocardiography (Figure 1C). Radionuclide ventriculography also showed impaired systolic and diastolic function of the LV with global ejection fraction 26% but normal systolic function of the RV. He also presented with frequent ventricular extrasystoles and non-sustained ventricular tachycardia and received an implantable cardioverter defibrillator. He has been under guideline-directed medical therapy for 10 years. He was in New York Heart Association classification II, and his ejection fraction did not improve much. The highest level of NT-pro-BNP was 600 ng/L.
Familial DCM was highly suspected, considering the family history. Therefore, molecular genetic studies were arranged and a novel mutation in the DSP gene was identified. With informed consent, we collected peripheral blood from the two patients and other family members (the proband’s second son and her younger sister), and genomic DNA was extracted for further mutation analysis. Whole-exome sequencing initially identified three potential variant candidates (laminin alpha-4, aminoacyl-tRNA synthetase 2, and DSP), and subsequent segregation analysis confirmed the DSP variant to be the culprit, with an autosomal dominant inheritance pattern. This unreported heterozygous deletion, c.6384delG (p.Glu2128SerfsTer18), in DSP (NM_001008844) leads to premature termination codon (PTC) formation (Figure 1D). The diagnosis of desmoplakin cardiomyopathy in these two patients was confirmed based on the clinical manifestations of LV systolic dysfunction, the electrocardiographic findings of frequent ventricular extrasystoles and non-sustained ventricular tachycardia, and the positive results of genetic analysis, with a novel heterozygous deletion mutation in DSP in this family.
Based on the Human Gene Mutation Database, more than 300 DSP mutations had been documented as of October 2020. These include nonsense/missense, insertion, deletion, and regulatory mutations, with diverse phenotypes (Figure 2). The unreported mutation identified in our study is a deletion mutation that is situated within the plakin repeat domain (PRD 1/A) of the tail domain. Consequential PTC formation leads to truncated desmoplakin protein or induces nonsense-mediated decay. Since that the C-terminus of desmoplakin is responsible for interactions with intermediate filaments, it is plausible that this mutation changes the structure of the carboxyl domain and further hinders anchorage between desmoplakin and intermediate filaments.
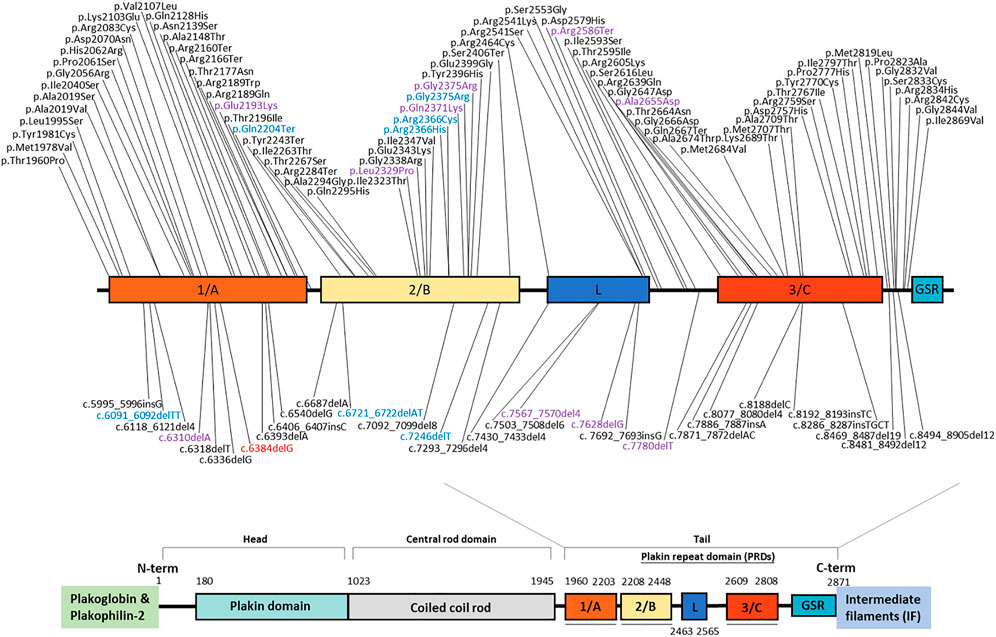
FIGURE 2. Schematic of desmoplakin protein domains labeled with reported mutations within the tail domain. The mutation identified in our study is marked in red. Mutations with different colors account for different phenotypes (black: heart only; blue: skin only; purple: heart and skin). Variants above the schematic are missense or nonsense mutations; those below the schematic are indel mutations.
