 R. Villafuerte-De la Cruz1†
R. Villafuerte-De la Cruz1† O. F. Chacon-Camacho2,3†
O. F. Chacon-Camacho2,3† A. C. Rodriguez-Martinez4N. Xilotl-De Jesus3R. Arce-Gonzalez3
A. C. Rodriguez-Martinez4N. Xilotl-De Jesus3R. Arce-Gonzalez3 C. Rodriguez-De la Torre1J. E. Valdez-Garcia1A. Rojas-Martinez1,5*
C. Rodriguez-De la Torre1J. E. Valdez-Garcia1A. Rojas-Martinez1,5* J. C. Zenteno3,6*
J. C. Zenteno3,6*- 1Tecnologico de Monterrey, Escuela de Medicina y Ciencias de la Salud, Monterrey, Mexico
- 2Carrera Médico Cirujano, Facultad de Estudios Superiores Iztacala, Universidad Nacional Autónoma de México, Mexico City, Mexico
- 3Genetics Department, Institute of Ophthalmology “Conde de Valenciana”, Mexico City, Mexico
- 4Department of Ophthalmology, University Hospital and Faculty of Medicine, Autonomous University of Nuevo Leon (UANL), Monterrey, Mexico
- 5Institute for Obesity Research, Tecnologico de Monterrey, Escuela de Medicina y Ciencias de la Salud, Monterrey, Mexico
- 6Biochemistry Department, Faculty of Medicine, National Autonomous University of Mexico (UNAM), Mexico City, Mexico
Inherited retinal diseases (IRDs) represent a spectrum of clinically and genetically heterogeneous disorders. Our study describes an IRD patient carrying ABCA4 and USH2A pathogenic biallelic mutations as a result of paternal uniparental disomy (UPD) in chromosome 1. The proband is a 9-year-old girl born from non-consanguineous parents. Both parents were asymptomatic and denied family history of ocular disease. Clinical history and ophthalmologic examination of the proband were consistent with Stargardt disease. Whispered voice testing disclosed moderate hearing loss. Next-generation sequencing and Sanger sequencing identified pathogenic variants in ABCA4 (c.4926C>G and c.5044_5058del) and USH2A (c.2276G>T). All variants were present homozygously in DNA from the proband and heterozygously in DNA from the father. No variants were found in maternal DNA. Further analysis of single nucleotide polymorphisms confirmed paternal UPD of chromosome 1. This is the first known patient with confirmed UPD for two recessively mutated IRD genes. Our study expands on the genetic heterogeneity of IRDs and highlights the importance of UPD as a mechanism of autosomal recessive disease in non-consanguineous parents. Moreover, a long-term follow-up is essential for the identification of retinal features that may develop as a result of USH2A-related conditions.
Introduction
Inherited retinal dystrophies (IRDs) are a group of phenotypically and genetically heterogeneous diseases affecting up to 4.5 million people worldwide and leading to disabling impairment of vision in affected individuals (Hohman, 2017). IRDs can be classified according to its progression (progressive or stationary), the presence or absence of concurrent extraocular anomalies (syndromic or non-syndromic), and the predominantly affected photoreceptor cell (rod dystrophy, cone dystrophy, or generalized dystrophies) (Berger et al., 2010). Recent advances in molecular genetic diagnostic techniques, such as next-generation sequencing (NGS), have led to the identification of the underlying genetic cause in a majority of IRD patients (Chiang et al., 2015; Bernardis et al., 2016). In addition, molecular diagnosis through NGS has allowed the recognition of the ethnic-specific mutational spectrum in IRDs (Avela et al., 2018; Zenteno et al., 2020) and has unmasked complex genotypes which were previously associated with diagnostic odysseys (Jones et al., 2017; González-Del Pozo et al., 2020).
ABCA4 and USH2A are two of the most frequently mutated genes in IRD patients worldwide. Biallelic mutations in ABCA4, a gene located at 1p22.1 and encoding the retina-specific transmembrane protein involved in the transport of retinoids in the visual cycle, cause Stargardt disease (STGD), a recessive macular dystrophy regarded as one of the most prevalent inherited retinal disorders in humans (Tsang and Sharma, 2018). To date, about 1700 different pathogenic variants in ABCA4 have been identified in STGD and other related retinal phenotypes collectively known as ABCA4-associated retinopathies (Stenson et al., 2020). Most disease-causing ABCA4 variants are single-nucleotide substitutions that predict missense changes in the protein. On the other hand, biallelic mutations in USH2A gene (located at 1q41), which encodes for usherin, a basement membrane protein in the inner ear and retina, underlie ∼20% of cases of non-syndromic retinitis pigmentosa, ∼7% of non-syndromic hearing loss, and up to 80% of cases of Usher syndrome (USH) type II (Koenekoop et al., 1999; McGee et al., 2010; Del Castillo et al., 2022). USH is an autosomal recessive disorder characterized by visual loss due to retinitis pigmentosa, sensorineural hearing impairment, and variable vestibular dysfunction. Around 1800 different USH2A pathogenic variants have been described to date (Stenson et al., 2020), including nonsense and missense mutations, splicing variants, small deletions and insertions, small indels, and large rearrangements (Gao et al., 2021).
