
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 22 July 2022
Sec. Human and Medical Genomics
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.940776
This article is part of the Research Topic The Genetics of Neurodevelopmental Disorders View all 11 articles
 Yu Ding1†
Yu Ding1† Jiande Chen2†
Jiande Chen2† Yijun Tang1Li-Na Chen3
Yijun Tang1Li-Na Chen3 Ru-En Yao3,4,5
Ru-En Yao3,4,5 Tingting Yu3,4,5
Tingting Yu3,4,5 Yong Yin2
Yong Yin2 Xiumin Wang1
Xiumin Wang1 Jian Wang3,4,5*
Jian Wang3,4,5* Niu Li3,4,5*
Niu Li3,4,5*SOX11 is a transcription factor belonging to the sex determining region Y-related high-mobility group box family that plays a vital role in early embryogenesis and neurogenesis. De novo variants in SOX11 have been initially reported to cause a rare neurodevelopmental disorder, mainly referred to Coffin-siris syndrome 9 (CSS9, OMIM# 615866) which is characterized with growth deficiency, intellectual disability (ID), microcephaly, coarse facies, and hypoplastic nails of the fifth fingers and/or toes. A recent large-scale cohort study suggests that SOX11 variation would result in a clinically and molecularly distinct disease from CSS. Here, we describe three unrelated Chinese cases with variable phenotype, mainly involving developmental delay, ID, short statute, microcephaly, facial deformities (i.e., prominent forehead, arched eye brow, flat nasal bridge, broad nose and short philtrum), and cryptorchidism. Whole-exome sequencing (WES) revealed three novel heterozygous variants in the SOX11 gene, including two missense variants of c.337T>C (p.Y113H) and c.425C>G (p.A142G), and one nonsense variant of c.820A>T (p. K142*). Luciferase reporting assay shows that the two missense variants impair the transcriptional activity of the SOX11 target gene GDF5. Additionally, WES uncovered a 4,300 kb deletion involving the region of 1q24.2-q25.1 (hg19,chr1:169,433,149-173,827,682) in patient 1, which also contributes to the condition of the patient. In summary, this is the first report of Chinese cases with de novo variants of SOX11. Our study partially supports the previous observation that the phenotype caused by SOX11 variants somewhat differs from classical CSS.
The sex determining region Y (SRY)-related high-mobility group (HMG) box (SOX) family encodes a group of transcription factors that play essential roles in cell fate decisions during many developmental processes (Sarkar and Hochedlinger, 2013). Thus far, a total of eight SOX subgroups (A, B1/B2, C, D, E, F, G, and H) with twenty members have been identified, all of which harbor a highly conserved DNA-binding HMG domains (Bowles et al., 2000). It is well known that dysfunction of the SOX proteins plays a critical role in the occurrence and development of multiple malignant tumors (i.e., hepatocellular carcinoma) via transcriptional activation or suppression of distinct downstream targets or signaling pathways (de la Rocha et al., 2014; Grimm et al., 2020; Luo et al., 2022). In addition, germline variants in twelve of SOX members can lead to kinds of human genetic disorders, including SRY (OMIM# 480000), SOX2 (OMIM# 184429), SOX3 (OMIM# 313430), SOX4 (OMIM# 184430), SOX5 (OMIM# 604975), SOX6 (OMIM# 607257), SOX8 (OMIM# 605923), SOX9 (OMIM# 608160), SOX10 (OMIM# 602229), SOX11 (OMIM# 600898), SOX17 (OMIM# 610928), and SOX18 (OMIM# 601618) (Angelozzi and Lefebvre, 2019).
The SOX11 gene belongs to subgroup C of SOX family, which locates at 2p25.2 and contains only one exon to encode a small protein comprising 441 amino acids (NP _003099.1; UCSC database, http://genome.ucsc.edu). In 2014, Tsurusaki et al. first demonstrated that de novo variants in SOX11 can result in Coffin-siris syndrome 9 (CSS9, OMIM#615866) in two patients and animal models (Tsurusaki et al., 2014). CSS is usually characterized with growth deficiency, intellectual disability (ID), microcephaly, coarse facies, and hypoplastic nails of the fifth fingers and/or toes (Kosho et al., 2014; Sokpor et al., 2017). In 2016, Hempel et al. confirmed the neural phenotype related to CSS9 in ten patients with de novo single nucleotide variants (SNVs) of SOX11 or microdeletions of the chromosome 2p25.2 containing SOX11 (Hempel et al., 2016). Along with the identification of new patients, several new clinical features were reported, including coarctation of the aorta (Okamoto et al., 2018), cleft palate (Khan et al., 2018), and glaucoma (Diel et al., 2021). More recently, a large cohort study revealed that developmental delay (DD) or ID, microcephaly, short stature, and low body weight were common characteristics in patients with SOX11 variants. In addition, ocular malformations (oculomotor apraxia, coloboma, and microphthalmia) and hypogonadotropic hypogonadism were also reported in patients with SOX11 variation (Al- Jawahiri et al., 2022). These studies indicate phenotypic complexity from SOX11 variation, and a full understanding of such disease requires the accumulation of additional cases from different ethnic groups.
Here, we reported three unrelated Chinese patients with distinct de novo variants in SOX11 gene which were identified by whole-exome sequencing (WES). Phenotypic analysis showed that most of the clinical features of the three patients were consistent with those reported, but there were still some differences. Additionally, the impact of the two missense variants (Y113H and A142G) on SOX11 protein was investigated by in vitro experiments.
Three unrelated patients (two males and one female) born to physically healthy and non-consanguineous parents were enrolled in this study. Written informed consent has been obtained from each family.