Mutations in human DSP can present as abnormalities in the skin, hair, and heart, emphasizing the importance of desmoplakin against continuous mechanical stress (Garrod & Chidgey, 2008). Of note, no skin or hair abnormalities were noted in our two cases, and no such defects were mentioned in other family members. This newly identified mutation seems to manifest a heart-only phenotype, based on the clinical history. To confirm the phenotypic landscape, a longer follow-up period for this family or observation from any other cases with this mutation is needed. The mutation itself did not add more information on the clinical phenotype, because previous studies (Mahoney et al., 2010; Pigors et al., 2015) demonstrated that individual DSP mutations are scattered throughout the coding sequence and that there is no clear association between mutation location, type, and clinical presentation (Figure 2).
In the present study, familial disease was suspected because the proband’s son was diagnosed with DCM at age 24. We took a thorough clinical history of the family and performed genetic analyses. A novel pathogenic DSP mutation with autosomal dominant inheritance was identified, with the clinical presentation of LV dysfunction and ventricular ectopy that are typical of desmoplakin cardiomyopathy (Smith et al., 2020). The findings are consistent with the current concept that DSP mutations seem to cause a unique form of cardiomyopathy with a high prevalence of LV involvement (Norman et al., 2005; Bhonsale et al., 2015; Pigors et al., 2015; Helio et al., 2020), especially if the patient is a carrier of a DSP nonsense mutation (Sen-Chowdhry et al., 2007; Lopez-Ayala et al., 2014; Castelletti et al., 2017).
DCM is the most frequent cause of heart transplantation, with approximately 20%–25% of transplant recipients having direct evidence of familial disease (Cuenca et al., 2016). A retrospective study using a large, contemporary, nationwide database that aimed to investigate the outcome of familial DCM patients after heart transplantation (Khayata et al., 2019) demonstrated that these patients have a higher risk of early rejection but are more likely to survive than are patients with ischemic cardiomyopathy (ICM). These findings could be explained by less hepatic or renal dysfunction in familial DCM patients and more comorbidities in ICM patients (Grimm et al., 2015; Shore et al., 2015).
Certain types of DCM have unique presentations. For example, patients with LMNA mutations possess higher rates of conduction block, ventricular arrhythmia, and sudden cardiac death (Schultheiss et al., 2019). Genetic study helps physicians to define the clinical course of this complicated disease and to identify patients who need more tailored therapies. Also, patients with DCM may have a clinically indolent phase during the early stage of the disease (Kinnamon et al., 2017). With the advent of next-generation sequencing technology (Yeh, 2019), we are able to provide better genetic counseling for patients and their families. Apart from confirming both the clinical diagnosis and the inheritance pattern, genetic study is important in terms of evaluating the risk for future offspring as well as achieving the goal of early diagnosis by enabling early optimal treatment to delay the disease progression and to prevent severe complications (McMurray, 2015; Fatkin et al., 2017; Kinnamon et al., 2017; Rivero et al., 2017).