Uniparental disomy (UPD), defined as the presence in a diploid genome of a chromosome pair derived from one progenitor, is a rare cause of recessively inherited disorders (Yamazawa et al., 2010; Erger et al., 2018). To the best of our knowledge, only 2 STGD and 3 Usher syndrome cases have been demonstrated to occur due to chromosome 1 UPD (Rivolta et al., 2002; Fingert et al., 2006; Riveiro-Alvarez et al., 2007; Fu et al., 2020; Wang et al., 2021).
In this work, we describe the clinical and molecular findings in a patient carrying simultaneous ABCA4 and USH2A biallelic mutations as a result of paternal UPD of chromosome 1. To the best of our knowledge, this is the first known patient with confirmed UPD for two recessively mutated IRD genes.
Material and methods
Clinical examination
The protocol was approved by the Institutional Review Board of the Institute of Ophthalmology “Conde de Valenciana”, Mexico City. All procedures followed the tenets of the Helsinki Declaration, and the patient’s parents gave written consent for their inclusion in the study. The participating proband underwent a detailed clinical history and a full ophthalmological evaluation. Additionally, color fundus photography (CFP), fundus autofluorescence (FAF) imaging (Zeiss Visucam NM, Carl Zeiss, Germany), spectral domain optical coherence tomography (Cirrus HD-OCT, Carl Zeiss Meditec AG, Jena, Germany), and electrophysiological assessment (full-field and multifocal electroretinography) were performed. Parents were asymptomatic and consented to donate peripheral blood for DNA analyses.
DNA isolation and next-generation sequencing
Genomic DNA (gDNA) was extracted from peripheral blood leukocytes using the QIAamp DNA Blood kit (QIAGEN, Hilden, Germany). gDNA quantification and purity of samples were measured using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA). All exon regions of the Invitae Inherited Retinal Disorders Panel (293 genes) (Invitae, San Francisco, CA) were sequenced. Briefly, gDNA was enriched for target regions using a hybridization-based protocol and sequenced using Illumina technology (Illumina, San Diego, CA). All targeted regions were sequenced with ≥ ×50 depth or were supplemented with additional analysis. Reads were aligned to a reference sequence GRCh37/19 human reference genome, and sequenced changes were identified and interpreted in the context of a single clinical relevant transcript, indicated as follows. Enrichment and analysis focus on the coding sequence of the indicated transcripts, 20 bp of flanking intronic regions, and other specific genomic regions demonstrated to be causative of disease at the time of the assay design. This assay achieves >99% analytical sensitivity and specificity for single-nucleotide variants, and insertions and deletions <15 bp in length. Designation of pathogenic/likely pathogenic variants was carried out following the American College of Genetics and Genomic (ACMG) guidelines (Richards et al., 2015). Exonic deletions and duplications (CNVs) were called using an in-house algorithm (Invitae) that determines the copy number at each target by comparing the read depth for each target in the proband sequence with both mean read depth distributions, obtained from a set of clinical samples.
Primer design and PCR
For PCR amplification of ABCA4 and USH2A exons 35-36 and 13, respectively, each 25 μL reaction contained 1X buffer, 200 ng of genomic DNA, 0.2 mM of each deoxynucleotide triphosphate, 2U Taq polymerase, 1 mM of forward and reverse primers, and 1.5 mM of MgCl2. The primers are the following: for ABCA4; 35F forward 5′-GCAGCGTCTCAGATGTCCTC-3′ and 35R reverse 3′-CGGTGGTGAGAATCCTCTCA-5′; 36F forward 5′-GTATCTTCTCCTCCTTCTGC-3′ and 36R reverse 3′-ACACACAAGCTCCACCTTG-5′; and for USH2A, 13F forward 5′-GCAGTAGCATTGTTTGTGTCTC-3′ and 13R reverse 3′-ATTTGTAGAAGCCACAAACC-5'. The PCR temperature program included 30 cycles of denaturation at 97°C for 1 min, annealing at 60°C for 1 min, and extension at 72°C for 1 min. Sanger sequencing was performed with the BrilliantDye Terminator v1.1 Cycle Sequencing Kit (NimaGen BV, Netherlands), adding about 15 ng of template DNA, 3.5 ml of the 5X sequencing buffer, 1 ml BrilliantDye v1.1 rr premix, 1 ml primer, and 13.5 ml water in each reaction (20 ml total) and using a temperature program that included 25 cycles of denaturation at 96°C for 10 s, annealing at 50°C for 5 s, and extension at 60°C for 4 min. Samples were analyzed in a Spectrum Compact CE System (Promega Corporation, WI, United States).
Chromosome 1 SNP analysis
Variant call format (VCF) files obtained from gene panel sequencing (Invitae) and containing the full list of variants identified (including pathogenic, likely pathogenic, VUS, likely benign, and benign variants) were analyzed. Subsequently, variants occurring in genes located at chromosome 1 were manually selected and verified for their zygosity status. Only variants sequenced with ≥50 depth were included in the SNP analysis.
Sanger sequencing of ABCA4 and USH2A genes
Specific oligonucleotide primers were designed by amplification of exons 35 and 36 of ABCA4 gene (NM_000350.3), and exon 13 of USH2A (NM_206933.4). Sanger nucleotide analysis was performed by BigDye Terminator v1.1 chemistry on a Spectrum Compact CE System (Promega Corporation, WI). Confirmatory Sanger sequencing of the pathogenic variants was carried out in DNAs from the proband and her parents.