WES was performed in all three patients as previously described (Wang et al., 2014; Wang et al., 2016). Briefly, genomic DNA was extracted from patients’ peripheral blood and was then sheared to create fragments of 150 to 200 bp. Sequencing library was prepared using the SureSelect XT Human All Exon V6 kit (Agilent Technologies, Santa Clara, CA, United States), and sequencing was performed by the Illumina NovaSeq 6000 System (Illumina, San Diego, CA, United States). After base calling, quality assessment, and alignment of the sequence reads to the reference human genome (GRCh37, dbSNP135), all single nucleotide variants (SNVs) and indels were saved as VCF format file which was then uploaded to the QIAGEN Clinical Insight (QCI) Interpret Translational tool (https://apps.qiagenbioinformatics.cn/) for filtering and annotation. The SOX11 variants identified by WES were validated by Sanger sequencing using the ABI 3700 sequencer (Applied Biosystems, Foster City, CA, United States), in indicated patient and their parents (sequencing primers were available upon request).
HEK293T cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% (v/v) fetal bovine serum (Sigma-Aldrich, St. Louis, MO, United States) and 1% penicillin/streptomycin (Thermo Fisher Scientific, Waltham, MA, United States) in a 5% CO2 incubator at 37°C.
The wild-type (WT) and mutant SOX11 cDNA were synthesized by BGI (Shenzhen, China), and were then cloned into pcDNA3.1(+)-N-MYC vector, respectively. The GDF5 promoter 5′-flanking sequence (−448/+319) was synthesized and cloned into the pGL3-basic vector. Plasmid DNA was transfected into cells using Lipofectamine™ LTX reagent (Thermo Fisher Scientific).
HEK293T cells were cultured on cover slips in 12-well plate at 80% confluence prior for transfection. 24 h after transfection with the WT or mutant SOX11 constructs, cells were washed by 1 x PBS and fixed using 4% paraformaldehyde for 15 min at room temperature. Samples were then washed with 1 × PBS three times and blocked in the blocking buffer (1x PBS/5% goat serum/0.3% Triton X-100) for 1 h. Coverslips were incubated with mouse anti-Myc monoclonal antibody (Cell Signaling Technology, Danvers, MA, United States) at 4°C overnight. The cover slips were then mounted on microscope slides using ProLong® Gold Antifade reagent with DAPI (Cell Signaling Technology) and analyzed using a Leica DM6000 fluorescence microscope (Leica Microsystems, Wetzlar, Germany).
HEK293T cells were seeded into 6-well plate and transfected with the indicated SOX11 constructs. 24 h after transfection, cells were washed with ice-cold 1x PBS and lysed with SDS lysis buffer (100 mM pH6.8 Tris-HCl, 10% Glycerol, 1% SDS). Whole cell lysates were separated on 10% SDS-PAGE gels, transferred to polyvinylidene difluoride (PVDF) membranes. After blocking in 5% non-fat milk in TBS-T for 1 h, membranes were then incubated overnight at 4°C with mouse anti-Myc monoclonal antibody (Cell Signaling Technology) and mouse anti-beta-Actin monoclonal antibody (Sigma-Aldrich). Proteins were detected using a chemiluminescence system with a horseradish peroxidase conjugated secondary antibody.
GDF5-luc plasmid was used to monitor the regulatory role of SOX11 in modulating the transcription. Briefly, HEK293T cells were seeded into 96-well plate at a density of 3×104 per well. 24 h after culture, the cells were co-transfected with 28 ng GDF5-luc, 2 ng pRL-SV40, and 70 ng control or SOX11 expression vectors. 24 h after transfection, luciferase activity was determined by the Dua-Glo luciferase assay system (Promega Corporation, Madison, WI, United States), and normalized to renilla luciferase.
Patient 1 was a male who was born at full-term gestation with a birth weight of 2100 g (−3.1 SD) by spontaneous vaginal delivery. At the stage of pregnancy, intrauterine growth restriction (IUGR) was noticed. At 3-year old, he was referred to our hospital due to growth retardation. Physical examination showed a height of 79 cm (−4.7 SD) and weight of 9.6 kg (−3.3 SD). His hair was sparse and the anterior hairline was high. He presented with facial deformities consisting of prominent forehead, arched eye brows, flat nasal bridge and broad nose, low-set ears, short philtrum, and micrognathia (Figure 1A). He had left cryptorchidism, short fingers, and clinodactyly of the 5th finger. In addition, finger joint laxity was also noticed in the patient (Figure 1B). X-ray showed delay bone age which was 1.5-year old. No DD or ID was found in the patient. During the last follow up, his parents denied the child have mental development problems and refused further assessment, though learning difficulties were mentioned. No special was recorded for the development of the gross motor skills of the patient. The levels of growth hormone, IGF-1, IGF-BP3, LH and FSH were normal. Cranial magnetic resonance imaging (MRI) showed his pituitary gland was thin and oblate. Abdominal ultrasound was normal.
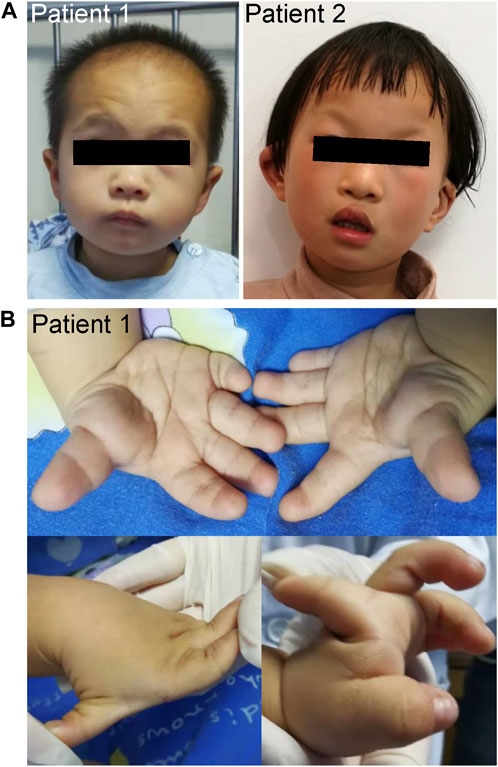
FIGURE 1. Clinical characteristics of the patients. (A) Facial photographs of patients 1 and 2. (B) Patient 1 had short fingers, clinodactyly of the fifth finger, and abnormal knuckles activity.
