 Christine Greil
Christine Greil- Department of Hematology, Oncology and Stem Cell Transplantation, Faculty of Medicine, Medical Center-University of Freiburg, Freiburg, Germany
To sustain genomic stability by correct DNA replication and mitosis, cell cycle progression is tightly controlled by the cyclic activity of cyclin-dependent kinases, their binding to cyclins in the respective phase and the regulation of cyclin levels by ubiquitin-dependent proteolysis. The spindle assembly checkpoint plays an important role at the metaphase-anaphase transition to ensure a correct separation of sister chromatids before cytokinesis and to initiate mitotic exit, as an incorrect chromosome distribution may lead to genetically unstable cells and tumorigenesis. The ubiquitin ligase anaphase-promoting complex or cyclosome (APC/C) is essential for these processes by mediating the proteasomal destruction of cyclins and other important cell cycle regulators. To this end, it interacts with the two regulatory subunits Cdh1 and Cdc20. Both play a role in tumorigenesis with Cdh1 being a tumor suppressor and Cdc20 an oncogene. In this review, we summarize the current knowledge about the APC/C-regulators Cdh1 and Cdc20 in tumorigenesis and potential targeted therapeutic approaches.
The Role of Cdh1 and Cdc20 in Cell Cycle Regulation
To ensure a correct DNA-duplication and distribution of chromosomes to the daughter cells, the cell cycle is controlled by a complex interaction of different intra- and extracellular factors with proliferation-promoting or -inhibiting effects. The correct sequence of cell cycle phases is regulated by the cyclic activity of cyclin-dependent kinases (Cdk) and their binding to regulatory cyclins (Figure 1) (Evans et al., 1983; King et al., 1996; Miller and Cross, 2001). Before forming of the pre-replication complex (pre-RC) at the origin of replication during the initiation step of DNA replication, the cyclin activity has to be low in G1-phase. DNA replication is initiated with increasing cyclin activity at the G1/2-transition. The high cyclin activity prevents a repeated formation of the pre-RC to ensure only one replication round per cell cycle. Finally, a decreasing cyclin activity allows mitotic exit (King et al., 1996).
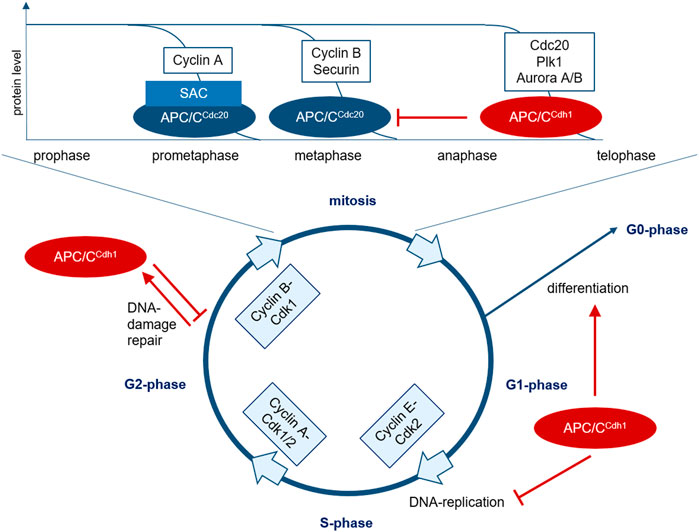
FIGURE 1. The role of APC/C in cell cycle regulation. The transition between the cell cycle phases is regulated by the cyclic activity of cyclin-Cdk complexes. In mitotic entry, cyclin B-Cdk1 plays a crucial role. In early prometaphase, chromosomes bind to the mitotic spindle and improperly connected kinetochores lead to SAC-activation. At first, this partially inhibits substrate recruitment of APC/C activated via Cdc20, but various prometaphase proteins such as cyclin A can be further marked for degradation by SAC-APC/CCdc20. After correct attachment of all chromosomes to the mitotic spindle, the SAC is inactivated, leading to degradation of additional proteins such as cyclin B and securin via APC/CCdc20 in metaphase, mediating chromosome segregation and initiating mitotic exit. Cyclin B-degradation leads to Cdk1 inhibition, resulting in dephosphorylation and activation of Cdh1. APC/CCdh1 then initiates the degradation of various proteins during ana- and telophase such as Cdc20, Plk1, and Aurora A/B. At the end of mitosis, APC/CCdh1 inactivates APC/CCdc20 and regulates anaphase spindle dynamics and cytokinesis. APC/CCdh1 is thus activated from late mitosis and controls the decision between proliferation and differentiation in G1-phase. In G2-phase, APC/CCdh1 can be activated in response to DNA-damage to prevent mitotic entry and allow DNA-repair.SAC, spindle assembly checkpoint; APC/C, anaphase promoting complex; Plk, Polo-like Kinase; Cdk, cyclin-dependent kinase.
The regulation of cyclin activity and thus proceeding in the cell cycle is regulated via synthesis and degradation of these proteins, phosphorylation, Cdk-inhibition, and the localization of the enzyme complexes within in the cell. The ubiquitin-dependent proteolysis of cyclins and other regulatory proteins by the proteasome plays a key role in this context (Hershko and Ciechanover, 1998).
There are two important ubiquitin ligases: the Skp1-, Cullin-, and F-box-protein containing-complex (SCF-complex) regulating entry into S-phase via degradation of Cdk-inhibitors in G1-phase, and the anaphase-promoting E3 ubiquitin ligase complex or cyclosome (APC/C) that mediates the separation of sister chromatids and mitotic exit by the degradation of regulatory proteins like securin and cyclin B (Harper et al., 2002; Peters, 2006).
Cell cycle checkpoints are control mechanisms to ensure its proper progression. The spindle assembly checkpoint (SAC) plays an important role at the metaphase-anaphase transition and prevents the separation of chromatids until each chromosome is correctly attached to the mitotic spindle to avoid chromosome missegregation (Figure 1). If a cell is not able to resolve an error detected at this checkpoint it initiates programmed cell death (apoptosis). A dysfunctional checkpoint can lead to aneuploidy and tumorigenesis (Greil et al., 2016; Zhang et al., 2014). Several studies have proven the particular significance of the SAC for genetic stability (Scully, 2010; Greil et al., 2016; Lawrence et al., 2015).
An accurate connection of the kinetochores to the microtubules of the mitotic spindle ensures the correct separation of the sister chromatids (Foley and Kapoor, 2013). This interaction is monitored by the SAC to prevent a premature separation and thus chromosomal instability (Musacchio and Salmon, 2007; Musacchio, 2015). Single unattached kinetochores keep the SAC active and mediate the assembly of the mitotic checkpoint complex (MCC), that inhibits the APC/C and thereby stabilizes its substrates and prevents chromosome separation and mitotic exit. The MCC is composed of Mad2, BubR1, Bub3, and the APC/C-co-activator Cdc20 (Sudakin et al., 2001). The inhibitory signal from unattached kinetochores leads to a conformational change of Mad2 and subsequently Cdc20 that thereby is able to bind BubR1 and Bub3 (Skinner et al., 2008; Han et al., 2013), can no longer activate the APC/C and thus inhibits anaphase-entry (Peters, 2006).
A correct arrangement of all chromosomes with bipolar binding to the mitotic spindle in the metaphase plate leads to SAC-silencing and facilitates mitotic exit when Cdc20 is released after MCC-degradation and can activate the APC/C (Zhang et al., 2007). Thus, the APC/C plays a crucial role in the regulation of the M/G1-transition. It consists of 13 subunits and its activity is regulated by the two co-activators Cdc20 and Cdh1 recruiting specific substrates. Their interaction is regulated by phosphorylation and temporally related to the M- and G1-phase (Morgan, 1999; Harper et al., 2002; Peters, 2006; Sullivan and Morgan, 2007; Pines, 2011; Greil et al., 2016). Major substrates of the APC/C are the mitotic cyclins A and B, mitotic kinases such as Aurora A and B and Plk1, proteins involved in chromosome separation as securin and replication proteins like Cdc6. The two co-activators Cdc20 and Cdh1 themselves are degraded APC/C-mediated (Figure 1) (Peters, 2006).