Results
Clinical features
The index case is a 9-year-old Mexican girl who presented with a 6-year history of uncorrected visual impairment and right hypertropia. This was accompanied by light sensitivity and dark-to-light adaptation difficulties. Her past ocular history was remarkable for occlusion therapy from age 3–6 years. She was the product of the first pregnancy of non-consanguineous parents. Family history disclosed three miscarriages and one instance of stillbirth in her otherwise healthy mother. Her parents denied a personal or family history of ocular diseases.
On ophthalmologic examination, best corrected visual acuity on the right eye was 20/200 (1.00 logMAR) and left eye was 20/160 (0.90 logMAR). The right pupillary diameter was 3 mm, while the left pupil diameter was 1.5 mm. There was no afferent pupillary defect, and both pupils were reactive to light and accommodation. Orthoptic assessment showed right hypertropia, and her extraocular movements were within normal limits. The color vision assessment, evaluated with the Ishihara test, was unremarkable. No anomalies were identified in the anterior segment at slit lamp examination. Funduscopic examination revealed a bilateral vermillion appearance. No evidence of bone spicule pigmentation was observed. Color fundus photography revealed a macular bull’s eye appearance with small pisciform yellow-white flecks scattered within the posterior pole (Figure 1A). Fundus autofluorescence (FAF) revealed a central area of macular hypoautofluorescence along with retinal flecks extending centrifugally and appearing hypoautofluorescent in the center and hyperautofluorescent in the periphery. The peripapillary sparing retina should be noted, as demonstrated by fundus autofluorescence (Figure 1B). High-resolution spectral domain optical coherence tomography (SD-OCT) demonstrated decreased retinal thickness, particularly at the foveola with the loss of inner and outer photoreceptor segment layers and hyper-reflective deposits below the RPE corresponding to flecks (Figure 2). Electroretinogram records show subnormal rod and cone responses with severely reduced amplitude and implicit time, especially evident in flicker response (Figure 3). General audiological examination using the patient’s responses to whispered questions and the general hearing test revealed moderate hearing loss, with no speech impairment. Parents refused formal audiological tests to be performed in the proband. Retinal phenotypic features and electrophysiological studies in our patient were consistent with STGD.

FIGURE 1. (A) Color fundus photography revealed a macular bull’s eye appearance with small pisciform yellow-white flecks scattered within the posterior pole on both eyes. (B) Fundus autofluorescence (FAF) revealed a central area of macular hypoautofluorescence along with retinal flecks extending centrifugally and appearing hypoautofluorescent in the center and hyperautofluorescent in the periphery. The peripapillary sparing retina should be noted, as demonstrated by fundus autofluorescence.
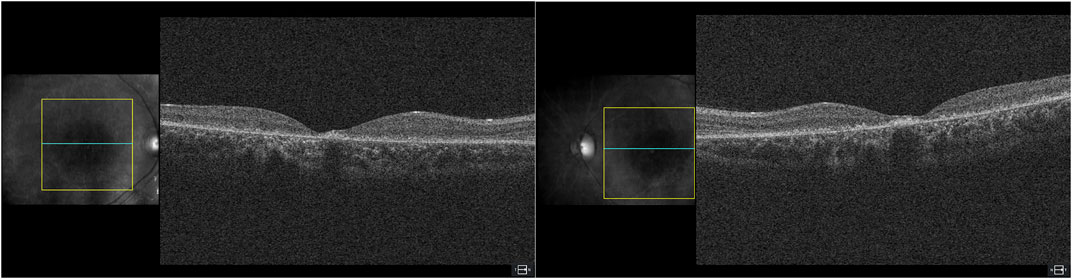
FIGURE 2. High-resolution spectral domain optical coherence tomography (SD-OCT) demonstrated decreased retinal thickness, particularly at the foveola, with loss of inner and outer photoreceptor segment layers and hyper-reflective deposits below the RPE corresponding to flecks.