In conclusion, we reported the case of a Taiwanese woman with familial DCM and a novel DSP mutation. Our findings broaden the genetic spectrum of this disease entity and strengthen the notion that a detailed family history, including genetic study, can contribute to the early detection and treatment of inherited diseases.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
The studies involving human participants were reviewed and approved by the Institutional Review Board of National Cheng Kung University Hospital (A-BR-104-052). The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Y-HC: experiment performing, case collection, original manuscript drafting, figures arrangement, manuscript revision. PL: case and clinical data collection, original manuscript drafting. J-LL: figures arrangement, manuscript revision. H-YH: bioinformatic processing and interpretation. C-KH: project design, supervision. C-HH: project design, supervision.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Azaouagh, A., Churzidse, S., Konorza, T., and Erbel, R. (2011). Arrhythmogenic right ventricular cardiomyopathy/dysplasia: Arrhythmogenic right ventricular cardiomyopathy/dysplasia: a review and update. review and update. ClinClin. Res. Cardiol. Res. Cardiol. 100 (5), 383–39494. doi:10.1007/s00392-011-0295-2
Bhonsale, A., Groeneweg, J. A., James, C. A., Dooijes, D., Tichnell, C., and Jongbloed, J. D., (2015). Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. EurEur. Heart J. Heart J. 36 (14), 847–85555. doi:10.1093/eurheartj/ehu509
Castelletti, S., Vischer, A. S., Syrris, P., Crotti, L., Spazzolini, C., and Ghidoni, A., (2017). Desmoplakin missense and non-missense mutations in arrhythmogenic right ventricular cardiomyopathy: Genotype-phenotype correlation. IntInt. J. Cardiol. J. Cardiol. 249, 268–273. doi:10.1016/j.ijcard.2017.05.018
Cuenca, S., Ruiz-Cano, M. J., Gimeno-Blanes, J. R., Jurado, A., Salas, C., and Gomez-Diaz, I., (2016). Genetic basis of familial dilated cardiomyopathy patients undergoing heart transplantation. JJ. Heart Lung Transplant. Heart Lung Transpl. 35 (5), 625–63535. doi:10.1016/j.healun.2015.12.014
Fatkin, D., Johnson, R., McGaughran, J., Weintraub, R. G., Atherton, J. J., and Group, C. G. C. W. (2017). Position Position Statement on the Diagnosis and Management of Familial Dilated Cardiomyopathy.tatement on the diagnosis and management of familial dilated cardiomyopathy. Heart Lung CircHeart Lung Circ. 26 (11), 1127–1132. doi:10.1016/j.hlc.2017.04.021
Garrod, D., and Chidgey, M. (2008). Desmosome structure, composition and function. BiochimBiochim. Biophys. Acta Biophys. Acta 1778 (3), 572–58787. doi:10.1016/j.bbamem.2007.07.014
Grimm, J. C., Shah, A. S., Magruder, J. T., Kilic, A., Valero, V., and Dungan, S. P., (2015). MELD-XI MELD-XI Score Predicts Early Mortality in Patients After Heart Transplantation.core predicts early mortality in patients after heart transplantation. AnnAnn. Thorac. Surg. Thorac. Surg. 100 (5), 1737–174343. doi:10.1016/j.athoracsur.2015.07.026
Helio, K., Kangas-Kontio, T., Weckstrom, S., Vanninen, S. U. M., Aalto-Setala, K., and Alastalo, T. P., (2020). DSP p.(Thr2104Glnfs*12) variant presents variably with early onset severe arrhythmias and left ventricular cardiomyopathy. BMC MedBMC Med. Genet. Genet. 21 (1), 19. doi:10.1186/s12881-020-0955-z
Hershberger, R. E., Givertz, M. M., Ho, C. Y., Judge, D. P., Kantor, P. F., and McBride, K. L., (2018). Genetic evaluation of cardiomyopathy: Genetic evaluation of cardiomyopathy: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). clinical practice resource of the American College of medical genetics and genomics (ACMG). GenetGenet. Med. Med. 20 (9), 899–909. doi:10.1038/s41436-018-0039-z
