
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 24 June 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.938814
This article is part of the Research Topic Advancing Our Understanding of the Genetic and Functional Basis of Skeletal Dysplasia View all 15 articles
 Tarık Kırkgöz1
Tarık Kırkgöz1 Behzat Özkan1Filiz Hazan2Sezer Acar1*Özlem Nalbantoğlu1Beyhan Özkaya1Melike Ataseven Kulalı3
Behzat Özkan1Filiz Hazan2Sezer Acar1*Özlem Nalbantoğlu1Beyhan Özkaya1Melike Ataseven Kulalı3 Semra Gürsoy4
Semra Gürsoy4 Shiro Ikegawa5
Shiro Ikegawa5 Long Guo5*
Long Guo5*Dysosteosclerosis (DOS) is a rare sclerosing bone dysplasia characterized by unique osteosclerosis of the long tubular bones and platyspondyly. DOS is inherited in an autosomal recessive manner and is genetically and clinically heterogeneous. To date, four individuals with DOS who have five different TNFRSF11A mutations have been reported. Based on their data, it is hypothesized that mutations producing aberrant mutant RANK proteins (missense or truncated or elongated) cause DOS, while null mutations lead to osteopetrosis, autosomal recessive 7 (OPTB7). Herein, we present the fifth case of TNFRSF11A-associated DOS with a novel homozygous frame-shift mutation (c.19_31del; p.[Arg7CysfsTer172]). The mutation is predicted to cause nonsense mutation-mediated mRNA decay (NMD) in all RANK isoform transcripts, resulting in totally null allele. Our findings suggest genotype-phenotype relationship in TNFRSF11A-associated OPTB7 and DOS remains unclear, and that the deficiency of TNFRSF11A functions might cause DOS, rather than osteopetrosis. More data are necessary to understand the phenotypic spectrum caused by TNFRSF11A mutations.
Dysosteosclerosis (DOS) is a rare form of dense bone disease characterized by osteosclerosis and platyspondyly (Spranger et al., 1968). Its features are short stature, recurrent fractures, optic atrophy, cranial nerve palsy, developmental delay, flattened fingernails, skin related complications, and failure of tooth eruption (MIM %224300). Irregular osteosclerosis, flattened diffusely dense vertebral bodies, sclerotic skull, radiolucent sub-metaphyseal portions of the long tubular bones with sclerotic diaphysis are radiological features of DOS (Houston et al., 1978; Elçioglu et al., 2002; Whyte et al., 2010; Guo et al., 2018). Moreover, metaphyseal osteosclerosis and platyspondyly are the characteristic and important guiding findings in differentiating DOS from other types of sclerosing bone dysplasia (Howaldt et al., 2019).
DOS is inherited in an autosomal recessive manner and is genetically and clinically heterogeneous. Four disease genes for DOS have been described to date (Campeau et al., 2012; Guo et al., 2018; Guo et al., 2019; Howaldt et al., 2019; Uludağ Alkaya et al., 2021). SLC29A3 (solute carrier family 29 member 3) was the first identified gene, followed by TNSFR11A (tumor necrosis factor receptor superfamily member 11a; also known as RANK (receptor activator of nuclear factor kappa B)), TCIRG1 (T cell immune regulator 1) and CSF1R (colony stimulating factor 1 receptor) (Campeau et al., 2012; Guo et al., 2018; Guo et al., 2019; Howaldt et al., 2019; Uludağ Alkaya et al., 2021). Notably, TNSFR11A and TCIRG1 mutations are also reported in osteopetrosis, autosomal recessive 7 (OPTB7) and osteopetrosis, autosomal recessive 1 (OPTB1), respectively (Frattini et al., 2000; Guerrini et al., 2008; Pangrazio et al., 2012).
To date, four DOS individuals with five different TNFRSF11A mutations (2 missense/nonsense, 2 splice-site, 1 deletion) have been reported (Guo et al., 2018; Xue et al., 2019; Xue et al., 2020; Xue et al., 2021a; Xue et al., 2021b). According to the previous hypothesis for genotype-phenotype relationship, aberrant mutant RANK proteins (missense or truncated or elongated) cause DOS, while null mutations lead to OPTB7 (Guo et al., 2018; Xue et al., 2019; Xue et al., 2020; Xue et al., 2021a). Herein, we present the fifth case of TNFRSF11A-associated DOS in a 19-month-old boy, in whom we identified a novel homozygous mutation in TNFRSF11A (c.19_31del; p.[Arg7CysfsTer172]). Unlike the previously reported DOS mutations, the mutation is predicted to cause nonsense mutation-mediated mRNA decay (NMD) in all RANK isoform transcripts, which leads us to re-consider the phenotype and genotype relationship of the TNFRSF11A mutation.