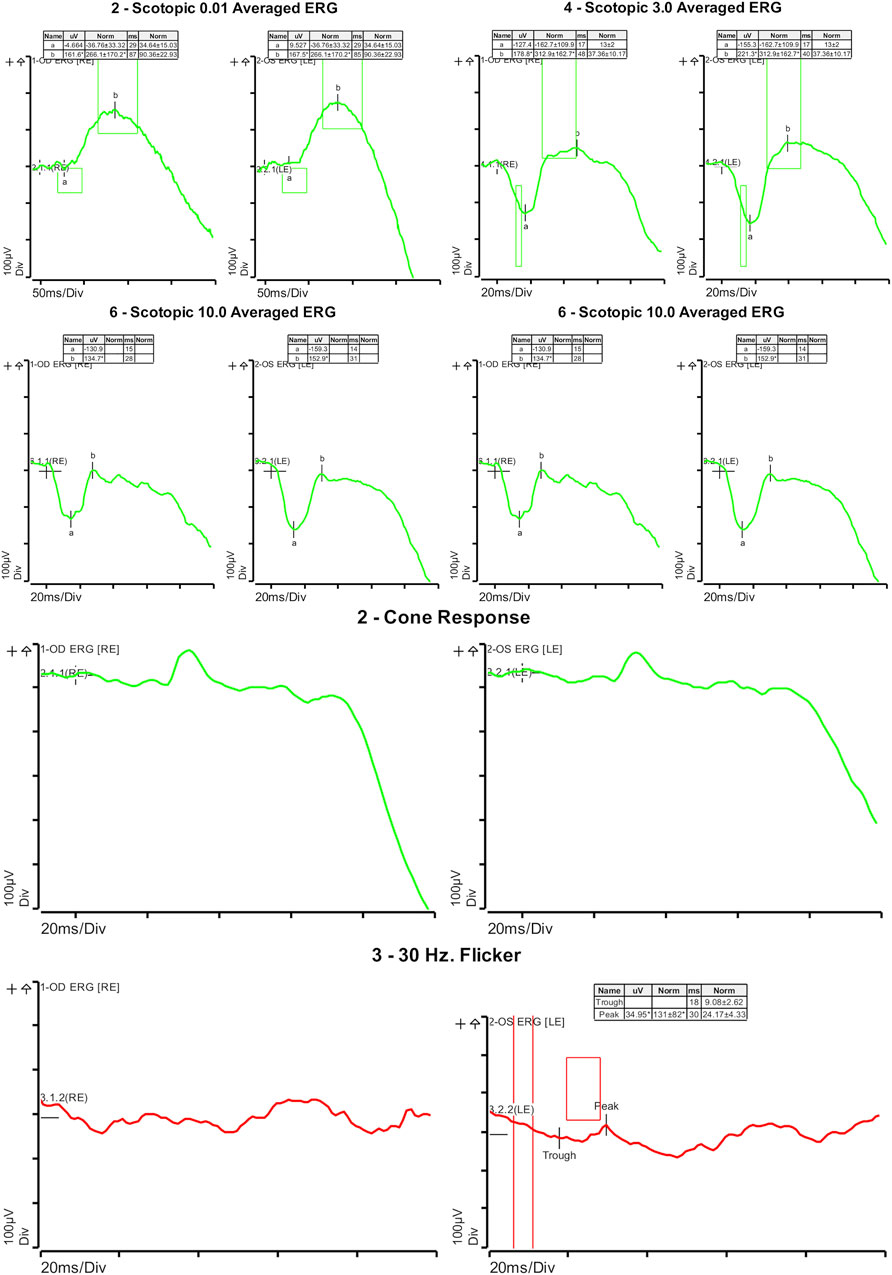
FIGURE 3. Electroretinogram with subnormal rod response and severe reduction of cone response.
Molecular analysis
Sequencing of a panel of 293 retinal dystrophy genes in DNA from the index case identified homozygosity for a pathogenic ABCA4 complex allele and homozygosity for a pathogenic variant in USH2A. The ABCA4 complex allele corresponded to a c.4926C>G transversion that predicts a p.Ser1642Arg missense mutation (Figures 4A1–A3) and c.5044_5058del that predicts an in-frame deletion of five amino acids (p.Val1682_Val1686del) in the ABCA4 protein (Figures 4B1–B3). The pathogenic variant identified in the USH2A gene was a c.2276G>T transversion that predicts an amino acid replacement of cysteine to phenylalanine at codon 759 of the protein (p.Cys759Phe) (Figures 4C1–C3). All three variants were present homozygously in DNA from the proband (Figures 4A3–C3), heterozygously in DNA from the proband’s father (Figures 4A2–C2), and were absent from maternal DNA. The pedigree and allele segregation is shown in Supplementary Figure S1. The ABCA4 and USH2A variants have already been reported as pathogenic in a number of reports (Allikmets at al., 1997; Chacón-Camacho et al., 2013; Porto et al., 2017; Salles et al., 2017; DuPont et al., 2018; Pérez-Carro et al., 2018). NGS data files were analyzed for SNPs located at chromosome 1. A total of 24 SNPS occurring in 11 different genes, situated from 1p22.1 to 1q41 were identified. All 24 SNPs in proband’s DNA were present in the homozygous state, supporting the occurrence of paternal chromosome 1 UPD (Supplementary Table S1).
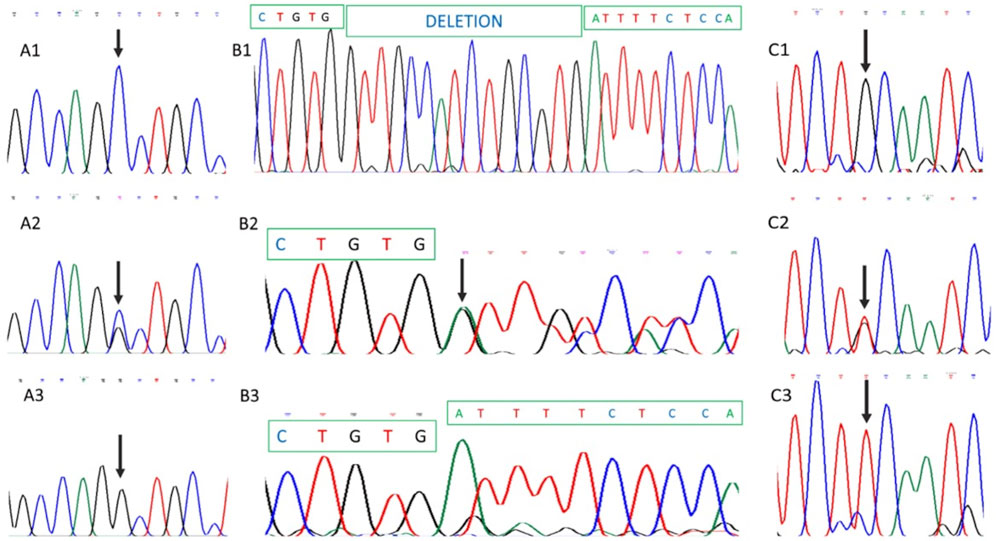
FIGURE 4. Partial DNA Sanger sequencing of ABCA4 and USH2A genes. (A1), (B1), and (C1) show partial DNA sequences from the healthy mother. (A1) and (A2) correspond to ABCA4 partial sequences, while (C1) shows the USH2A sequence. As shown, no pathogenic variants were identified in maternal DNA. (A2), (B2), and (C2) show paternal DNA analysis demonstrating heterozygosity for ABCA4 and USH2A pathogenic variants. (A3), (B3), and (C3) show homozygosity for ABCA4 (A3 and B3) and USH2A (C3) mutations in proband’s DNA. Involved nucleotides are arrowed.