APC/CCdc20 mediates separation of sister chromatids by ubiquitin-mediated degradation of securin. Moreover, it initiates cyclin B-degradation and thereby leads to Cdk1-inactivation (Wäsch and Cross, 2002; Peters, 2006; Musacchio and Salmon, 2007; Schnerch et al., 2012a; Greil et al., 2016; Schrock et al., 2020). After Cdk1-inactivation, the second regulator Cdh1 is able to bind and activate the APC/C. It is active during late mitosis and early G1-phase and controls different cell cycle-regulators to ensure a stable G1-phase after mitotic exit and thereby optimal conditions prior to DNA-replication in the following S-phase and progression in the cell cycle towards either differentiation or division (Figure 1) (Wäsch and Cross, 2002; Qiao et al., 2010; Wäsch et al., 2010). Moreover, APC/CCdh1 is activated during DNA-damage response in G2-phase to prevent mitotic entry and initiate DNA-repair processes (Figure 1) (Sudo et al., 2001; Bassermann et al., 2008).
The Role of Cdc20 and Cdh1 in Tumorigenesis
The strict regulation of cell growth and division within the cell cycle ensures that each daughter cell receives a complete set of chromosomes. An incorrect chromosomal separation leads to aneuploidy and thus possibly to the loss of tumor suppressor genes or the overexpression of oncogenes (Jallepalli and Lengauer, 2001). Additionally, an inaccurate DNA-replication can lead to mutations with subsequent loss of function of tumor suppressors (Rouse and Jackson, 2002). Mutations in different cell cycle regulators can consecutively result in various mutations leading to genomic instability, the main feature of malignant cells (Negrini et al., 2010). The APC/C-co-activator Cdh1 is considered a potential tumor suppressor (Wang et al., 2015) and it is downregulated in different tumor entities (Engelbert et al., 2008). Heterozygous Cdh1-knockdown (kd) mice have been shown to develop tumors more frequently (García-Higuera et al., 2008). Loss of Cdh1 leads to inefficient proliferation, the accumulation of chromosomal aberrations (Wheeler et al., 2008), elevated sensitivity to DNA-damage (Ishizawa et al., 2011; de Boer et al., 2016) and development of various tumor entities (García-Higuera et al., 2008). After Cdh1-downregulation different APC/CCdh1–substrates such as cyclin A and B, Aurora A and Plk1 can accumulate. Persistent residual levels of the mitotic cyclins A and B at mitotic exit lead to a disturbed DNA-replication (Diffley, 2004), a delayed subsequent mitotic entry of cells with incompletely replicated DNA, may cause disturbed mitosis, stabilization of p53/p21 and finally genomic instability (Engelbert et al., 2008). However, it is not fully understood how Cdh1 inactivation leads to genomic instability (Wäsch and Engelbert, 2005; García-Higuera et al., 2008). The stabilization of mitotic cyclins in G1-phase may lead to premature and prolonged S-phase (Diffley, 2004; Greil et al., 2016; Choudhury et al., 2016). In the following mitosis, the chromosomes that are defective or not completely replicated in the disturbed S-phase lead to further aberrations and ultimately to the malignant transformation of the cell. By stabilization of the mitotic cyclins A and B in the nucleus, Cdh1-kd subsequently leads to a reduced accumulation and chromatin binding of the pre-RC-component MCM4 (Greil et al., 2016). MCM4-accumulation is not restored after additional inhibition of Cdk1, thus, other kinases may also play a role in this context (Wheeler et al., 2008; Greil et al., 2016). Cdh1-kd may lead to a prolonged S-phase because after reduced formation of pre-RC complexes, DNA-replication starts from fewer replication origins in G1-phase (Ayuda-Duran et al., 2014; Greil et al., 2016). Stabilization of cyclin A and B with persistent cyclin A and B-dependent Cdk1-activity and cyclin A and E-dependent Cdk2-activation in G1-phase after stabilization of the SCF-component Skp2 and degradation of the Cdk-inhibitors p27 and p21 may contribute to premature S-phase entry after Cdh1-kd (Bashir et al., 2004; Wäsch and Engelbert, 2005; Yuan et al., 2014). Incomplete DNA-replication after Cdh1-kd can cause double-strand breaks during chromosome condensation and segregation (Greil et al., 2016). As a consequence of this disturbed replication and defective chromosome segregation, mitotic aberrations occur leading to anaphase bridges, micronuclei, impaired cytokinesis, and polyploid cells (Neelsen et al., 2013; Greil et al., 2016). Tetraploid cells formed by re-fusion of daughter cells after insufficient division show supernumerary centrosomes that can either lead to multipolar mitoses or, after clustering to ensure bipolarity, to merotelic chromosome junctions and lagging chromosomes (Ganem et al., 2009; Greil et al., 2016). The APC/C plays a role in this centrosomal clustering and Cdh1-kd can lead to multipolar mitotic spindles by stabilizing the Eg5 motor protein (Drosopoulos et al., 2014). These disturbed processes result in either mitotic catastrophe and apoptosis or accumulation of chromosomal aberrations and tumorigenesis. Downstream of Cdh1-kd, stabilization of topoisomerase 2α has also been described, probably contributing to genomic instability (Eguren et al., 2014). However, the mechanism leading to genomic instability is currently not fully understood (Tavormina et al., 2002). Despite increasing genomic instability, Cdh1-deficient cells can survive and malignant transformation occurs only after a longer latency period and additional mutations (García-Higuera et al., 2008; Skaar and Pagano, 2008).
Interestingly, Cdh1 overexpression may also promote tumorigenesis: High levels of Cdh1 lead to a prolonged G1-phase, delayed entry in S-phase and increased degradation of geminin (Sørensen et al., 2000). Thereby, the pre-RC component Cdt1 is no longer inhibited, which may lead to repeated DNA replication rounds and thus to aneuploidy (Wohlschlegel et al., 2000).
Cdc20 overexpression prevents the SAC from inhibiting the APC/C, leading to mitotic slippage (Bonaiuti et al., 2018). Persistent SAC-activation due to an incorrect spindle assembly may induce mitotic arrest (Kapanidou et al., 2017). Misregulation of the APC/C may allow cells to pass this cell cycle checkpoint and to proliferate uncontrolled (Riffell et al., 2009), leading to chromosomal aberrations (Zhu et al., 2014) and probably resistance to chemotherapeutic agents that interfere with the microtubules of the mitotic spindle (Liu et al., 2019). Thus, Cdc20 acts as an oncogene (Schrock et al., 2020). A correlation between higher Cdc20 expression and poorer prognosis has been demonstrated in various malignant tumors such as breast or non-small cell lung cancer (NSCLC) (Kato et al., 2012; Karra et al., 2014). Increased levels of Cdc20 lead to a dysregulated cell cycle by overwhelming the inhibitory capacity of the SAC (Izawa and Pines, 2015), thus APC/C-activation despite an active SAC may cause premature mitotic exit, resulting in dysregulated proliferation and tumorigenesis (Bonaiuti et al., 2018). A dysfunction of the APC/C itself can lead to accumulation of Cdc20 due to its inefficient degradation. Cdc20 overexpression is accompanied by the overexpression of various other genes associated with APC/C impairment in diverse cancers (Zhang et al., 2019), including overexpression of other APC/C substrates, indicating that impairment of the APC/C and not specifically Cdc20 overexpression is important for tumorigenesis.