A 19-month-old boy was consulted to pediatric endocrinology unit for hypocalcemia and short stature. He was the third baby to healthy consanguineous Turkish parents who had healthy boys and two abortions. Family history was unremarkable with no affected family members. The prenatal course was uneventful. The proband was born at term by normal Cesarean section with a birth weight of 3,200 g (+0.02 standard deviation score (SDS)). He had a history of hypocalcemic convulsion at day 4, and was treated with intravenous calcium and vitamin D supplementation. The patient was discharged to home with phenobarbital therapy at day 14. From 4th month, he was hospitalized three times due to lower respiratory tract infection.
On the physical examination at age 19 months, his height, weight and head circumference were at −3.1, −2.7, and +0.2 SDSs, respectively. Midface hypoplasia, edematous eyelids, down slanting palpebral fissures, long eyelashes, long philtrum, high arched palate, low set ears, and micrognathia were detected. A prominent forehead, open anterior fontanelle (4 × 4 cm), pectus carinatum, café-au-lait spot (4 × 3 cm) on anterior thorax, and bowing of femora and ulnae were noticed. Ophthalmological examination revealed optic atrophy and horizontal nystagmus. Moderate delay in developmental milestones was observed.
On biochemical evaluation, serum calcium was 7.4 mg/dl (Normal (N): 9.0–11), phosphorus was 4.4 mg/dl (N: 4–6.5), ALP was 84 U/L (N: 116–450), albumin was 4.1 g/L (N: 3.5-5.5), PTH was 102.3 ng/L (N: 15–88), and 25-OH vitamin D was 27 μg/L (N: 30–100). Liver-kidney function tests, thyroid function tests and ions were normal. Skeletal survey showed diffuse osteosclerosis of the craniofacial, axial and appendicular skeletons, especially in diaphyseal areas of the long tubular bones (Figures 1A,D–F). In the evaluation of the vertebral structures, end plate irregularities, mildly reduced height (platyspondyly) and thoracic and lumbar scoliosis were observed (Figures 1B,C). Pelvic bones showed sclerosis, especially in the iliac bodies (Figure 1D). Calcium replacement (50 mg/kg/day) and maintenance dose vitamin D (400 U/day) treatments were started, and the calcium value was normalized within a week. After normalization of serum calcium level, the treatment was discontinued and biochemical parameters remained normal in the follow-up. After his clinical improvement at age 21 months, he was discharged to follow up in the outpatient clinic.
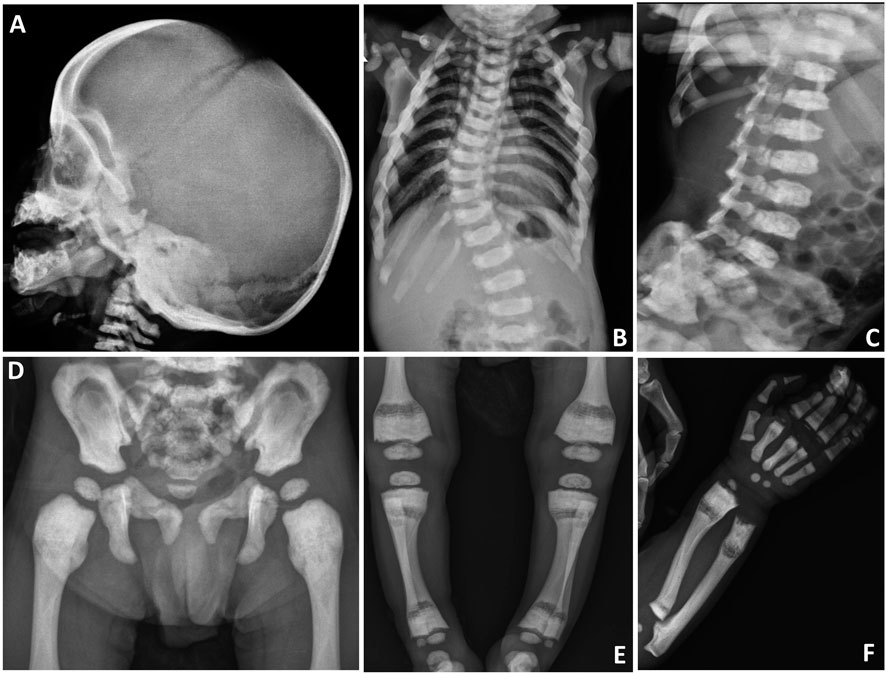
FIGURE 1. Skeletal survey of the patient. (A) Sclerosis of the craniofacial bones, especially at the skull base. (B) Scoliosis at the thoracic level. (C) Plate irregularities and mildly reduced height of the vertebral corpus (platyspondyly). (D) Sclerosis at the pelvic bones, especially in the iliac bodies. (E) Radiolucency at the metadiaphyseal junction and sclerosis at the end of long bones. (F) Sclerosis of the radius, ulna, metacarpal bones, and proximal phalanges. Radiolucency at the metadiaphyseal junction of radius and ulna is also noted.