Discussion
The frequency of UPD for any human chromosome has been estimated to be approximately 1 in 3,500 live births (Robinson, 2000). UPD can be classified according to parental origin or according to the size of the genomic region affected (Yamazawa et al., 2010). Two different processes may contribute to the UPD phenotypic effects and human diseases: the first of them involves a pathology if there is an underlying gene with genomic imprinting, while the second is related to the unmasking of a deleterious recessive gene with a pathogenic mutation, which in disomic chromosomes leads to homozygosity and consequently to recessively inherited diseases (Liehr, 2014).
In our case, concurrent homozygous pathogenic variants in both ABCA4 and USH2A genes were identified in the proband, heterozygous in the paternal DNA, and absent in the maternal DNA. Additionally, SNP analysis indicated that 24 SNPs located in 11 loci along chromosome 1 were homozygous in the patient’s DNA. In conjunction, these data supported the occurrence of paternal UPD for chromosome 1 as the source of simultaneous homozygous mutations in ABCA4 and USH2A. Various instances of recessive diseases associated with paternally derived UPD for chromosome 1 have been demonstrated, including infantile hypophosphatasia (ALPl gene) (Watanabe et al., 2014), neuronal ceroid lipofuscinosis-1 (PPT1) (Nilda et al., 2016; Travaglini et al., 2017), and atypical Hutchinson–Gilford progeria syndrome (ZMPSTE24) (Cassini et al., 2018) among others (Zeng et al., 2006; Miura et al., 2000; Manoli et al., 2010).
The simultaneous occurrence of ABCA4 and USH2A recessive mutations in a single patient has not been previously reported and, to the best of our knowledge, only five affected patients with UPD for either ABCA4 or USH2A gene have been described. In 2006, a 15-year-old female diagnosed with STGD was identified to carry a homozygous c.4139C>T (p.Pro1380Leu) ABCA4 mutation. Parental DNA analyses showed that only her father was the carrier for the mutation, and genotyping of 24 short tandem repeats (STRs) on chromosome 1 demonstrated paternal UPD (Fingert et al., 2006). In 2007, a female with a clinical diagnosis of STGD was demonstrated to carry a homozygous c.3386G>T (p. Arg1129Leu) pathogenic variant in ABCA4, and analysis of microsatellite markers along the entire chromosome 1 showed that the isodisomy spanned a 4.4-Mb segment that included the ABCA4 gene (Riveiro-Alvarez et al., 2007).
On the other hand, in a female patient with non-syndromic retinitis pigmentosa, a paternal UPD for the telomeric region of chromosome 1, including the USH2A gene, was demonstrated (Rivolta et al., 2002). In 2020, UPD for chromosome 1 was identified in a boy with non-syndromic hearing loss, and through STR analysis and homozygosity mapping, it was demonstrated that his homozygous USH2A variant arose from maternal UPD (Fu et al., 2020). Finally, a recent report described a case of paternal UPD resulting in homozygous mutations in both USH2A and AGL (glycogen storage disease type III) genes in a 4-year-old girl with congenital deafness, learning difficulties, and enlarged liver. No ocular anomalies were observed (Wang et al., 2021). This latter case is similar to our case since two pathogenic recessive mutations associated with two different diseases were unmasked by UPD for chromosome 1. However, our case is unique as chromosome 1 UPD resulted in homozygous pathogenic variants in two loci associated with IRD. Interestingly, but representing a disease mechanism other than uniparental isodisomy, a patient with compound heterozygous pathogenic variants in both ABCA4 and USH2A genes has also recently been described (Liu et al., 2020). In the patient described by the authors, the clinical findings represented a combination of both retinal phenotypes.