Hershberger, R. E., Hedges, D. J., and Morales, A. (2013). Dilated cardiomyopathy: Dilated cardiomyopathy: the complexity of a diverse genetic architecture.he complexity of a diverse genetic architecture. NatNat. Rev. Cardiol. Rev. Cardiol. 10 (9), 531–54747. doi:10.1038/nrcardio.2013.105
Howe, K. L., Achuthan, P., Allen, J., Allen, J., Alvarez-Jarreta, J., and Amode, M. R., (2021). Ensembl 2021. Nucleic Acids ResNucleic Acids Res. 49 (D1), D884–d891. doi:10.1093/nar/gkaa942
Khayata, M., Al-Kindi, S. G., and Oliveira, G. H. (2019). Contemporary characteristics and outcomes of adults with familial dilated cardiomyopathy listed for heart transplantation. World JWorld J. Cardiol. Cardiol. 11 (1), 38–46. doi:10.4330/wjc.v11.i1.38
Kinnamon, D. D., Morales, A., Bowen, D. J., Burke, W., Hershberger, R. E., and Consortium*, D. C. M. (2017). Toward Toward Genetics-Driven Early Intervention in Dilated Cardiomyopathy: Design and Implementation of the DCM Precision Medicine Study.enetics-driven early intervention in dilated cardiomyopathy: Design and implementation of the DCM precision medicine study. CircCirc. Cardiovasc. Genet. Cardiovasc Genet. 10 (6), e001826. doi:10.1161/CIRCGENETICS.117.001826
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., and Homer, N., (2009). The The Sequence Alignment/Map format and SAMtools.equence alignment/map format and SAMtools. Bioinformatics 25 (16), 2078–20799. doi:10.1093/bioinformatics/btp352
Lopez-Ayala, J. M., Gomez-Milanes, I., Sanchez Munoz, J. J., Ruiz-Espejo, F., Ortiz, M., and Gonzalez-Carrillo, J., (2014). Desmoplakin truncations and arrhythmogenic left ventricular cardiomyopathy: Desmoplakin truncations and arrhythmogenic left ventricular cardiomyopathy: characterizing a phenotype.haracterizing a phenotype. Europace 16 (12), 1838–184646. doi:10.1093/europace/euu128
Mahoney, M. G., Sadowski, S., Brennan, D., Pikander, P., Saukko, P., and Wahl, J., (2010). Compound heterozygous desmoplakin mutations result in a phenotype with a combination of myocardial, skin, hair, and enamel abnormalities. JJ. Invest. Dermatol. Invest. Dermatol 130 (4), 968–97878. doi:10.1038/jid.2009.357
McMurray, J. J. (2015). Improving outcomes in heart failure: Improving outcomes in heart failure: a personal perspective. personal perspective. EurEur. Heart J. Heart J. 36 (48), 3467–347070. doi:10.1093/eurheartj/ehv565
Norman, M., Simpson, M., Mogensen, J., Shaw, A., Hughes, S., and Syrris, P., (2005). Novel mutation in desmoplakin causes arrhythmogenic left ventricular cardiomyopathy. Circulation 112 (5), 636–64242. doi:10.1161/CIRCULATIONAHA.104.532234
Petretta, M., Pirozzi, F., Sasso, L., Paglia, A., and Bonaduce, D. (2011). Review and metaanalysis of the frequency of familial dilated cardiomyopathy. AmAm. J. Cardiol. J. Cardiol. 108 (8), 1171–11766. doi:10.1016/j.amjcard.2011.06.022
Pigors, M., Schwieger-Briel, A., Cosgarea, R., Diaconeasa, A., Bruckner-Tuderman, L., and Fleck, T., (2015). Desmoplakin mutations with palmoplantar keratoderma, woolly hair and cardiomyopathy. Acta DermActa Derm. Venereol. Venereol. 95 (3), 337–34040. doi:10.2340/00015555-1974
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: BEDTools: a flexible suite of utilities for comparing genomic features. flexible suite of utilities for comparing genomic features. Bioinformatics 26 (6), 841–8422. doi:10.1093/bioinformatics/btq033
Rampazzo, A., Nava, A., Malacrida, S., Beffagna, G., Bauce, B., and Rossi, V., (2002). Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. AmAm. J. Hum. Genet. J. Hum. Genet. 71 (5), 1200–12066. doi:10.1086/344208
Rivero, F., Cuesta, J., Garcia-Guimaraes, M., Bastante, T., Alvarado, T., and Antuna, P., (2017). Time-related microcirculatory dysfunction in patients with takotsubo cardiomyopathy. JAMA Cardiol. 2 (6), 699–700.