Patient 2 was a 5.5-year old female who was born at full-term gestation with birth weight of 3500 g (0.7 SD) and birth length of 50 cm (0.2 SD) by cesarean section. She was able to sit at 6-month old and walk alone at 20-month old. At the age of 5-year old, she was found with severe speech impairment and moderate ID. Physical examination showed a height of 113 cm (−0.1 SD) and weight of 18.7 kg (−0.3 SD). She had prominent forehead, high hairline, sparse scalp hair, arched eye brows, short philtrum, flat nasal bridge and broad nose, hypoplastic right nasal alar, auricle malformation, everted upper lip, and cleft palate and lip (Figure 1A). X-ray of the bone age, cranial MRI, and the abdominal ultrasound were normal.
Patient 3 was an 8-year old male patient born at full-term gestation by cesarean section. He was admitted to the Endocrinology department due to a slow increase in height, which was less than 4 cm/year for 4 years. His birth weight was 3200 g (−0.3 SD) and birth length was 49 cm (−0.8 SD). Physical examination showed a height of 118 cm (−2.2 SD) and weight of 17 kg (−2.5 SD). He had prominent forehead, high hairline, flat nasal bridge, and bilateral cryptorchidism. Specialist assessment suggested he had DD and mild ID. X-ray showed a two-year delay in his bone age. The levels of growth hormone, IGF-1, IGF-BP3, LH and FSH were normal. Testosterone levels had a good response after HCG stimulation, increasing from less than 0.01–0.28 ng/ml. The cranial MRI and abdominal ultrasound were normal.
We compared the clinical phenotypes of our patients with those previously reported (Tsurusaki et al., 2014; Hempel et al., 2016; Khan et al., 2018; Okamoto et al., 2018; Sekiguchi et al., 2019; Diel et al., 2021; Wakim et al., 2021; Al- Jawahiri et al., 2022; Cho et al., 2022), which have been summarized by Al-Jawahiri et al. (Al- Jawahiri et al., 2022). Although the clinical features of the three Chinese patients were variable, they still fell within the spectrum of SOX11 syndrome (Table 1). In summary, patients 1 and 2 had highly consistent facial deformities, comprising of prominent forehead, arched eye brow, flat nasal bridge, broad nose, short philtrum, and abnormal ears. In comparison, the facial deformity of patient 3 was much milder. Sparse scalp hair and high hairline were also noticed in both patients 1 and 2. Patients 2 and 3 had DD and ID, and both male patients had short stature and cryptorchidism. In addition, patient 1 harbored the 5th finger clinodactyly and microcephaly was only found in patient 3.
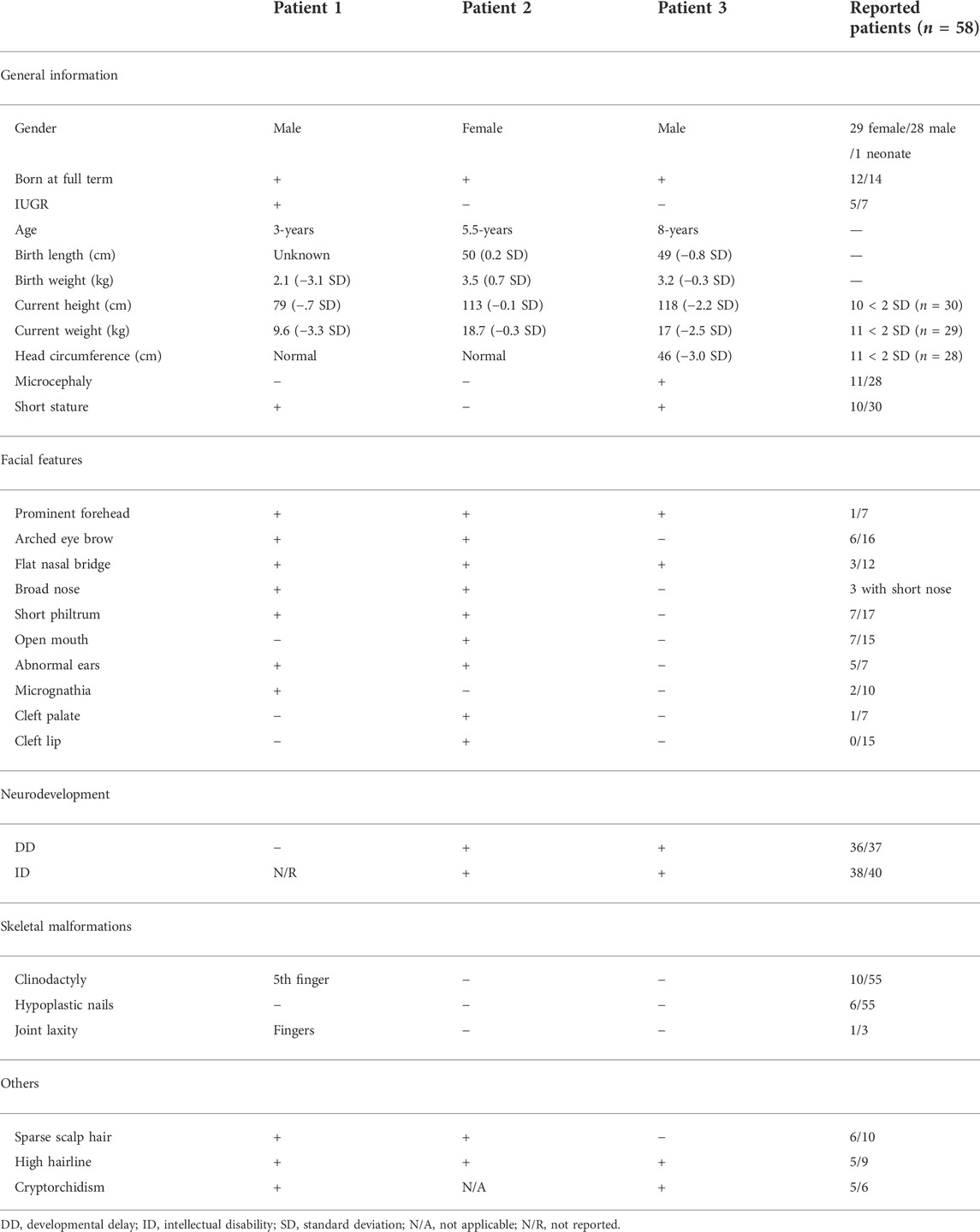
TABLE 1. Phenotypic comparison of our patient with reported patients.