The pathogenic variants identified in our patient have been previously described in the literature. The ABCA4 c.4926C>G/c.5044_5058del variants have been demonstrated to occur in cis form, and thus, it is considered a complex ABCA4 allele, as demonstrated in Brazilian STGD families (Porto et al., 2017; Salles et al., 2017). However, they can also occur as isolated pathogenic variants in individuals with STGD or cone dystrophy (Allikmets at al., 1997; Chacón-Camacho et al., 2013). c.2276G>T (p.759Phe) in USH2A is a common pathogenic variant identified in individuals with Usher syndrome type II or with isolated recessive retinitis pigmentosa (DuPont et al., 2018; Pérez-Carro et al., 2018). A previous study evaluated the ush2a p.(Cys771Phe) zebrafish model and identified the reduced usherin expression at the photoreceptor periciliary membrane, as well as increased rhodopsin levels in the photoreceptor cell body and decreased ERG b-wave amplitudes (Reurink et al., 2022).
Interestingly, no clinical evidence of Usher syndrome was observed in the retinal examination of our patient despite carrying a homozygous pathogenic mutation in USH2A. Mutations in USH2A are identified in 57–79% of cases of Usher syndrome (Koenekoop et al., 1999) but only in 10–15% of cases of non-syndromic retinitis pigmentosa (Bonnet et al., 2011). Although a formal hearing test was refused by the family, we found clinical evidence of hearing loss, which may be attributable to biallelic mutations in USH2A, whereas no retinitis pigmentosa features were observed in this girl; thus, it cannot be ruled out that a rod-cone dystrophy could develop at older age.
Finally, the recognition of biallelic pathogenic USH2A mutations in this STGD patient raises the need for frequent ophthalmological examinations for a potential early identification of retinitis pigmentosa manifestations.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding authors.
Author contributions
All authors contributed to the study conception and design. Clinical and ophthalmologic evaluations of the patient and her family were performed by RV, OC, AR-M, NX, RA-G, CR, and JZ. Data collection and analysis were performed by RV, OC, CR, JV, AR, and JZ. The first draft of the manuscript was written by OC and RV. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Funding
This work was financed with funds from the participating academic centers. Initial genetic studies were performed using the Inherited Retinal Disease (IRD) program from Invitae Corp. (San Francisco, CA). This study received funding from Invitae Corporation. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication.
Acknowledgments
The authors specially acknowledge Invitae Corporation for providing access to their IRD program for our patients. This work has been possible because of the scholarship given by Tecnologico de Monterrey to RV for a PhD.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.949437/full#supplementary-material
References
Allikmets, R., Singh, N., Sun, H., Shroyer, N. F., Hutchinson, A., Chidambaram, A., et al. (1997). A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat. Genet. 15, 236–246. doi:10.1038/ng0397-236
Avela, K., Sankila, E. M., Seitsonen, S., Kuuluvainen, L., Barton, S., Gillies, S., et al. (2018). A founder mutation in CERKL is a major cause of retinal dystrophy in Finland. Acta Ophthalmol. 96, 183–191. doi:10.1111/aos.13551
Berger, W., Kloeckener-Gruissem, B., and Neidhardt, J. (2010). The molecular basis of human retinal and vitreoretinal diseases. Prog. Retin. Eye Res. 29 (5), 335–375. doi:10.1016/j.preteyeres.2010.03.004
Bernardis, I., Chiesi, L., Tenedini, E., Artuso, L., Percesepe, A., Artusi, V., et al. (2016). Unravelling the complexity of inherited retinal dystrophies molecular testing: added value of targeted next-generation sequencing. Biomed. Res. Int. 2016, 6341870. doi:10.1155/2016/6341870
Bonnet, C., Grati, M., Marlin, S., Levilliers, J., Hardelin, J. P., Parodi, M., et al. (2011). Complete exon sequencing of all known Usher syndrome genes greatly improves molecular diagnosis. Orphanet J. Rare Dis. 6, 21. doi:10.1186/1750-1172-6-21
Cassini, T. A., Robertson, A. K., Bican, A. G., Cogan, J. D., Hannig, V. L., Newman, J. H., et al. (2018). Phenotypic heterogeneity of ZMPSTE24 deficiency. Am. J. Med. Genet. A 176, 1175–1179. doi:10.1002/ajmg.a.38493
Chacón-Camacho, O. F., Granillo-Alvarez, M., Ayala-Ramírez, R., and Zenteno, J. C. (2013). ABCA4 mutational spectrum in Mexican patients with stargardt disease: identification of 12 novel mutations and evidence of a founder effect for the common p.A1773V mutation. Exp. Eye Res. 109, 77–82. doi:10.1016/j.exer.2013.02.006
Chiang, J. P. W., Lamey, T., McLaren, T., Thompson, J. A., Montgomery, H., and De Roach, J. (2015). Progress and prospects of next-generation sequencing testing for inherited retinal dystrophy. Expert Rev. Mol. Diagn. 15, 1269–1275. doi:10.1586/14737159.2015.1081057
Del-Castillo, I., Morin, M., Domínguez-Ruiz, M., and Moreno-Pelayo, M. (2022). Genetic etiology of non-syndromic hearing loss in Europe. Hum. Genet. 141, 683–696. doi:10.1007/s00439-021-02425-6