The Role of Cdh1 in Cell Differentiation
The APC/CCdh1 is one of the most important cell cycle regulators in G1/G0-phase, where the decision between entry into a new division cycle or terminal differentiation after cell cycle exit is made. Thus, it does not only play a role in maintaining genomic stability but also in regulating cell differentiation. Its role in differentiation processes has been described in several cell types, such as neurons, myocytes and hepatocytes (Wirth et al., 2004; Li et al., 2007; Delgado-Esteban et al., 2013). In hematopoietic stem cells (HSC), a tightly controlled cell cycle regulation is crucial for the balance between differentiation and self-renewal and impairment can lead to leukemogenesis and clonal expansion of leukemic blasts. Cdh1-expression is high in human CD34-positive HSC and declines after initiation of differentiation and Cdh1-kd inhibits myeloid differentiation, contributes to B-cell development and the preservation of immature HSC without affecting proliferation or viability (Ewerth et al., 2019). The significantly decreased Cdh1-expression in blasts of acute myeloid leukemia (AML) as compared to HSC may be a possible cause of their differentiation block (Ewerth et al., 2016). In contrast, acute promyelocytic leukemia (APL) blasts are resistant to differentiation block mediated by low Cdh1-expression. Here, Cdh1-depletion leads to a marked decrease in cell viability upon all-trans retinoic acid (ATRA)-induced differentiation (Ewerth et al., 2016). Thus, low levels of Cdh1 may enhance the therapeutic effect of ATRA. In APL, the differentiation block is primarily caused by differentiation genes downregulated via the PML-RARα fusion gene and modulation of ubiquitination via APC/CCdh1 appears ineffective. By inducing myeloid differentiation, ATRA can lead to long-term remissions in APL, depending on the risk constellation in combination with other agents (Sanz et al., 2009). In contrast, differentiation by PMA is controlled by both gene regulation and proteasomal degradation and PMA-stimulation of non-APL myeloid blasts leads to an increased Cdh1-expression (Li et al., 2014). After Cdh1-kd, differentiation in these cells may be delayed by Id2 stabilization but proliferation is not disrupted. In contrast, differentiation in APL is not perturbed after Cdh1-kd. Thus, ATRA here possibly leads to differentiated but genomically unstable cells due to low Cdh1 expression, with a consequently increased apoptosis rate (Ewerth et al., 2016). In solid tumor cells, Cdh1-kd led to a higher susceptibility to replication stress by DNA-damage-inducing chemotherapy (Wheeler et al., 2008) which may also explain the high efficacy of anthracyclines in combination with ATRA in APL with low Cdh1-expression. Cdh1 levels are probably controlled post-transcriptionally in AML by SCF-mediated proteolysis (Fukushima et al., 2013). The SCF-subunit Skp2 plays a role in tumorigenesis and -growth, Skp2-overexpression has also been demonstrated in AML (Min et al., 2004). In Skp2-kd cells, elevated Cdh1 levels were detected (Ewerth et al., 2016). Conversely, Skp2 is also a Cdh1 substrate: As described above, Cdh1-mediated Skp2-degradation in early G1-phase leads to stabilization of the Cdk-inhibitors p21 and p27; after Cdh1 inactivation in late G1-phase, Skp2 stabilization then leads to degradation of p21, p27, and Cdh1 and to S-phase entry (Bashir et al., 2004; Wäsch and Engelbert, 2005; Yuan et al., 2014).
Therapeutic Approaches Targeting the APC/C
Treatment with spindle poisons like taxanes and vinca alkaloids which inhibit the assembly of the mitotic spindle belong to the most common chemotherapeutic approaches in various malignant solid tumors (Dominguez-Brauer et al., 2015). Paclitaxel stabilizes the microtubule polymer and protects it from disassembly. It could be shown that high doses lead to a sustained mitotic arrest after SAC-activation (Weaver, 2014): due to the suppression of microtubule dynamics, chromosomes are unable to achieve a metaphase spindle configuration which prevents the SAC from being satisfied (Stukenberg and Burke, 2015), leading to continuous APC/C-inhibition with persistent high levels of cyclin B and mitotic arrest to provide additional time to repair the spindle damage (Figure 2). If this attempt fails, SAC-induced mitotic arrest triggers either apoptosis in mitosis via the mitochondrial pathway (Topham and Taylor, 2013), leads to reversion to the G0-phase and dormancy without cell division or results in mitotic slippage with mitotic exit prior to a successful cell division and thus to G1-entry of tetraploid cells. These cells may either die right after G1-entry or re-enter the cell cycle leading to proliferation of cells with an aberrant genotype (Fujiwara et al., 2005; Gascoigne and Taylor, 2008). However, other studies have suggested that therapeutically relevant concentrations of paclitaxel may not lead to mitotic arrest but to multipolar spindles and thus to chromosome missegregation and increased cell death in the interphase that follows the perturbed mitosis (Zasadil et al., 2014; Zeng et al., 2019). It was shown in several tumor cell lines that spindle apparatus damage can still result in mitotic exit without apoptosis in mitosis (Shi et al., 2008). For example in breast cancer cells, in vivo achievable paclitaxel concentrations led to multipolar spindles, incorrect chromosome distribution and postmitotic cell death (Zasadil et al., 2014; Alves et al., 2018).
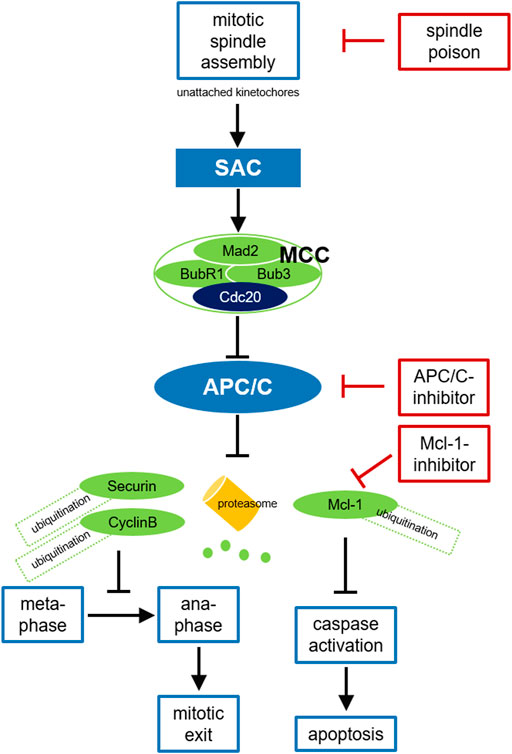
FIGURE 2. Cancer treatment targeting the APC/C. Due to disturbed microtubule kinetics after spindle poison treatment with resulting defective chromosome attachment and free kinetochores, the SAC cannot be satisfied and the MCC is formed. After binding to Mad2, BubR1, and Bub3 in the MCC Cdc20 can no longer activate the APC/C. APC/C inhibition leads to stabilization of securin and cyclin B, preventing chromosome segregation and mitotic exit. Prolonged mitotic arrest triggers Mcl-1-degradation, thus caspase activation and apoptosis in mitosis. SAC-deficient malignant cells may survive treatment with spindle poisons due to residual APC/C activity and slow cyclin B-degradation. Inhibition of APC/C or the proteasome can prevent this mitotic slippage, consolidate the mitotic block and enhance the antitumor effect. Mcl-1 inhibition may additionally promote apoptosis. SAC, spindle assembly checkpoint; MCC, mitotic checkpoint complex; APC/C, anaphase promoting complex.
The mechanism mediating whether a cell survives prolonged mitosis after spindle damage or enters apoptosis is not fully understood. A model with two independent competing signaling pathways is discussed: During mitotic arrest, pro-apoptotic signals continuously increase while cyclin B levels decrease. If the cyclin B level falls below the threshold that allows mitotic escape first, mitotic slippage occurs; if the threshold for apoptosis initiation is crossed first, mitotic cell death is initiated (Gascoigne and Taylor, 2008; Topham and Taylor, 2013). Thus, two mechanisms may contribute to the survival of tumor cells despite perturbed mitotic spindle assembly and despite subsequent mitotic arrest and thus to reduced efficacy of spindle poisons (Gascoigne and Taylor, 2008): a predominance of mitotic slippage or a dysregulation of the apoptosis signaling pathway.