WES identified three heterozygous SOX11 variants, including the missense variant of c.425C>G, p.A142G in patient 1, the nonsense variant of c.820A>T, p.K274* in patient 2, and the missense variant of c.337T>C, p.Y113H in patient 3. Sanger sequencing confirmed these variants in the patients (Figure 2A), and also revealed the de novo status of each variant from the results that their parents were WT in SOX11 gene. All of the three variants were not reported in previous cases and not included in the known public database (i.e., gnomAD, HGMD and ClinVar), suggesting they were novel.
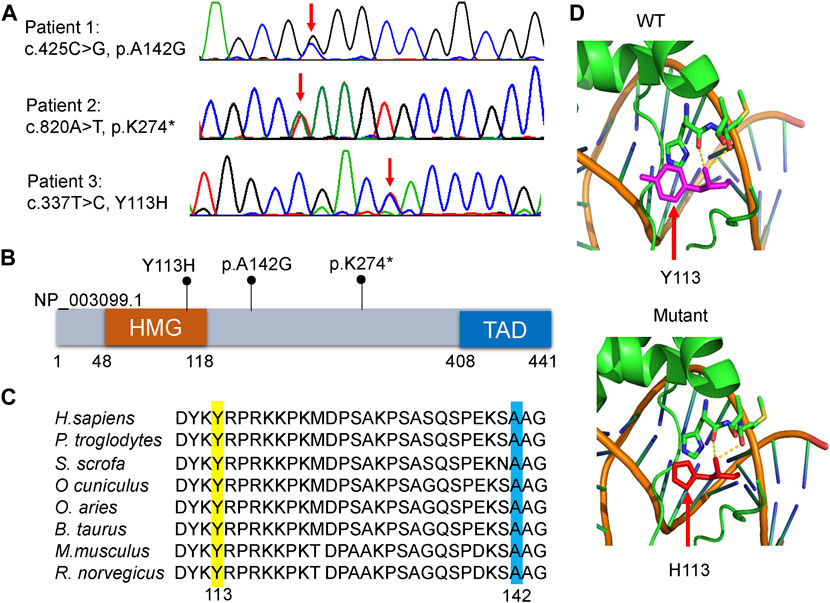
FIGURE 2. Molecular characteristics of the patients. (A) Sanger sequencing revealed that each patient harbored a heterozygous variant in SOX11 gene (NM_003108.4). Red arrows indicate the variant base. (B) Distribution schematic of the three variants of SOX11 gene identified in this study. Of them, the variant of Y113H localize to the high-mobility group (HMG) domain. TAD, transactivating domain. (C) Inter-species amino acid sequence alignment to show the missense variants of Y113H (yellow) and A142G (blue) within a highly conserved region of the protein. (D) Solved and predicted three-dimensional models of WT and mutant (Y113H) SOX11. The crystal structure was simulated using the mouse Sox4 HMG domain.
The K274* is not located in the last exon or within the 3′-most 50 nucleotides of SOX11, so it is very likely to lead to nonsense-mediated mRNA decay (Abou Tayoun et al., 2018). The Y113 residue locates at the HMG domain of SOX11 protein (Figure 2B), which is highly conserved in multiple species (Figure 2C). Crystal structure analysis using the mouse Sox4 HMG domain showed the Y113H variant may not alter the secondary structure of SOX11, but is likely to affect its ability to bind DNA (Figure 2D). By contrast, the other two variants of A142G and K274* locate downstream of the HMG domain (Figure 2B), and the A142 residue is also highly conserved (Figure 2C). In addition, the two missense variants of Y113H and A142G are predicted to be deleterious according to various in silico tools including PolyPhen-2, MutationTaster2, CADD, and ClinPred (Table 2).
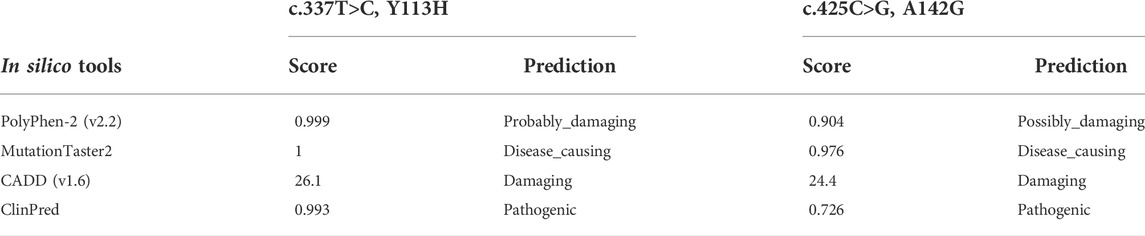
TABLE 2. Pathogenicity Predictions for Y113H and A142G variants of SOX11.