One month after the discharge, he was hospitalized to the pediatric intensive care unit of another hospital with complaints of fever, cough, vomiting, and weight loss. In the initial evaluation, lower respiratory tract infection and sepsis were considered and appropriate fluid and antibiotic therapy was initiated. When respiratory findings worsened and respiratory acidosis became evident, he was intubated and connected to a mechanical ventilator. Cranial magnetic resonance imaging revealed severe hydrocephalus and therefore a ventro-peritoneal shunt was inserted. Despite the treatments, his clinical condition deteriorated and he died after 4 months of follow-up in the pediatric intensive care unit at age 26 months.
After written informed consents, blood samples were obtained from the patient, parents, and brothers. Genomic DNA was extracted from leukocytes using the MagPurix kit (Zinexts Life Science Corp., New Taipei City 235, Taiwan), according to manufacturer’s instructions. For the molecular genetic evaluation, a Custom Target Capture-based Osteopetrosis gene panel (Celemix Inc., Seoul, Korea), which was designed according to the before-2018 ENMC classification was used.
The variant identified by the targeted sequencing was confirmed by Sanger sequencing. The region of genome including the mutations was amplified by PCR and sequenced both strands. Exon-specific Sanger sequencing was performed using 5′-GAGCTTGGGCACCACCTG-3′ and 5′-TCCGCTCCCCAAAACTCC-3′ primers on Applied Biosystems 3500 Genetic Analyzer. The sequences were evaluated by two different sequencing programs; i.e., CLC Genomics Workbench 3 sequencing program (Qiagen) and Chromas lite Software (Technelysium, South Brisbane QLD, Australia).
The variant was evaluated by dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/), 1000 genomes (http://www.1000genomes.org/), ExAC (http://exac.broadinstitute.org/), gnomAD (http://gnomad.broadinstitute.org/), ESP6500 (http://evs.gs.washington.edu/EVS/), Human Gene Mutation Database (HGMD; http://portal.biobase‐international.com/hgmd/pro/start.php), and Mutation Taster (http://mutationtaster.org).
By the target sequence, a novel homozygous frameshift variant (NM_003839.3: c.19_31del; p.Arg7CysfsTer172) in TNFRSF11A was detected. The variant was confirmed by Sanger sequencing (Figure 2). The parents were both heterozygous for the variant (Figure 2). This variant was interpreted as “Likely Pathogenic” according to American College of Medical Genetics criteria (ACGM) (Richards et al., 2015). This variant did not exist in any available databases including dbSNP, 1000 genomes, ExAC, gnomAD, ESP6500, and HGMD. According to an in silico analysis with Mutation Taster, this variant was predicted to affect signal peptide and protein structure, and causes NMD.