Nevertheless, tumor cells can escape a SAC-mediated mitotic arrest by mitotic slippage (Brito and Rieder, 2006; Gascoigne and Taylor, 2008). Even if the SAC cannot be passed after spindle damage, residual background APC/C-activity may result in slow but continuous cyclin B-degradation via the ubiquitin-proteasome system (Brito and Rieder, 2006; Schnerch et al., 2012b). If the cyclin B level falls below a critical limit, mitotic arrest can no longer be maintained although the SAC has not been satisfied. This prevents initiation of apoptosis after prolonged mitosis and may reduce the efficacy of spindle poisons, comparable to a premature mitotic exit due to a weakened or abolished SAC (Gascoigne and Taylor, 2008). Consequently, preventing exit from mitosis by APC/C-inhibition when the SAC is not satisfied should enhance the antitumor effect (Figure 2). Some data indicate that the APC/C and its coactivator Cdc20 could be targets for oncological therapies. Cdc20-kd leads to mitotic arrest and apoptosis in various tumor cell lines (Huang et al., 2009). In 2014, the efficacy of direct APC/C-inhibitors was described for the first time: By binding Cdc20, apcin competitively prevents substrate recognition. The efficacy of apcin in blocking mitotic exit is synergistically amplified by proTAME (tosyl-L-arginine methyl ester), a prodrug that is converted to TAME by an intracellular esterase and blocks Cdc20 binding to the APC/C at a different site (Sackton et al., 2014). The combination of both molecules (pT/A) thus leads to a significant stabilization of APC/C-substrates, prevents mitotic exit in tumor cells and promotes apoptosis (Sackton et al., 2014). APC/C-inhibitors have not yet been tested in clinical trials, but preclinical data suggest a promising therapeutic approach: For example, it has been shown that proTAME leads to enhanced mitotic arrest and apoptosis in paclitaxel-treated cell lines of various solid tumors like ovarian cancer (Zeng et al., 2010; Giovinazzi et al., 2013; Sinnott et al., 2014; Raab et al., 2019). pT/A can reduce the viability of glioblastoma cells (De et al., 2019) and shows also activity in hematologic neoplasms like multiple myeloma and different types of non-Hodgkin lymphoma (Lub et al., 2016; Maes et al., 2019). Since slow cyclin B-degradation is mediated by the proteasome, preventing mitotic slippage by inhibiting proteasomal degradation seems reasonable. Indeed, a stable mitotic block was demonstrated after treatment with a proteasome inhibitor (Brito and Rieder, 2006; Schnerch et al., 2012b). Programmed cell death via the intrinsic signaling pathway is triggered by cellular stress following defective cell division or by insufficient repair mechanisms and is essential for the destruction of degenerate or potentially harmful cells (Green and Llambi, 2015). In response to DNA-damage, pro-apoptotic members of the Bcl-2 family such as BAD and Bim are activated and can thus neutralize anti-apoptotic members such as Bcl-2 itself, Bcl-xL, and Mcl-1, leading to mitochondrial outer membrane permeabilization via the pore-forming proteins BAX and BAK, to the release of various effector proteins such as cytochrome C, to caspase activation and cleavage of cellular proteins (Kalkavan and Green, 2018). Indeed, it has been shown that cell death during a mitotic arrest is initiated via this mitochondrial pathway (Gascoigne and Taylor, 2008). An imbalance between members of the Bcl-2 family may result in inefficient apoptosis (Delbridge et al., 2016). Because of the complexity of these signaling pathways, it is challenging to identify the key regulators that trigger apoptosis during prolonged mitosis. During prolonged mitotic arrest, nearly all Bcl-2 family proteins and caspases are posttranscriptionally modified (Topham and Taylor, 2013). The protein Mcl-1, which is overexpressed in many tumors, seems to play a central role here (Akgul, 2009; Beroukhim et al., 2010; Harley et al., 2010). Mcl-1 levels are regulated in a cell cycle-dependent manner, being highest in G2-phase. During mitosis, Mcl-1 is degraded after Cdk1/cyclin B-mediated phosphorylation via APC/CCdc20 (Figure 2). Due to the slow degradation of this anti-apoptotic protein, cells arrested in mitosis after therapy with spindle poisons can initiate apoptosis (Harley et al., 2010). Conversely, stabilization of Mcl-1 during mitotic arrest, for example by mutation of phosphorylation sites, prevents initiation of apoptosis after antimitotic therapy. In various tumors it was shown, that small molecules inhibiting anti-apoptotic proteins such as Mcl-1 reactivate the apoptosis signaling pathway (Wäsch, 2011; Leverson et al., 2015; Raab et al., 2019).
Besides conventional chemotherapy, those small molecules targeting signaling pathways that can be deregulated in tumor cells currently play an increasing role, as they may be more effective and cause fewer side effects. Inhibitors directed against cell cycle regulators such as Plk1 are in preclinical development and are tested in clinical trials (Gutteridge et al., 2016; Zeidan et al., 2020). Mitotic block after antimitotic therapy with classical spindle poisons or with such small molecules should be consolidated by preventing mitotic slippage through slow cyclin B-degradation via APC/C- or proteasome inhibition, thus enhancing the antitumor effect. Lower levels of the APC/C-coactivator Cdc20 reduced cyclin B-degradation even after mitotic arrest by spindle poison treatment (Schnerch et al., 2017). The effect of antimitotic therapy could be enhanced by additional proteasome inhibition in AML (Schnerch et al., 2017). It was suggested that the proteasome inhibitor bortezomib leads to G2/M-arrest in myeloid blasts and thus acts synergistically with antimitotic agents (Colado et al., 2008; Bucur et al., 2013). Nevertheless, it was shown that bortezomib acts primarily in mitosis, prevents slow cyclin B degradation and cyclin B overexpression enhances mitotic arrest after volasertib (Schnerch et al., 2017). Delayed mitotic progression after cyclin B overexpression was described (Gascoigne and Taylor, 2008) and a similar effect in volasertib-treated cells by adding bortezomib (Schnerch et al., 2017). Paclitaxel and bortezomib have also been used sequentially in solid tumors at low concentrations achievable in vivo(Weaver, 2014; Weyburne et al., 2017). In vitro, a similar effect of proteasome inhibitors on solid tumor cells as on myeloma cells was observed (Garnett et al., 2012). The sequential combination of antimitotic agents with proteasome inhibition enhances cell death in different solid tumor cells, but during interphase and not through the presumed effect of a consolidated mitotic arrest with subsequent apoptosis in mitosis (Greil et al., 2021). In clinical trials, neither bortezomib alone nor a combination with doxorubicin has been shown to have a therapeutic effect in lung and breast cancer (Dou and Zonder, 2014), perhaps because the bortezomib concentration achievable in vivo inhibits only one of the three subunits of the proteasome (Weyburne et al., 2017).
Consistent with the presumed synergistic effect on mitotic exit, APC/C-inhibition alone or combined with a taxane can lead to strong mitotic cell death in certain tumor cells, but in others with high expression of the anti-apoptotic regulator Mcl-1, cell death is induced in interphase (Zeng et al., 2010; Raab et al., 2020; Greil et al., 2021). Here, combined APC/C- and Mcl-1-inhibition is highly lethal but also in interphase (Greil et al., 2021). Despite an initially increased apoptosis rate and reduced cell number shortly after exposure, the substances do not always lead to a durable response in a cell type-specific manner (Greil et al., 2021). This difference between short-term effect and long-term survival seems to be explained by mitotic slippage (Zeng et al., 2019): These cells escape mitotic block and thus evade cell death shortly after spindle toxin therapy, but their long-term survival is compromised by increasing genomic instability after disrupted cell division.
Conclusion
The APC/C either has an oncogenic function after binding to its co-activator Cdc20, or works as a tumor suppressor when bound to Cdh1. Thus, dysfunction of both the APC/CCdh1 and APC/CCdc20 and loss of Cdh1 itself lead to dysregulation of the cell cycle, aneuploidy and increased genomic instability and contribute to tumorigenesis and probably drug resistance.
Targeting the APC/C has shown potent anti-tumor capacity and the combination of spindle poisons with a proteasome inhibitor or direct inhibitors of the APC/C and Mcl-1 seems a promising approach to improve treatment response in different solid tumors, even though they act entity-dependent at different cell cycle phases. Taken together, the current state of knowledge provides a compelling rationale to further pursue on the role of the APC/C in tumor development and treatment.