To further evaluate the impact of the Y113H and A142G variants on SOX11 function, we first performed luciferase assay using a reporter construct containing a fragment of GDF5 promoter which has been described previously (Tsurusaki et al., 2014). Compared to the with WT SOX11, both Y113H and A142G mutants result in decreased transcriptional activities (Figure 3A). In vitro experiments in HEK293T cells revealed that both missense variants increased SOX11 expression (Figure 3B), which were also observed in previous studies (Tsurusaki et al., 2014; Hempel et al., 2016). Immunofluorescence analysis showed that the WT and two missense mutant SOX11 protein localized in the nucleus (Figure 3C).
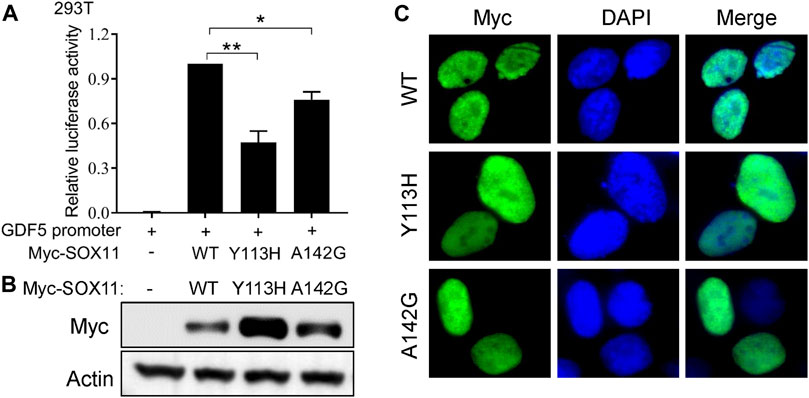
FIGURE 3. Functional study of the SOX11 missense variants. (A) Transcriptional activity of the GDF5 promoter was determined by luciferase reporter assays in HEK293T cells after co-transfection of the WT or the mutant Myc-SOX11 and the GDF5 promoter reporter construct. Data are presented as mean values ±s.e.m. from three independent experiments. *p < 0.05, **p < 0.01, two-tailed Student’s t-test. (B) The expression level of the WT and the mutant (Y113H and A142G) Myc-SOX11 were evaluated by immunoblotting in HEK293T cells. (C) Immunofluorescence analysis to show the subcellular localization of the WT and the mutant (Y113H and A142G) Myc-SOX11.
It is estimated that near 5% of patients can have more than one molecular findings, and may cause more serious phenotypic features (Posey et al., 2017). We routinely analyze copy number variation (CNV) of each patient by comparing the read-depth with the WES data from the other samples of the same batch (Yao et al., 2017; Yao et al., 2019). As shown in Figure 4A, patient 1 harbored a 4,300 kb deletion involving the region of 1q24.2-q25.1 (hg19,chr1:169,433,149-173,827,682), which includes multiple disease-causing genes, such as EEF1AKNMT (OMIM#617987), FASLG (OMIM#134638), FM O 3 (OMIM#136132), GORAB (OMIM#607983), MYOC (OMIM#601652), PIGC (OMIM#601730), PRRX1 (OMIM#167420), SLC19A2 (OMIM#603941), and TNFSF4 (OMIM# 603,594) (Figure 4B). Deletion of this genomic region has been described in series of patients (Burkardt et al., 2011; Chatron et al., 2015). According to the clinical interpretation of CNV by the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen) (Riggs et al., 2020), the microdeletion of 1q24.2-q25.1 in this patient was classified as pathogenic. No questionable CNVs were identified in the other two patients.
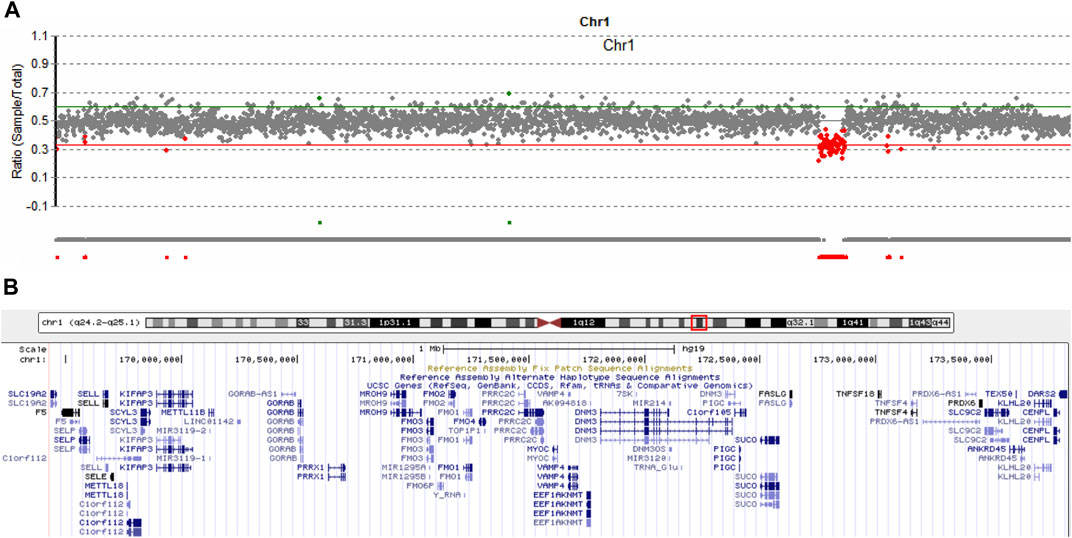
FIGURE 4. Copy number variation analysis in patient 1. (A) Sequencing depth analysis using the WES data showed patient 1 harbored 1q24.2-q25.1 deletion (hg19, chr1:169,433,149-173,827,682). (B) Involving genes in the 1q24.2-q25.1 region.