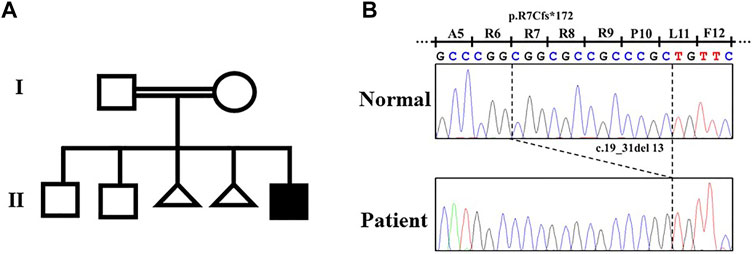
FIGURE 2. Pedigree of the family with dysosteosclerosis and the TNFRSF11A variant. (A) Pedigree. (B) Electropherograms of the Sanger sequence analysis.
DOS and OPTB7 are inherited as autosomal recessive traits and are clinically very similar. However, they can be differentiated by the remarkable radiological features of DOS, including platyspondyly and enlarged lower metaphyseal parts of tubular bones with punctate densities and radiolucency (Spranger et al., 1968; Houston et al., 1978; Elçioglu et al., 2002; Whyte et al., 2010; Guo et al., 2018). OPTB7 is caused by loss-of-function mutations of TNFRSF11A, which are missense mutations or deleterious mutations leading to NMD (Guerrini et al., 2008; Pangrazio et al., 2012; Xue et al., 2021b). TNFRSF11A-associated DOS is caused by splice-site or frameshift mutations which are capable of producing concurrent truncated or long RANK proteins (Table 1). Five TNFRSF11A isoforms encoding five different proteins have been identified (Figure 3). It has been speculated that the clinical difference of DOS and OPTB7 may relate to the different effects of TNFRSF11A mutations in different TNFRSF11A isoform transcripts (Guo et al., 2018). The variants identified in the first three reported cases of TNFRSF11A-associated DOS cause NMD in some transcript isoforms, while simultaneously producing truncated or elongated RANK proteins in the remaining transcript isoforms according to the results of RT-PCR for the patient-derived cells and the exon trapping assay for cell lines (Guo et al., 2018; Xue et al., 2019; Xue et al., 2020). The functions of these truncated or elongated RANK proteins remain unclear. In a mutant mouse model with a nine-amino-acid insertion in Tnfrsf11a, the homozygotes develop osteopetrosis while the heterozygotes show osteolytic lesions. The abnormal RANK proteins in the mutant mice accumulated in Golgi apparatus and increased osteoclastogenesis by activating the unfolded protein response (Alonso et al., 2021). The findings suggest that the truncated or elongated RANK proteins generated in the first three cases of TNFRSF11A-associated DOS probably results in gain-of-function, which would be associated with the DOS phenotype. However, in the fourth case, a single amino acid substitution in TNFRSF11A was identified, suggesting that phenotype-genotype association is not easily predictable in TNFRSF11A-related bone diseases (Table 1) (Xue et al., 2021a).