DuPont, M., Jones, E. M., Xu, M., and Chen, R. (2018). Investigating the disease association of USH2A p.C759F variant by leveraging large retinitis pigmentosa cohort data. Ophthalmic Genet. 39, 291–292. doi:10.1080/13816810.2017.1418388
Erger, F., Burau, K., Elsässer, M., Zimmermann, K., Moog, U., and Netzer, C. (2018). Uniparental isodisomy as a cause of recessive mendelian disease: a diagnostic pitfall with a quick and easy solution in medium/large NGS analyses. Eur. J. Hum. Genet. 26, 1392–1395. doi:10.1038/s41431-018-0195-2
Fingert, J. H., Eliason, D. A., Phillips, N. C., Lotery, A. J., Sheffield, V. C., and Stone, E. M. (2006). Case of stargardt disease caused by uniparental isodisomy. Arch. Ophthalmol. 124, 744–745. doi:10.1001/archopht.124.5.744
Fu, J., Shen, S., Cheng, J., Lv, H., and Fu, J. (2020). A case of Usher syndrome type IIA caused by a rare USH2A homozygous frameshift variant with maternal uniparental disomy (UPD) in a Chinese family. J. Cell. Mol. Med. 24, 7743–7750. doi:10.1111/jcmm.15405
Gao, F. J., Wang, D. D., Chen, F., Sun, H. X., Hu, F. Y., Xu, P., et al. (2021). Prevalence and genetic-phenotypic characteristics of patients with USH2A mutations in a large cohort of Chinese patients with inherited retinal disease. Br. J. Ophthalmol. 105, 87–92. doi:10.1136/bjophthalmol-2020-315878
González-Del Pozo, M., Fernández-Suárez, E., Martín-Sánchez, M., Bravo-Gil, N., Méndez-Vidal, C., Rodríguez-De La Rúa, E., et al. (2020). Unmasking Retinitis Pigmentosa complex cases by a whole genome sequencing algorithm based on open-access tools: hidden recessive inheritance and potential oligogenic variants. J. Transl. Med. 18, 73–12. doi:10.1186/s12967-020-02258-3
Hohman, T. C. (2017). “Hereditary retinal dystrophy,” in Handbook of experimental pharmacology. Editors S. Whitcup, and D. Azar (Denmark: Springer Cham), 337–367.
Jones, K. D., Wheaton, D. K., Bowne, S. J., Sullivan, L. S., Birch, D. G., Chen, R., et al. (2017). Next-generation sequencing to solve complex inherited retinal dystrophy: a case series of multiple genes contributing to disease in extended families. Mol. Vis. 23, 470–481.
Koenekoop, R., Arriaga, M., Trzupek, K. M., and Lentz, J. (1999). Usher syndrome type II. Genereviews. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1341/(Accessed 03 03, 2022).
Liehr, T. (2014). Uniparental disomy (Upd) in clinical genetics a guide for clinicians and patients. Heidelberg: Springler.
Liu, X. Z., Tao, T. C., Qi, H., Feng, S. N., Chen, N. N., Zhao, L., et al. (2020). Simultaneous expression of two pathogenic genes in four Chinese patients affected with inherited retinal dystrophy. Int. J. Ophthalmol. 13 (2), 220–230. doi:10.18240/ijo.2020.02.04
Manoli, I., Golas, G., Westbroek, W., Vilboux, T., Markello, T. C., Introne, W., et al. (2010). Chediak-Higashi syndrome with early developmental delay resulting from paternal heterodisomy of chromosome 1. Am. J. Med. Genet. A 152, 1474–1483. doi:10.1002/ajmg.a.33389
McGee, T. L., Seyedahmadi, B. J., Sweeney, M. O., Dryja, T. P., and Berson, E. L. (2010). Novel mutations in the long isoform of the USH2A gene in patients with usher syndrome type II or non-syndromic retinitis pigmentosa. J. Med. Genet. 47, 499–506. doi:10.1136/jmg.2009.075143
Miura, Y., Hiura, M., Torigoe, K., Numata, O., Kuwahara, A., Matsunaga, M., et al. (2000). Complete paternal uniparental isodisomy for chromosome 1 revealed by mutation analyses of the TRKA (NTRK1) gene encoding a receptor tyrosine kinase for nerve growth factor in a patient with congenital insensitivity to pain with anhidrosis. Hum. Genet. 107, 205–209. doi:10.1007/s004390000369
Niida, Y., Yokoi, A., Kuroda, M., Mitani, Y., Nakagawa, H., and Ozaki, M. (2016). A girl with infantile neuronal ceroid lipofuscinosis caused by novel PPT1 mutation and paternal uniparental isodisomy of chromosome 1. Brain Dev. 38, 674–677. doi:10.1016/j.braindev.2016.01.004
Pérez-Carro, R., Blanco-Kelly, F., Galbis-Martínez, L., García-García, G., Aller, E., García-Sandoval, B., et al. (2018). Unravelling the pathogenic role and genotype-phenotype correlation of the USH2A p.(Cys759Phe) variant among Spanish families. PLoS One 13, e0199048–18. doi:10.1371/journal.pone.0199048
Porto, F. B. O., Jones, E. M., Branch, J., Soens, Z. T., Maia, I. M., Sena, I. F. G., et al. (2017). Molecular screening of 43 brazilian families diagnosed with leber congenital amaurosis or early-onset severe retinal dystrophy. Genes 8, 3555–E418. doi:10.3390/genes8120355
Reurink, J., de Vrieze, E., Li, C. H. Z., van Berkel, E., Broekman, S., Aben, M., et al. (2022). Scrutinizing pathogenicity of the USH2A c.2276 G > T; p.(Cys759Phe) variant. NPJ Genom. Med. 7, 37–39. doi:10.1038/s41525-022-00306-z
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Riveiro-Alvarez, R., Valverde, D., Lorda-Sanchez, I., Trujillo-Tiebas, M. J., Cantalapiedra, D., Vallespin, E., et al. (2007). Partial paternal uniparental disomy (UPD) of chromosome 1 in a patient with Stargardt disease. Mol. Vis. 13, 96–101.