In this study, we reported three novel SOX11 variants (Y113H, A142G, and K274*) in Chinese patients. According to the clinical interpretation of genetic variants by the ACMG/AMP 2015 guideline (Richards et al., 2015), The K274* nonsense variant was classified as pathogenic (PVS1+PS2+PM2), In vitro experiments show that Y113H and A142G has no effect on the nuclear localization of SOX11 protein, but impairs the transcriptional activity of the target gene GDF5. Therefore, the two missense variants are also classified as pathogenic (PS2+PS3+PM2+PP3). Interestingly, rare missense variants in SOX11 appear to increase its protein expression levels. Such observation is particularly pronounced when the missense variants locate in the HMG domain, including the Y113H variant in our patient and the previously reported variants of K50N, S60P, Y116C and P120H (Tsurusaki et al., 2014; Hempel et al., 2016). It is noticed that variants in the HMG domain more severely impair the transcriptional regulatory activity of SOX11 than outside the HMG domain (A142G in this study and A176E in ref. 14). These results suggest that the increased protein expression caused by missense variants in HMG domain is a compensatory effect after functional loss. We here provide functional evidence to explain the phenomenon observed by Al-Jawahiri et al. in the gnomAD database that there are far fewer missense variants in the HMG box than outside the HMG domain, including the N-terminal, central, and transactivating domains in SOX11 (Al- Jawahiri et al., 2022).
In addition to the A142G variant in SOX11, patient 1 yet harbored microdeletion of 1q24.2-q25.1, which has been described in multiple patients (Burkardt et al., 2011; Chatron et al., 2015). The clinical features resulting from this CNV highly overlap with SOX11-related syndrome. For example, Chatron et al. summarized 18 patients with 1q24-q25 deletions and suggested that common clinical phenotypes include IUGR, short stature, microcephaly, delayed bone age, ID, hypertelorism, dysplastic ears, micro- and retrognathia, short hands and feet, brachydactyly, and fifth finger clinodactyly (Chatron et al., 2015). At the moment, it was difficult to determine the contribution of SOX11 A142 variant and 1q24-q25 deletion to the phenotype of patient 1.
SOX11 is widely expressed and has an important regulatory role in tissue remodelling during early embryogenesis. Conventional knockout of Sox11 in mice lead to birth death with severe developmental defects, involving in the malformations in brain, skeleton, eyes, spleen, lung, stomach, pancreas, and heart (Sock et al., 2004; Kato et al., 2015). In particular, SOX11 is critical for neurogenesis, as it can drive the differentiation of embryonic stem cells into neuronal progenitor cells and the generation of mature neurons and glial cells (Bergsland et al., 2011; Kavyanifar et al., 2018). Such roles of SOX11 in neurodevelopment have been validated in Sox11 conditional knockout mice (Wang et al., 2013), sox11 knockdown xenopus laevis (Hempel et al., 2016), and the SOX11+/− heterozygous human embryonic stem cell models (Turan et al., 2019). These molecular mechanism studies can well explain that patients with SOX11 variants are mainly manifested as DD and/or ID. Consistently, patients 2 and 3 in this study had both DD and ID. However, patients with neither DD nor ID were also reported in previous studies (i.e., case 9 who harbored H109P missense variant in the HMG domain in ref. 14), which may also be true in patient 1 of this study. These indicate a high degree of heterogeneity in clinical features caused by SOX11 variants.
Typical CSS, which is also referred to as BAFopathy, is caused by variants in the subunit of BAF complex, including ARID1A (OMIM#603024), ARID1B (OMIM#614556), SMARCA4 (OMIM#603254), SMARCB1 (OMIM#601607), ARID2 (OMIM#609539), and SMARCE1 (OMIM#603111) (Bogershausen and Wollnik, 2018). Due to the clinical phenotypic similarity, SOX11 was considered as the new causative gene of CSS (Tsurusaki et al., 2014; Hempel et al., 2016). However, with the number of patients increased, the differences between patients with SOX11 variation and CSS gradually became apparent. Al-Jawahiri et al. revealed that SOX11 syndrome has distinct clinical features from ARID1B-related CSS. Firstly, SOX11 mutant patients with microcephaly tend to be associated with oculomotor apraxia or abnormal eye morphology, while the ARID1B-related CSS patients usually have a coarse face. Secondly, deformity of the fifth fingers is an identifiable feature of CSS but do not appear to be specific to SOX11 syndrome. Thirdly, nearly one in five of the SOX11 mutant patients had hypogonadotrophic hypogonadism which was rare reported in CSS patients. Additionally, they uncovered a distinctive pattern of blood DNA methylation in the patients with SOX11 variants, and thus generated a SOX11 variation-related episignature model which was also failed to identify the BAFopathy complex samples. These results strongly suggest SOX11 syndrome and CSS were two groups of clinically and molecularly distinct diseases (Al- Jawahiri et al., 2022). In our study, none of the patients had coarse facies and hypoplastic nails, which are the core features of CSS. Moreover, the phenotypic summary of reported patients showed only about one-third of patients had microcephaly, and one-tenth had nail dysplasia (Table 1), also suggesting differences between SOX11-related phenotypes and classical CSS.