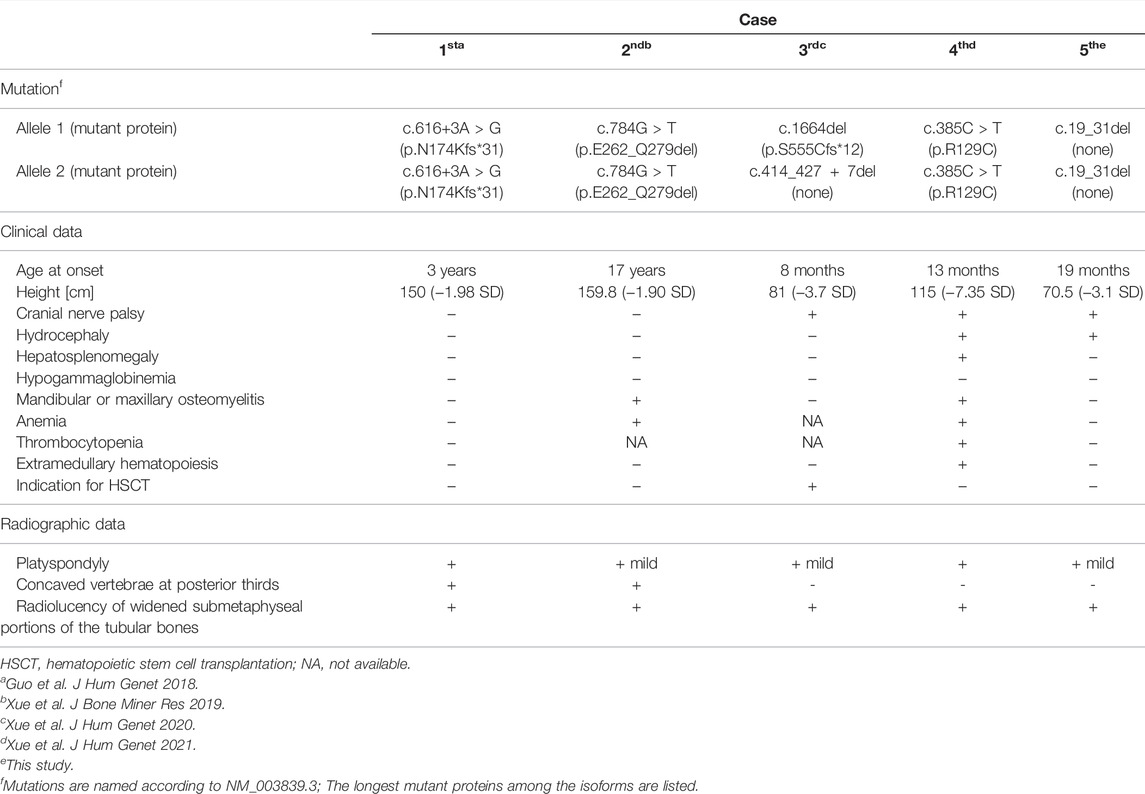
TABLE 1. Clinical and radiographic findings of TNFRSF11A-associated dysosteosclerosis.
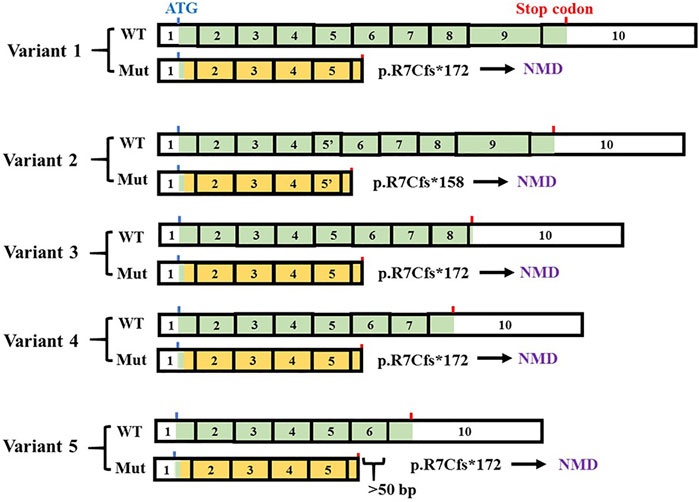
FIGURE 3. TNFRSF11A isoforms and the effect of the variant (c.19_31del). Five variant transcripts are in NCBI database (variant 1: NM_003839.3; variant 2: NM_001278268.1, variant 3: NM_001270949.1, variant 4: NM_001270950.1, variant 5: NM_001270951.1). The exons are numbered based on the longest variant (variant 1). The changed parts of coding sequence are labeled in orange. The positions of the first ATG and the stop codons were indicated by blue and red bars, respectively. c.19_31del produces no transcripts due to nonsense mutation-mediated mRNA decay (NMD).
The variant we identified in this study leads to frameshift and premature termination in exon 6 (Figure 3), thus being predicted to be a null mutation due to NMD. Contrary to the previous hypothesis, the clinical findings of our patient were quite compatible with DOS, while the variant causes NMD in all transcript isoforms (Figure 3). These conflicting findings suggest that the relationship between genotype and phenotype in TNFRSF11A-associated DOS cases is complicated and further studies are needed. It would also be interesting to explore the similar issue further for TCRIG1 (Howaldt et al., 2019).
Our study reports the first case of DOS caused by TNFRSF11A null mutations, which are previously considered to cause OPTB7. We re-evaluated the phenotypes of the OPTB7 patients with null mutations. Until now, six patients carrying seven mutations causing NMD in all or some of the transcripts have been reported (Table 2) (Guerrini et al., 2008; Pangrazio et al., 2012; Silveira et al., 2021; Xu et al., 2021). Their radiographic data critical to differentiate DOS are not publicly available, except for a case reported by (Xu et al., 2021). Its skeletal phenotype is more compatible with the diagnosis of DOS rather than OP, since the platyspondyly with concaved vertebrae at posterior thirds and the radiolucency of widened sub-metaphyseal portions of the tubular bones are evident (Xu et al., 2021). Based on the findings on the case and our case, it could be hypothesized that the deficient TNFRSF11A functions causes a broad phenotypic spectrum covering DOS and OP. The remaining RANK function of the TNFRSF11A mutations may be an important factor that decides the radiographic features of the patients.