Author Contributions
Conceptualization: CG and RW; Literature research: CG; Draft preparation: CG and RW; Review and editing: CG, ME, and RW. All authors have read and agreed to the published version of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Akgul, C. (2009). Mcl-1 Is a Potential Therapeutic Target in Multiple Types of Cancer. Cell. Mol. Life Sci. 66, 1326–1336. doi:10.1007/s00018-008-8637-6
Alves, R. C., Fernandes, R. P., Eloy, J. O., Salgado, H. R. N., and Chorilli, M. (2018). Characteristics, Properties and Analytical Methods of Paclitaxel: A Review. Crit. Rev. Anal. Chem. 48, 110–118. doi:10.1080/10408347.2017.1416283
Ayuda-Duran, P., Devesa, F., Gomes, F., Sequeira-Mendes, J., Avila-Zarza, C., Gomez, M., et al. (2014). The CDK Regulators Cdh1 and Sic1 Promote Efficient Usage of DNA Replication Origins to Prevent Chromosomal Instability at a Chromosome Arm. Nucleic Acids Res. 42, 7057–7068. doi:10.1093/nar/gku313
Bashir, T., Dorrello, N. V., Amador, V., Guardavaccaro, D., and Pagano, M. (2004). Control of the SCFSkp2-Cks1 Ubiquitin Ligase by the APC/CCdh1 Ubiquitin Ligase. Nature 428, 190–193. doi:10.1038/nature02330
Bassermann, F., Frescas, D., Guardavaccaro, D., Busino, L., Peschiaroli, A., and Pagano, M. (2008). The Cdc14B-Cdh1-Plk1 Axis Controls the G2 DNA-Damage-Response Checkpoint. Cell 134, 256–267. doi:10.1016/j.cell.2008.05.043
Beroukhim, R., Mermel, C. H., Porter, D., Wei, G., Raychaudhuri, S., Donovan, J., et al. (2010). The Landscape of Somatic Copy-Number Alteration across Human Cancers. Nature 463, 899–905. doi:10.1038/nature08822
Bonaiuti, P., Chiroli, E., Gross, F., Corno, A., Vernieri, C., Štefl, M., et al. (2018). Cells Escape an Operational Mitotic Checkpoint through a Stochastic Process. Curr. Biol. 28, 28–37. e7. doi:10.1016/j.cub.2017.11.031
Brito, D. A., and Rieder, C. L. (2006). Mitotic Checkpoint Slippage in Humans Occurs via Cyclin B Destruction in the Presence of an Active Checkpoint. Curr. Biol. 16, 1194–1200. doi:10.1016/j.cub.2006.04.043
Bucur, O., Stancu, A. L., Goganau, I., Petrescu, S. M., Pennarun, B., Bertomeu, T., et al. (2013). Combination of Bortezomib and Mitotic Inhibitors Down-Modulate Bcr-Abl and Efficiently Eliminates Tyrosine-Kinase Inhibitor Sensitive and Resistant Bcr-Abl-Positive Leukemic Cells. PLOS ONE 8, e77390. doi:10.1371/journal.pone.0077390
Choudhury, R., Bonacci, T., Arceci, A., Lahiri, D., Mills, C. A., Kernan, J. L., et al. (2016). APC/C and SCF Cyclin F Constitute a Reciprocal Feedback Circuit Controlling S-phase Entry. Cell Rep. 16, 3359–3372. doi:10.1016/j.celrep.2016.08.058
Colado, E., Alvarez-Fernandez, S., Maiso, P., Martin-Sanchez, J., Vidriales, M. B., Garayoa, M., et al. (2008). The Effect of the Proteasome Inhibitor Bortezomib on Acute Myeloid Leukemia Cells and Drug Resistance Associated with the CD34+ Immature Phenotype. Haematologica 93, 57–66. doi:10.3324/haematol.11666
de Boer, H. R., Guerrero Llobet, S., and van Vugt, M. A. T. M. (2016). Controlling the Response to DNA Damage by the APC/C-Cdh1. Cell. Mol. Life Sci. 73, 949–960. doi:10.1007/s00018-015-2096-7
De, K., Grubb, T. M., Zalenski, A. A., Pfaff, K. E., Pal, D., Majumder, S., et al. (2019). Hyperphosphorylation of CDH1 in Glioblastoma Cancer Stem Cells Attenuates APC/CCDH1 Activity and Pharmacologic Inhibition of APC/CCDH1/CDC20 Compromises Viability. Mol. Cancer Res. 17, 1519–1530. doi:10.1158/1541-7786.MCR-18-1361
Delbridge, A. R. D., Grabow, S., Strasser, A., and Vaux, D. L. (2016). Thirty Years of BCL-2: Translating Cell Death Discoveries into Novel Cancer Therapies. Nat. Rev. Cancer 16, 99–109. doi:10.1038/nrc.2015.17
Delgado-Esteban, M., García-Higuera, I., Maestre, C., Moreno, S., and Almeida, A. (2013). APC/C-Cdh1 Coordinates Neurogenesis and Cortical Size during Development. Nat. Commun. 4, 2879. doi:10.1038/ncomms3879
Diffley, J. F. X. (2004). Regulation of Early Events in Chromosome Replication. Curr. Biol. 14, R778–R786. doi:10.1016/j.cub.2004.09.019
Dominguez-Brauer, C., Thu, K. L., Mason, J. M., Blaser, H., Bray, M. R., and Mak, T. W. (2015). Targeting Mitosis in Cancer: Emerging Strategies. Mol. Cell 60, 524–536. doi:10.1016/j.molcel.2015.11.006
Dou, Q., and Zonder, J. (2014). Overview of Proteasome Inhibitor-Based Anti-cancer Therapies: Perspective on Bortezomib and Second Generation Proteasome Inhibitors versus Future Generation Inhibitors of Ubiquitin-Proteasome System. Ccdt 14, 517–536. doi:10.2174/1568009614666140804154511
Drosopoulos, K., Tang, C., Chao, W. C. H., and Linardopoulos, S. (2014). APC/C Is an Essential Regulator of Centrosome Clustering. Nat. Commun. 5, 3686. doi:10.1038/ncomms4686
Eguren, M., Álvarez-Fernández, M., García, F., López-Contreras, A. J., Fujimitsu, K., Yaguchi, H., et al. (2014). A Synthetic Lethal Interaction between APC/C and Topoisomerase Poisons Uncovered by Proteomic Screens. Cell Rep. 6, 670–683. doi:10.1016/j.celrep.2014.01.017
Engelbert, D., Schnerch, D., Baumgarten, A., and Wäsch, R. (2008). The Ubiquitin Ligase APCCdh1 Is Required to Maintain Genome Integrity in Primary Human Cells. Oncogene 27, 907–917. doi:10.1038/sj.onc.1210703
Evans, T., Rosenthal, E. T., Youngblom, J., Distel, D., and Hunt, T. (1983). Cyclin: A Protein Specified by Maternal MRNA in Sea Urchin Eggs that Is Destroyed at Each Cleavage Division. Cell 33, 389–396. doi:10.1016/0092-8674(83)90420-8
Ewerth, D., Kreutmair, S., Schmidts, A., Ihorst, G., Follo, M., Wider, D., et al. (2019). APC/CCdh1 Regulates the Balance between Maintenance and Differentiation of Hematopoietic Stem and Progenitor Cells. Cell. Mol. Life Sci. 76, 369–380. doi:10.1007/s00018-018-2952-3
Ewerth, D., Schmidts, A., Hein, M., Schnerch, D., Kvainickas, A., Greil, C., et al. (2016). Suppression of APC/CCdh1 Has Subtype Specific Biological Effects in Acute Myeloid Leukemia. Oncotarget 7, 48220–48230. doi:10.18632/oncotarget.10196
Foley, E. A., and Kapoor, T. M. (2013). Microtubule Attachment and Spindle Assembly Checkpoint Signalling at the Kinetochore. Nat. Rev. Mol. Cell Biol. 14, 25–37. doi:10.1038/nrm3494
Fujiwara, T., Bandi, M., Nitta, M., Ivanova, E. V., Bronson, R. T., and Pellman, D. (2005). Cytokinesis Failure Generating Tetraploids Promotes Tumorigenesis in P53-Null Cells. Nature 437, 1043–1047. doi:10.1038/nature04217
Fukushima, H., Ogura, K., Wan, L., Lu, Y., Li, V., Gao, D., et al. (2013). SCF-mediated Cdh1 Degradation Defines a Negative Feedback System that Coordinates Cell-Cycle Progression. Cell Rep. 4, 803–816. doi:10.1016/j.celrep.2013.07.031
Ganem, N. J., Godinho, S. A., and Pellman, D. (2009). A Mechanism Linking Extra Centrosomes to Chromosomal Instability. Nature 460, 278–282. doi:10.1038/nature08136
García-Higuera, I., Manchado, E., Dubus, P., Cañamero, M., Méndez, J., Moreno, S., et al. (2008). Genomic Stability and Tumour Suppression by the APC/C Cofactor Cdh1. Nat. Cell Biol. 10, 802–811. doi:10.1038/ncb1742
Garnett, M. J., Edelman, E. J., Heidorn, S. J., Greenman, C. D., Dastur, A., Lau, K. W., et al. (2012). Systematic Identification of Genomic Markers of Drug Sensitivity in Cancer Cells. Nature 483, 570–575. doi:10.1038/nature11005
Gascoigne, K. E., and Taylor, S. S. (2008). Cancer Cells Display Profound Intra- and Interline Variation Following Prolonged Exposure to Antimitotic Drugs. Cancer Cell 14, 111–122. doi:10.1016/j.ccr.2008.07.002
Giovinazzi, S., Bellapu, D., Morozov, V. M., and Ishov, A. M. (2013). Targeting Mitotic Exit with Hyperthermia or APC/C Inhibition to Increase Paclitaxel Efficacy. Cell Cycle 12, 2598–2607. doi:10.4161/cc.25591
Green, D. R., and Llambi, F. (2015). Cell Death Signaling. Cold Spring Harb. Perspect. Biol. 7, a006080. doi:10.1101/cshperspect.a006080
Greil, C., Felthaus, J., Follo, M., Ihorst, G., Ewerth, D., Schüler, J., et al. (2021). Targeting Mitotic Exit in Solid Tumors. Am. J. Cancer Res. 11, 3698–3710.