In conclusion, our study identified three novel SOX11 variants and elucidated the damage effect of two missense variants on SOX11 protein via in vitro experiments. This is the first report of Chinese patients with SOX11-related CSS9. Our study provides new evidence to support the observation that the clinical phenotype caused by SOX11 variants is highly heterogeneous and differs from classical CSS to some extent.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Shanghai Children’s Medical Center. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
NL and JW conceived the study. YD, JC, YT, YY, and XW were responsible for recruiting patients and collection of clinical features. YD, JC, and L-NC performed the in vitro experiments. R-EY and TY were responsible for the sequencing work. JC and NL drafted the manuscript, tables, and figures. All authors read and approved the final manuscript.
This research was sponsored by the grant from Shanghai “Rising Stars of Medical Talents” Youth Development Program (NL), Shanghai Pujiang Program (20PJ1409900 to NL), and the Project of Clinical Research Plan of Shanghai Hospital Development Center (Grant No. SHDC2020CR3042B to JW).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We are deeply grateful to the patients and their families for participating in this study.
Abou Tayoun, A. N., Pesaran, T., DiStefano, M. T., Oza, A., Rehm, H. L., Biesecker, L. G., et al. (2018). Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 39 (11), 1517–1524. doi:10.1002/humu.23626
Al-Jawahiri, R., Foroutan, A., Kerkhof, J., McConkey, H., Levy, M., Haghshenas, S., et al. (2022). SOX11 variants cause a neurodevelopmental disorder with infrequent ocular malformations and hypogonadotropic hypogonadism and with distinct DNA methylation profile. Genet. Med. 24, 1261–1273. doi:10.1016/j.gim.2022.02.013
Angelozzi, M., and Lefebvre, V. (2019). SOXopathies: growing family of developmental disorders due to SOX mutations. Trends Genet. 35 (9), 658–671. doi:10.1016/j.tig.2019.06.003
Bergsland, M., Ramskold, D., Zaouter, C., Klum, S., Sandberg, R., Muhr, J., et al. (2011). Sequentially acting Sox transcription factors in neural lineage development. Genes. Dev. 25 (23), 2453–2464. doi:10.1101/gad.176008.111
Bogershausen, N., and Wollnik, B. (2018). Mutational landscapes and phenotypic spectrum of SWI/SNF-related intellectual disability disorders. Front. Mol. Neurosci. 11, 252. doi:10.3389/fnmol.2018.00252
Bowles, J., Schepers, G., and Koopman, P. (2000). Phylogeny of the SOX family of developmental transcription factors based on sequence and structural indicators. Dev. Biol. 227 (2), 239–255. doi:10.1006/dbio.2000.9883
Burkardt, D. D., Rosenfeld, J. A., Helgeson, M. L., Angle, B., Banks, V., Smith, W. E., et al. (2011). Distinctive phenotype in 9 patients with deletion of chromosome 1q24-q25. Am. J. Med. Genet. A 155A (6), 1336–1351. doi:10.1002/ajmg.a.34049
Chatron, N., Haddad, V., Andrieux, J., Desir, J., Boute, O., Dieux, A., et al. (2015). Refinement of genotype-phenotype correlation in 18 patients carrying a 1q24q25 deletion. Am. J. Med. Genet. A 167A (5), 1008–1017. doi:10.1002/ajmg.a.36856
Cho, C. Y., Tsai, W. Y., Lee, C. T., Liu, S. Y., Huang, S. Y., Chien, Y. H., et al. (2022). Clinical and molecular features of idiopathic hypogonadotropic hypogonadism in taiwan: a single center experience. J. Formos. Med. Assoc. 121 (1 Pt 1), 218–226. doi:10.1016/j.jfma.2021.03.010
de la Rocha, A. M., Sampron, N., Alonso, M. M., and Matheu, A. (2014). Role of SOX family of transcription factors in central nervous system tumors. Am. J. Cancer Res. 4 (4), 312–324.
Diel, H., Ding, C., Grehn, F., Chronopoulos, P., Bartsch, O., Hoffmann, E. M., et al. (2021). First observation of secondary childhood glaucoma in coffin-siris syndrome: a case report and literature review. BMC Ophthalmol. 21 (1), 28. doi:10.1186/s12886-020-01788-0
Grimm, D., Bauer, J., Wise, P., Kruger, M., Simonsen, U., Wehland, M., et al. (2020). The role of SOX family members in solid tumours and metastasis. Semin. Cancer Biol. 67 (Pt 1), 122–153. doi:10.1016/j.semcancer.2019.03.004
Hempel, A., Pagnamenta, A. T., Blyth, M., Mansour, S., McConnell, V., Kou, I., et al. (2016). Deletions and de novo mutations of SOX11 are associated with a neurodevelopmental disorder with features of coffin-siris syndrome. J. Med. Genet. 53 (3), 152–162. doi:10.1136/jmedgenet-2015-103393
Kato, K., Bhattaram, P., Penzo-Mendez, A., Gadi, A., and Lefebvre, V. (2015). SOXC transcription factors induce cartilage growth plate formation in mouse embryos by promoting noncanonical WNT signaling. J. Bone Min. Res. 30 (9), 1560–1571. doi:10.1002/jbmr.2504
Kavyanifar, A., Turan, S., and Lie, D. C. (2018). SoxC transcription factors: multifunctional regulators of neurodevelopment. Cell Tissue Res. 371 (1), 91–103. doi:10.1007/s00441-017-2708-7