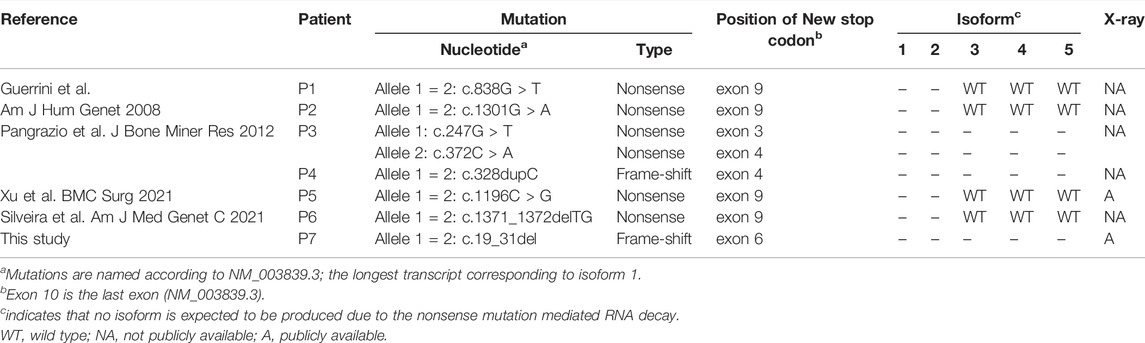
TABLE 2. Patients with null mutations of TNFRSF11A.
The previous four cases of TNFRSF11A-associated DOS show that the skeletal phenotypes are heterogeneous even if all cases meet the criteria of DOS. Generally, the young cases (Case 3 and 4) had a clear radiolucent band in the sub-metaphyseal region of tubular bones, which was widely splayed and sclerotic (Xue et al., 2020; Xue et al., 2021a). In contrast, the radiolucency in the enlarged lower metaphyseal parts was diffuse in the older cases (Case 1 and 2) (Guo et al., 2018; Xue et al., 2019). Moreover, although platyspondyly were found in all cases, concaved vertebrae at posterior thirds were present only in the older cases (Table 1) (Guo et al., 2018; Xue et al., 2019). In this study, our patient at age 19 months showed similarities to the previous young cases in both spinal and sub-metaphyseal changes (Figures 1C,E; Table 1). These results suggest the phenotypes of TNFRSF11A-associated DOS considerably evolve with age and form a continuous phenotypic spectrum. Long-term observation would contribute to the understanding of the evolving phenotypes.
In conclusion, our findings indicate that we are still far from establishing a genotype-phenotype relationship in TNFRSF11A-associated OPTB7 and DOS. Detailed and continuous evaluation on the radiographic data remains necessary to elucidate the phenotypic spectrum caused by TNFRSF11A mutations.
The datasets presented in this article are not readily available because study participants did not give full consent for releasing individual genomic data publicly. Requests to access the datasets should be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by the Ethical Committee of RIKEN. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
The study was designed by SA and LG. The draft was written by TK, SI, and LG. The patient samples and the clinical information were collected and summarized by BO, FH, SA, ON, BO, MK, and SI. The experiments were performed by TK, SG, and BO. The data were analyzed by SA, SI, and LG. All authors revised the manuscript and approved the final version.
This study is supported by the grants from the Japan Society for the Promotion of Science (SI, No. 18H02932 and 22H03207), the Japan Agency For Medical Research and Development (SI, No. 20bm0804006h0 and 20ek0109486h), the Japanese Society for Bone and Mineral Research (LG, the JSBMR Rising Stars Grant), and the Suzuken Memorial Foundation (LG).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank the patient and the family for participating in this study.