Greil, C., Krohs, J., Schnerch, D., Follo, M., Felthaus, J., Engelhardt, M., et al. (2016). The Role of APC/CCdh1 in Replication Stress and Origin of Genomic Instability. Oncogene 35, 3062–3070. doi:10.1038/onc.2015.367
Gutteridge, R. E. A., Ndiaye, M. A., Liu, X., and Ahmad, N. (2016). Plk1 Inhibitors in Cancer Therapy: From Laboratory to Clinics. Mol. Cancer Ther. 15, 1427–1435. doi:10.1158/1535-7163.MCT-15-0897
Han, J. S., Holland, A. J., Fachinetti, D., Kulukian, A., Cetin, B., and Cleveland, D. W. (2013). Catalytic Assembly of the Mitotic Checkpoint Inhibitor BubR1-Cdc20 by a Mad2-Induced Functional Switch in Cdc20. Mol. Cell 51, 92–104. doi:10.1016/j.molcel.2013.05.019
Harley, M. E., Allan, L. A., Sanderson, H. S., and Clarke, P. R. (2010). Phosphorylation of Mcl-1 by CDK1-Cyclin B1 Initiates its Cdc20-dependent Destruction during Mitotic Arrest. EMBO J. 29, 2407–2420. doi:10.1038/emboj.2010.112
Harper, J. W., Burton, J. L., and Solomon, M. J. (2002). The Anaphase-Promoting Complex: It's Not Just for Mitosis Any More. Genes Dev. 16, 2179–2206. doi:10.1101/gad.1013102
Hershko, A., and Ciechanover, A. (1998). The Ubiquitin System. Annu. Rev. Biochem. 67, 425–479. doi:10.1146/annurev.biochem.67.1.425
Huang, H.-C., Shi, J., Orth, J. D., and Mitchison, T. J. (2009). Evidence that Mitotic Exit Is a Better Cancer Therapeutic Target Than Spindle Assembly. Cancer Cell 16, 347–358. doi:10.1016/j.ccr.2009.08.020
Ishizawa, J., Kuninaka, S., Sugihara, E., Naoe, H., Kobayashi, Y., Chiyoda, T., et al. (2011). The Cell Cycle Regulator Cdh1 Controls the Pool Sizes of Hematopoietic Stem Cells and Mature Lineage Progenitors by Protecting from Genotoxic Stress. Cancer Sci. 102, 967–974. doi:10.1111/j.1349-7006.2011.01884.x
Izawa, D., and Pines, J. (2015). The Mitotic Checkpoint Complex Binds a Second CDC20 to Inhibit Active APC/C. Nature 517, 631–634. doi:10.1038/nature13911
Jallepalli, P. V., and Lengauer, C. (2001). Chromosome Segregation and Cancer: Cutting through the Mystery. Nat. Rev. Cancer 1, 109–117. doi:10.1038/35101065
Kalkavan, H., and Green, D. R. (2018). MOMP, Cell Suicide as a BCL-2 Family Business. Cell Death Differ. 25, 46–55. doi:10.1038/cdd.2017.179
Kapanidou, M., Curtis, N. L., and Bolanos-Garcia, V. M. (2017). Cdc20: At the Crossroads between Chromosome Segregation and Mitotic Exit. Trends Biochem. Sci. 42, 193–205. doi:10.1016/j.tibs.2016.12.001
Karra, H., Repo, H., Ahonen, I., Löyttyniemi, E., Pitkänen, R., Lintunen, M., et al. (2014). Cdc20 and Securin Overexpression Predict Short-Term Breast Cancer Survival. Br. J. Cancer 110, 2905–2913. doi:10.1038/bjc.2014.252
Kato, T., Daigo, Y., Aragaki, M., Ishikawa, K., Sato, M., and Kaji, M. (2012). Overexpression of CDC20 Predicts Poor Prognosis in Primary Non-small Cell Lung Cancer Patients. J. Surg. Oncol. 106, 423–430. doi:10.1002/jso.23109
King, R. W., Deshaies, R. J., Peters, J.-M., and Kirschner, M. W. (1996). How Proteolysis Drives the Cell Cycle. Science 274, 1652–1659. doi:10.1126/science.274.5293.1652
Lawrence, K. S., Chau, T., and Engebrecht, J. (2015). DNA Damage Response and Spindle Assembly Checkpoint Function throughout the Cell Cycle to Ensure Genomic Integrity. PLoS Genet. 11, e1005150. doi:10.1371/journal.pgen.1005150
Leverson, J. D., Zhang, H., Chen, J., Tahir, S. K., Phillips, D. C., Xue, J., et al. (2015). Potent and Selective Small-Molecule MCL-1 Inhibitors Demonstrate On-Target Cancer Cell Killing Activity as Single Agents and in Combination with ABT-263 (Navitoclax). Cell Death Dis. 6, e1590. doi:10.1038/cddis.2014.561
Li, C., Peart, N., Xuan, Z., Lewis, D. E., Xia, Y., and Jin, J. (2014). PMA Induces SnoN Proteolysis and CD61 Expression through an Autocrine Mechanism. Cell. Signal. 26, 1369–1378. doi:10.1016/j.cellsig.2014.03.006
Li, W., Wu, G., and Wan, Y. (2007). The Dual Effects of Cdh1/APC in Myogenesis. FASEB J. 21, 3606–3617. doi:10.1096/fj.07-8159com
Liu, X., Chen, Y., Li, Y., Petersen, R. B., and Huang, K. (2019). Targeting Mitosis Exit: A Brake for Cancer Cell Proliferation. Biochimica Biophysica Acta (BBA) - Rev. Cancer 1871, 179–191. doi:10.1016/j.bbcan.2018.12.007
Lub, S., Maes, A., Maes, K., De Veirman, K., De Bruyne, E., Menu, E., et al. (2016). Inhibiting the Anaphase Promoting Complex/Cyclosome Induces a Metaphase Arrest and Cell Death in Multiple Myeloma Cells. Oncotarget 7, 4062–4076. doi:10.18632/oncotarget.6768
Maes, A., Maes, K., De Raeve, H., De Smedt, E., Vlummens, P., Szablewski, V., et al. (2019). The Anaphase-Promoting Complex/Cyclosome: A New Promising Target in Diffuse Large B-Cell Lymphoma and Mantle Cell Lymphoma. Br. J. Cancer 120, 1137–1146. doi:10.1038/s41416-019-0471-0
Miller, M. E., and Cross, F. R. (2001). Cyclin Specificity: How Many Wheels Do You Need on a Unicycle? J. Cell Sci. 114, 1811–1820. doi:10.1242/jcs.114.10.1811
Min, Y. H., Cheong, J.-W., Lee, M. H., Kim, J. Y., Lee, S. T., Hahn, J. S., et al. (2004). Elevated S-phase Kinase-Associated Protein 2 Protein Expression in Acute Myelogenous Leukemia. Clin. Cancer Res. 10, 5123–5130. doi:10.1158/1078-0432.CCR-04-0136
Morgan, D. O. (1999). Regulation of the APC and the Exit from Mitosis. Nat. Cell Biol. 1, E47–E53. E53. doi:10.1038/10039
Musacchio, A., and Salmon, E. D. (2007). The Spindle-Assembly Checkpoint in Space and Time. Nat. Rev. Mol. Cell Biol. 8, 379–393. doi:10.1038/nrm2163