Rivolta, C., Berson, E. L., and Dryja, T. P. (2002). Paternal uniparental heterodisomy with partial isodisomy of chromosome 1 in a patient with retinitis pigmentosa without hearing loss and a missense mutation in the usher syndrome type II gene USH2A. Arch. Ophthalmol. 120, 1566–1571. doi:10.1001/archopht.120.11.1566
Robinson, W. P. (2000). Mechanisms leading to uniparental disomy and their clinical consequences. BioEssays 22, 452–459. doi:10.1002/(SICI)1521-1878(200005)22:5<452::AID-BIES7>3.0.CO;2-K
Salles, M. V., Motta, F. L., da Silva, E. D., Varela, P., Costa, K. A., Filippelli-Silva, R., et al. (2017). Novel complex ABCA4 alleles in Brazilian patients with stargardt disease: genotype-phenotype correlation. Invest. Ophthalmol. Vis. Sci. 58, 5723–5730. doi:10.1167/iovs.17-22398
Stenson, P. D., Mort, M., Ball, E. V., Chapman, M., Evans, K., Azevedo, L., et al. (2020). The human gene mutation database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum. Genet. 139, 1197–1207. doi:10.1007/s00439-020-02199-3
Travaglini, L., Aiello, C., Alesi, V., Loddo, S., Novelli, A., Tozzi, G., et al. (2017). Uniparental disomy of chromosome 1 unmasks recessive mutations of PPT1 in a boy with neuronal ceroid lipofuscinosis type 1. Brain Dev. 39, 182–183. doi:10.1016/j.braindev.2016.08.010
Tsang, S. H., and Sharma, T. (2018). Stargardt disease. Adv. Exp. Med. Biol. 1085, 139–151. doi:10.1007/978-3-319-95046-4_27
Wang, H., Huo, L., Wang, Y., Sun, W., and Gu, W. (2021). Usher syndrome type 2A complicated with glycogen storage disease type 3 due to paternal uniparental isodisomy of chromosome 1 in a sporadic patient. Mol. Genet. Genomic Med. 9, e1779–9. doi:10.1002/mgg3.1779
Watanabe, A., Satoh, S., Fujita, A., Naing, B. T., Orimo, H., and Shimada, T. (2014). Perinatal hypophosphatasia caused by uniparental isodisomy. Bone 60, 93–97. doi:10.1016/j.bone.2013.12.009
Yamazawa, K., Ogata, T., and Ferguson-Smith, A. C. (2010). Uniparental disomy and human disease: an overview. Am. J. Med. Genet. C Semin. Med. Genet. 154, 329–334. doi:10.1002/ajmg.c.30270
Zeng, W. Q., Gao, H., Brueton, L., Hutchin, T., Gray, G., Chakrapani, A., et al. (2006). Fumarase deficiency caused by homozygous P131R mutation and paternal partial isodisomy of chromosome 1. Am. J. Med. Genet. A 140, 1004–1009. doi:10.1002/ajmg.a.31186
Zenteno, J. C., García Montaño, L. A., Cruz-Aguilar, M., Ronquillo, J., Rodas Serrano, A., Aguilar Castul, L., et al. (2020). Extensive genic and allelic heterogeneity underlying inherited retinal dystrophies in Mexican patients molecularly analyzed by next generation sequencing. Mol. Genet. Genomic Med. 8, mgg3.1044. doi:10.1002/mgg3.1044
Keywords: uniparental disomy, retinal dystrophy, USH2A gene, ABCA4 gene, Stargardt disease, Usher syndrome
Citation: Villafuerte-De la Cruz R, Chacon-Camacho OF, Rodriguez-Martinez AC, Xilotl-De Jesus N, Arce-Gonzalez R, Rodriguez-De la Torre C, Valdez-Garcia JE, Rojas-Martinez A and Zenteno JC (2022) Case report: Disease phenotype associated with simultaneous biallelic mutations in ABCA4 and USH2A due to uniparental disomy of chromosome 1. Front. Genet. 13:949437. doi: 10.3389/fgene.2022.949437
Received: 20 May 2022; Accepted: 18 July 2022;
Published: 16 August 2022.
Edited by:
Enrico Baruffini, University of Parma, ItalyReviewed by:
Rando Allikmets, Columbia University, United StatesHongbin L. V., Southwest Medical University, China
Copyright © 2022 Villafuerte-De la Cruz, Chacon-Camacho, Rodriguez-Martinez, Xilotl-De Jesus, Arce-Gonzalez, Rodriguez-De la Torre, Valdez-Garcia, Rojas-Martinez and Zenteno. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: A. Rojas-Martinez, YXVndXN0by5yb2phc210ekB0ZWMubXg=; J. C. Zenteno, amN6ZW50ZW5vQGluc3RpdHV0b2Rlb2Z0YWxtb2xvZ2lhLm9yZw==
†These authors share first authorship