Khan, U., Study, D., Baker, E., and Clayton-Smith, J. (2018). Observation of cleft palate in an individual with SOX11 mutation: indication of a role for SOX11 in human palatogenesis. Cleft Palate. Craniofac. J. 55 (3), 456–461. doi:10.1177/1055665617739312
Kosho, T., Miyake, N., and Carey, J. C. (2014). Coffin-siris syndrome and related disorders involving components of the BAF (mSWI/SNF) complex: historical review and recent advances using next generation sequencing. Am. J. Med. Genet. C Semin. Med. Genet. 166C (3), 241–251. doi:10.1002/ajmg.c.31415
Luo, X., Ji, X., Xie, M., Zhang, T., Wang, Y., Sun, M., et al. (2022). Advance of SOX transcription factors in hepatocellular carcinoma: From role, tumor immune relevance to targeted therapy. Cancers (Basel) 14 (5), 1165. doi:10.3390/cancers14051165
Okamoto, N., Ehara, E., Tsurusaki, Y., Miyake, N., and Matsumoto, N. (2018). Coffin-Siris syndrome and cardiac anomaly with a novel SOX11 mutation. Congenit. Anom. 58 (3), 105–107. doi:10.1111/cga.12242
Posey, J. E., Harel, T., Liu, P., Rosenfeld, J. A., James, R. A., Coban Akdemir, Z. H., et al. (2017). Resolution of disease phenotypes resulting from multilocus genomic variation. N. Engl. J. Med. 376 (1), 21–31. doi:10.1056/NEJMoa1516767
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Riggs, E. R., Andersen, E. F., Cherry, A. M., Kantarci, S., Kearney, H., Patel, A., et al. (2020). Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the american college of medical genetics and genomics (ACMG) and the clinical genome Resource (ClinGen). Genet. Med. 22 (2), 245–257. doi:10.1038/s41436-019-0686-8
Sarkar, A., and Hochedlinger, K. (2013). The sox family of transcription factors: versatile regulators of stem and progenitor cell fate. Cell Stem Cell 12 (1), 15–30. doi:10.1016/j.stem.2012.12.007
Sekiguchi, F., Tsurusaki, Y., Okamoto, N., Teik, K. W., Mizuno, S., Suzumura, H., et al. (2019). Genetic abnormalities in a large cohort of coffin-siris syndrome patients. J. Hum. Genet. 64 (12), 1173–1186. doi:10.1038/s10038-019-0667-4
Sock, E., Rettig, S. D., Enderich, J., Bosl, M. R., Tamm, E. R., Wegner, M., et al. (2004). Gene targeting reveals a widespread role for the high-mobility-group transcription factor Sox11 in tissue remodeling. Mol. Cell. Biol. 24 (15), 6635–6644. doi:10.1128/MCB.24.15.6635-6644.2004
Sokpor, G., Xie, Y., Rosenbusch, J., and Tuoc, T. (2017). Chromatin remodeling BAF (SWI/SNF) complexes in neural development and disorders. Front. Mol. Neurosci. 10, 243. doi:10.3389/fnmol.2017.00243
Tsurusaki, Y., Koshimizu, E., Ohashi, H., Phadke, S., Kou, I., Shiina, M., et al. (2014). De novo SOX11 mutations cause coffin-siris syndrome. Nat. Commun. 5, 4011. doi:10.1038/ncomms5011
Turan, S., Boerstler, T., Kavyanifar, A., Loskarn, S., Reis, A., Winner, B., et al. (2019). A novel human stem cell model for coffin-siris syndrome-like syndrome reveals the importance of SOX11 dosage for neuronal differentiation and survival. Hum. Mol. Genet. 28 (15), 2589–2599. doi:10.1093/hmg/ddz089
Wakim, V., Nair, P., Delague, V., Bizzari, S., Al-Ali, M. T., Castro, C., et al. (2021). SOX11-related syndrome: report on a new case and review. Clin. Dysmorphol. 30 (1), 44–49. doi:10.1097/MCD.0000000000000348
Wang, J., Yu, T., Wang, Z., Ohte, S., Yao, R. E., Zheng, Z., et al. (2016). A new subtype of multiple synostoses syndrome is caused by a mutation in GDF6 that decreases its sensitivity to noggin and enhances its potency as a BMP signal. J. Bone Min. Res. 31 (4), 882–889. doi:10.1002/jbmr.2761
Wang, J., Zhang, W., Jiang, H., and Wu, B. L. (2014). Mutations in HFM1 in recessive primary ovarian insufficiency. N. Engl. J. Med. 370 (10), 972–974. doi:10.1056/NEJMc1310150
Wang, Y., Lin, L., Lai, H., Parada, L. F., and Lei, L. (2013). Transcription factor Sox11 is essential for both embryonic and adult neurogenesis. Dev. Dyn. 242 (6), 638–653. doi:10.1002/dvdy.23962
Yao, R., Yu, T., Qing, Y., Wang, J., and Shen, Y. (2019). Evaluation of copy number variant detection from panel-based next-generation sequencing data. Mol. Genet. Genomic Med. 7 (1), e00513. doi:10.1002/mgg3.513
Keywords: SOX11, coffin-siris syndrome, missense variants, functional study, phenotypic differences
Citation: Ding Y, Chen J, Tang Y, Chen L-N, Yao R-E, Yu T, Yin Y, Wang X, Wang J and Li N (2022) Identification and functional analysis of novel SOX11 variants in Chinese patients with Coffin-Siris syndrome 9. Front. Genet. 13:940776. doi: 10.3389/fgene.2022.940776
Received: 10 May 2022; Accepted: 04 July 2022;
Published: 22 July 2022.
Edited by:
Xiaoli Chen, Capital Institute of Pediatrics, ChinaCopyright © 2022 Ding, Chen, Tang, Chen, Yao, Yu, Yin, Wang, Wang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Niu Li, bGluaXUwNTA5QDE2My5jb20=; Jian Wang, TGFid2FuZ2ppYW5Ac2hzbXUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.