Musacchio, A. (2015). The Molecular Biology of Spindle Assembly Checkpoint Signaling Dynamics. Curr. Biol. 25, R1002–R1018. doi:10.1016/j.cub.2015.08.051
Neelsen, K. J., Zanini, I. M. Y., Herrador, R., and Lopes, M. (2013). Oncogenes Induce Genotoxic Stress by Mitotic Processing of Unusual Replication Intermediates. J. Cell Biol. 200, 699–708. doi:10.1083/jcb.201212058
Negrini, S., Gorgoulis, V. G., and Halazonetis, T. D. (2010). Genomic Instability - an Evolving Hallmark of Cancer. Nat. Rev. Mol. Cell Biol. 11, 220–228. doi:10.1038/nrm2858
Peters, J.-M. (2006). The Anaphase Promoting Complex/Cyclosome: A Machine Designed to Destroy. Nat. Rev. Mol. Cell Biol. 7, 644–656. doi:10.1038/nrm1988
Pines, J. (2011). Cubism and the Cell Cycle: The Many Faces of the APC/C. Nat. Rev. Mol. Cell Biol. 12, 427–438. doi:10.1038/nrm3132
Qiao, X., Zhang, L., Gamper, A. M., Fujita, T., and Wan, Y. (2010). APC/C-Cdh1. Cell Cycle 9, 3904–3912. doi:10.4161/cc.9.19.13585
Raab, M., Kobayashi, N. F., Becker, S., Kurunci‐Csacsko, E., Krämer, A., Strebhardt, K., et al. (2020). Boosting the Apoptotic Response of High‐grade Serous Ovarian Cancers with CCNE1 Amplification to Paclitaxel In Vitro by Targeting APC/C and the Pro‐survival Protein MCL‐1. Int. J. Cancer 146, 1086–1098. doi:10.1002/ijc.32559
Raab, M., Sanhaji, M., Zhou, S., Rödel, F., El-Balat, A., Becker, S., et al. (2019). Blocking Mitotic Exit of Ovarian Cancer Cells by Pharmaceutical Inhibition of the Anaphase-Promoting Complex Reduces Chromosomal Instability. Neoplasia 21, 363–375. doi:10.1016/j.neo.2019.01.007
Riffell, J. L., Zimmerman, C., Khong, A., McHardy, L. M., and Roberge, M. (2009). Effects of Chemical Manipulation of Mitotic Arrest and Slippage on Cancer Cell Survival and Proliferation. Cell Cycle 8, 3025–3038. doi:10.4161/cc.8.18.9623
Rouse, J., and Jackson, S. P. (2002). Interfaces between the Detection, Signaling, and Repair of DNA Damage. Science 297, 547–551. doi:10.1126/science.1074740
Sackton, K. L., Dimova, N., Zeng, X., Tian, W., Zhang, M., Sackton, T. B., et al. (2014). Synergistic Blockade of Mitotic Exit by Two Chemical Inhibitors of the APC/C. Nature 514, 646–649. doi:10.1038/nature13660
Sanz, M. A., Grimwade, D., Tallman, M. S., Lowenberg, B., Fenaux, P., Estey, E. H., et al. (2009). Management of Acute Promyelocytic Leukemia: Recommendations from an Expert Panel on Behalf of the European LeukemiaNet. Blood 113, 1875–1891. doi:10.1182/blood-2008-04-150250
Schnerch, D., Yalcintepe, J., Schmidts, A., Becker, H., Follo, M., Engelhardt, M., et al. (2012). Cell Cycle Control in Acute Myeloid Leukemia. Am. J. Cancer Res. 2, 508–528.
Schnerch, D., Follo, M., Krohs, J., Felthaus, J., Engelhardt, M., and Wäsch, R. (2012). Monitoring APC/C Activity in the Presence of Chromosomal Misalignment in Unperturbed Cell Populations. Cell Cycle 11, 310–321. doi:10.4161/cc.11.2.18737
Schnerch, D., Schüler, J., Follo, M., Felthaus, J., Wider, D., Klingner, K., et al. (2017). Proteasome Inhibition Enhances the Efficacy of Volasertib-Induced Mitotic Arrest in AML In Vitro and Prolongs Survival In Vivo. Oncotarget 8, 21153–21166. doi:10.18632/oncotarget.15503
Schrock, M. S., Stromberg, B. R., Scarberry, L., and Summers, M. K. (2020). APC/C Ubiquitin Ligase: Functions and Mechanisms in Tumorigenesis. Seminars Cancer Biol. 67, 80–91. doi:10.1016/j.semcancer.2020.03.001
Scully, R. (2010). The Spindle-Assembly Checkpoint, Aneuploidy, and Gastrointestinal Cancer. N. Engl. J. Med. 363, 2665–2666. doi:10.1056/NEJMe1008017
Shi, J., Orth, J. D., and Mitchison, T. (2008). Cell Type Variation in Responses to Antimitotic Drugs that Target Microtubules and Kinesin-5. Cancer Res. 68, 3269–3276. doi:10.1158/0008-5472.CAN-07-6699
Sinnott, R., Winters, L., Larson, B., Mytsa, D., Taus, P., Cappell, K. M., et al. (2014). Mechanisms Promoting Escape from Mitotic Stress-Induced Tumor Cell Death. Cancer Res. 74, 3857–3869. doi:10.1158/0008-5472.CAN-13-3398
Skaar, J. R., and Pagano, M. (2008). Cdh1: a Master G0/G1 Regulator. Nat. Cell Biol. 10, 755–757. doi:10.1038/ncb0708-755
Skinner, J. J., Wood, S., Shorter, J., Englander, S. W., and Black, B. E. (2008). The Mad2 Partial Unfolding Model: Regulating Mitosis through Mad2 Conformational Switching. J. Cell Biol. 183, 761–768. doi:10.1083/jcb.200808122
Sørensen, C. S., Lukas, C., Kramer, E. R., Peters, J.-M., Bartek, J., and Lukas, J. (2000). Nonperiodic Activity of the Human Anaphase-Promoting Complex-Cdh1 Ubiquitin Ligase Results in Continuous DNA Synthesis Uncoupled from Mitosis. Mol. Cell Biol. 20, 7613–7623. doi:10.1128/MCB.20.20.7613-7623.2000
Stukenberg, P. T., and Burke, D. J. (2015). Connecting the Microtubule Attachment Status of Each Kinetochore to Cell Cycle Arrest through the Spindle Assembly Checkpoint. Chromosoma 124, 463–480. doi:10.1007/s00412-015-0515-z
Sudakin, V., Chan, G. K. T., and Yen, T. J. (2001). Checkpoint Inhibition of the APC/C in HeLa Cells Is Mediated by a Complex of BUBR1, BUB3, CDC20, and MAD2. J. Cell Biol. 154, 925–936. doi:10.1083/jcb.200102093
Sudo, T., Ota, Y., Kotani, S., Nakao, M., Takami, Y., Takeda, S., et al. (2001). Activation of Cdh1-dependent APC Is Required for G1 Cell Cycle Arrest and DNA Damage-Induced G2 Checkpoint in Vertebrate Cells. EMBO J. 20, 6499–6508. doi:10.1093/emboj/20.22.6499