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., and Getz, G., (2011). Integrative genomics viewer. NatNat. Biotechnol. Biotechnol. 29 (1), 24–266. doi:10.1038/nbt.1754
Schultheiss, H. P., Fairweather, D., Caforio, A. L. P., Escher, F., Hershberger, R. E., and Lipshultz, S. E., (2019). Dilated cardiomyopathy. NatNat. Rev. Dis. Primers Rev. Dis. Prim. 5 (1), 32. doi:10.1038/s41572-019-0084-1
Sen-Chowdhry, S., Syrris, P., Ward, D., Asimaki, A., Sevdalis, E., and McKenna, W. J. (2007). Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation 115 (13), 1710–172020. doi:10.1161/CIRCULATIONAHA.106.660241
Shore, S., Grau-Sepulveda, M. V., Bhatt, D. L., Heidenreich, P. A., Eapen, Z. J., and Hernandez, A. F., (2015). Characteristics, Characteristics, Treatments, and Outcomes of Hospitalized Heart Failure Patients Stratified by Etiologies of Cardiomyopathy.reatments, and outcomes of hospitalized heart failure patients stratified by etiologies of cardiomyopathy. JACC. Heart Fail. 3 (11), 906–91616. doi:10.1016/j.jchf.2015.06.012
Siva, N. (2008). 1000 Genomes project. NatNat. Biotechnol. Biotechnol. 26 (3), 256. doi:10.1038/nbt0308-256b
Smith, E. D., Lakdawala, N. K., Papoutsidakis, N., Aubert, G., Mazzanti, A., and McCanta, A. C., (2020). Desmoplakin Desmoplakin Cardiomyopathy, a Fibrotic and Inflammatory Form of Cardiomyopathy Distinct From Typical Dilated or Arrhythmogenic Right Ventricular Cardiomyopathy.ardiomyopathy, a fibrotic and inflammatory form of cardiomyopathy distinct from typical dilated or arrhythmogenic right ventricular cardiomyopathy. Circulation 141 (23), 1872–1884. doi:10.1161/CIRCULATIONAHA.119.044934
Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B. C., and Remm, M., (2012). Primer3--new capabilities and interfaces. Nucleic Acids ResNucleic Acids Res. 40 (15), e115. doi:10.1093/nar/gks596
Wang, K., Li, M., and Hakonarson, H. (2010). AANNOVAR: functional annotation of genetic variants from high-throughput sequencing data.: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids ResNucleic Acids Res. 38 (16), e164. doi:10.1093/nar/gkq603
Yeh, J. K., Liu, W. H., Wang, C. Y., Lu, J. J., Chen, C. H., and Wu-Chou, Y. H., (2019). Targeted Targeted Next Generation Sequencing for Genetic Mutations of Dilated Cardiomyopathy.ext generation sequencing for genetic mutations of dilated cardiomyopathy. Acta CardiolActa Cardiol. Sin. Sin. 35 (6), 571–584. doi:10.6515/ACS.201911_35(6).20190402A
Keywords: familial dilated cardiomyopathy (FDC), desmoplakin gene (DSP), heart transplantation (HTx), whole exome sequencing, dilated cardiomyopathy (DCM)
Citation: Chang Y-H, Lin P, Lin J-L, Huang H-Y, Hsu C-K and Hsu C-H (2022) Case Report: A novel desmoplakin mutation in a taiwanese woman with familial dilated cardiomyopathy that necessitated heart transplantation. Front. Genet. 13:954931. doi: 10.3389/fgene.2022.954931
Received: 27 May 2022; Accepted: 08 September 2022;
Published: 23 September 2022.
Edited by:
Zahurul A. Bhuiyan, Centre Hospitalier Universitaire Vaudois (CHUV), SwitzerlandReviewed by:
Minoru Horie, Shiga University of Medical Science, JapanCopyright © 2022 Chang, Lin, Lin, Huang, Hsu and Hsu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chih-Hsin Hsu, Y2hpaGhzaW5oc3VAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.