Sullivan, M., and Morgan, D. O. (2007). Finishing Mitosis, One Step at a Time. Nat. Rev. Mol. Cell Biol. 8, 894–903. doi:10.1038/nrm2276
Tavormina, P. A., Come, M.-G., Hudson, J. R., Mo, Y.-Y., Beck, W. T., and Gorbsky, G. J. (2002). Rapid Exchange of Mammalian Topoisomerase IIα at Kinetochores and Chromosome Arms in Mitosis. J. Cell Biol. 158, 23–29. doi:10.1083/jcb.200202053
Topham, C. H., and Taylor, S. S. (2013). Mitosis and Apoptosis: How Is the Balance Set? Curr. Opin. Cell Biol. 25, 780–785. doi:10.1016/j.ceb.2013.07.003
Wang, L., Zhang, J., Wan, L., Zhou, X., Wang, Z., and Wei, W. (2015). Targeting Cdc20 as a Novel Cancer Therapeutic Strategy. Pharmacol. Ther. 151, 141–151. doi:10.1016/j.pharmthera.2015.04.002
Wäsch, R., and Cross, F. R. (2002). APC-dependent Proteolysis of the Mitotic Cyclin Clb2 Is Essential for Mitotic Exit. Nature 418, 556–562. doi:10.1038/nature00856
Wäsch, R., and Engelbert, D. (2005). Anaphase-Promoting Complex-dependent Proteolysis of Cell Cycle Regulators and Genomic Instability of Cancer Cells. Oncogene 24, 1–10. doi:10.1038/sj.onc.1208017
Wäsch, R., Robbins, J. A., and Cross, F. R. (2010). The Emerging Role of APC/CCdh1 in Controlling Differentiation, Genomic Stability and Tumor Suppression. Oncogene 29, 1–10. doi:10.1038/onc.2009.325
Wäsch, R. (2011). Targeting Mitotic Exit for Cancer Treatment. Expert Opin. Ther. Targets 15, 785–788. doi:10.1517/14728222.2011.577420
Weaver, B. A. (2014). How Taxol/Paclitaxel Kills Cancer Cells. MBoC 25, 2677–2681. doi:10.1091/mbc.E14-04-0916
Weyburne, E. S., Wilkins, O. M., Sha, Z., Williams, D. A., Pletnev, A. A., de Bruin, G., et al. (2017). Inhibition of the Proteasome β2 Site Sensitizes Triple-Negative Breast Cancer Cells to β5 Inhibitors and Suppresses Nrf1 Activation. Cell Chem. Biol. 24, 218–230. doi:10.1016/j.chembiol.2016.12.016
Wheeler, L. W., Lents, N. H., and Baldassare, J. J. (2008). Cyclin A-CDK Activity during G1 Phase Impairs MCM Chromatin Loading and Inhibits DNA Synthesis in Mammalian Cells. Cell Cycle 7, 2179–2188. doi:10.4161/cc.7.14.6270
Wirth, K. G., Ricci, R., Giménez-Abián, J. F., Taghybeeglu, S., Kudo, N. R., Jochum, W., et al. (2004). Loss of the Anaphase-Promoting Complex in Quiescent Cells Causes Unscheduled Hepatocyte Proliferation. Genes Dev. 18, 88–98. doi:10.1101/gad.285404
Wohlschlegel, J. A., Dwyer, B. T., Dhar, S. K., Cvetic, C., Walter, J. C., and Dutta, A. (2000). Inhibition of Eukaryotic DNA Replication by Geminin Binding to Cdt1. Science 290, 2309–2312. doi:10.1126/science.290.5500.2309
Yuan, X., Srividhya, J., De Luca, T., Lee, J.-h. E., and Pomerening, J. R. (2014). Uncovering the Role of APC-Cdh1 in Generating the Dynamics of S-phase Onset. MBoC 25, 441–456. doi:10.1091/mbc.E13-08-0480
Zasadil, L. M., Andersen, K. A., Yeum, D., Rocque, G. B., Wilke, L. G., Tevaarwerk, A. J., et al. (2014). Cytotoxicity of Paclitaxel in Breast Cancer Is Due to Chromosome Missegregation on Multipolar Spindles. Sci. Transl. Med. 6, 229ra43. doi:10.1126/scitranslmed.3007965
Zeidan, A. M., Ridinger, M., Lin, T. L., Becker, P. S., Schiller, G. J., Patel, P. A., et al. (2020). A Phase Ib Study of Onvansertib, a Novel Oral PLK1 Inhibitor, in Combination Therapy for Patients with Relapsed or Refractory Acute Myeloid Leukemia. Clin. Cancer Res. 26, 6132–6140. doi:10.1158/1078-0432.CCR-20-2586
Zeng, X., Sigoillot, F., Gaur, S., Choi, S., Pfaff, K. L., Oh, D.-C., et al. (2010). Pharmacologic Inhibition of the Anaphase-Promoting Complex Induces a Spindle Checkpoint-dependent Mitotic Arrest in the Absence of Spindle Damage. Cancer Cell 18, 382–395. doi:10.1016/j.ccr.2010.08.010
Zeng, X., Xu, W. K., Lok, T. M., Ma, H. T., and Poon, R. Y. C. (2019). Imbalance of the Spindle-Assembly Checkpoint Promotes Spindle Poison-Mediated Cytotoxicity with Distinct Kinetics. Cell Death Dis. 10, 1–15. doi:10.1038/s41419-019-1539-8
Zhang, D., Yin, S., Jiang, M.-X., Ma, W., Hou, Y., Liang, C.-G., et al. (2007). Cytoplasmic Dynein Participates in Meiotic Checkpoint Inactivation in Mouse Oocytes by Transporting Cytoplasmic Mitotic Arrest-Deficient (Mad) Proteins from Kinetochores to Spindle Poles. Reproduction 133, 685–695. doi:10.1530/rep.1.01167
Zhang, J., Wan, L., Dai, X., Sun, Y., and Wei, W. (2014). Functional Characterization of Anaphase Promoting Complex/Cyclosome (APC/C) E3 Ubiquitin Ligases in Tumorigenesis. Biochimica Biophysica Acta (BBA) - Rev. Cancer 1845, 277–293. doi:10.1016/j.bbcan.2014.02.001
Zhang, Y., Li, J., Yi, K., Feng, J., Cong, Z., Wang, Z., et al. (2019). Elevated Signature of a Gene Module Coexpressed with CDC20 Marks Genomic Instability in Glioma. Proc. Natl. Acad. Sci. U.S.A. 116, 6975–6984. doi:10.1073/pnas.1814060116
Keywords: mitotic exit, APC/C, Cdh1, mitotic slippage, spindle assembly checkpoint, antimitotic therapy
Citation: Greil C, Engelhardt M and Wäsch R (2022) The Role of the APC/C and Its Coactivators Cdh1 and Cdc20 in Cancer Development and Therapy. Front. Genet. 13:941565. doi: 10.3389/fgene.2022.941565
Received: 11 May 2022; Accepted: 08 June 2022;
Published: 27 June 2022.
Edited by:
Meenakshi Maitra, The University of Texas at Dallas, United StatesReviewed by:
Matthew Wong, Children’s Cancer Institute Australia, AustraliaCopyright © 2022 Greil, Engelhardt and Wäsch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ralph Wäsch, ralph.waesch@uniklinik-freiburg.de