Alonso, N., Wani, S., Rose, L., Van't Hof, R. J., Ralston, S. H., and Albagha, O. M. E. (2021). Insertion Mutation in Tnfrsf11a Causes a Paget's Disease-like Phenotype in Heterozygous Mice and Osteopetrosis in Homozygous Mice. J Bone & Mineral Res 36, 1376–1386. doi:10.1002/jbmr.4288
Campeau, P. M., Lu, J. T., Sule, G., Jiang, M.-M., Bae, Y., Madan, S., et al. (2012). Whole-exome Sequencing Identifies Mutations in the Nucleoside Transporter Gene SLC29A3 in Dysosteosclerosis, a Form of Osteopetrosis. Hum. Mol. Genet. 21, 4904–4909. doi:10.1093/hmg/dds326
Elçioglu, N. H., Vellodi, A., and Hall, C. M. (2002). Dysosteosclerosis: a Report of Three New Cases and Evolution of the Radiological Findings. J. Med. Genet. 39, 603–607. doi:10.1136/jmg.39.8.603
Frattini, A., Orchard, P. J., Sobacchi, C., Giliani, S., Abinun, M., Mattsson, J. P., et al. (2000). Defects in TCIRG1 Subunit of the Vacuolar Proton Pump Are Responsible for a Subset of Human Autosomal Recessive Osteopetrosis. Nat. Genet. 25, 343–346. doi:10.1038/77131
Guerrini, M. M., Sobacchi, C., Cassani, B., Abinun, M., Kilic, S. S., Pangrazio, A., et al. (2008). Human Osteoclast-Poor Osteopetrosis with Hypogammaglobulinemia Due to TNFRSF11A (RANK) Mutations. Am. J. Hum. Genet. 83, 64–76. doi:10.1016/j.ajhg.2008.06.015
Guo, L., Bertola, D. R., Takanohashi, A., Saito, A., Segawa, Y., Yokota, T., et al. (2019). Bi-Allelic CSF1R Mutations Cause Skeletal Dysplasia of Dysosteosclerosis-Pyle Disease Spectrum and Degenerative Encephalopathy with Brain Malformation. Am. J. Hum. Genet. 104, 925–935. doi:10.1016/j.ajhg.2019.03.004
Guo, L., Elcioglu, N. H., Karalar, O. K., Topkar, M. O., Wang, Z., Sakamoto, Y., et al. (2018). Dysosteosclerosis Is Also Caused by TNFRSF11A Mutation. J. Hum. Genet. 63, 769–774. doi:10.1038/s10038-018-0447-6
Houston, C., Gerrard, J., and Ives, E. (1978). Dysosteosclerosis. Am. J. Roentgenol. 130, 988–991. doi:10.2214/ajr.130.5.988
Howaldt, A., Nampoothiri, S., Quell, L.-M., Ozden, A., Fischer-Zirnsak, B., Collet, C., et al. (2019). Sclerosing Bone Dysplasias with Hallmarks of Dysosteosclerosis in Four Patients Carrying Mutations in SLC29A3 and TCIRG1. Bone 120, 495–503. doi:10.1016/j.bone.2018.12.002
Pangrazio, A., Cassani, B., Guerrini, M. M., Crockett, J. C., Marrella, V., Zammataro, L., et al. (2012). RANK‐dependent Autosomal Recessive Osteopetrosis: Characterization of Five New Cases with Novel Mutations. J. Bone Min. Res. 27, 342–351. doi:10.1002/jbmr.559
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Silveira, K. C., Kanazawa, T. Y., Silveira, C., Lacarrubba‐Flores, M. D. J., Carvalho, B. S., and Cavalcanti, D. P. (2021). Molecular Diagnosis in a Cohort of 114 Patients with Rare Skeletal Dysplasias. Am. J. Med. Genet. 187, 396–408. doi:10.1002/ajmg.c.31937
Spranger, J., Albrecht, C., Rohwedder, H.-J., and Wiedemann, H.-R. (1968). Die Dysosteosklerose - eine Sonderform der generalisierten Osteosklerose. Fortschr Röntgenstr 109, 504–512. doi:10.1055/s-0029-1228480
Uludağ Alkaya, D., Akpınar, E., Bilguvar, K., and Tüysüz, B. (2021). Resolution of Sclerotic Lesions of Dysosteosclerosis Due to Biallelic SLC29A3 Variant in a Turkish Girl. Am. J. Med. Genet. A 185, 2271–2277. doi:10.1002/ajmg.a.62198
Whyte, M. P., Wenkert, D., McAlister, W. H., Novack, D. V., Nenninger, A. R., Zhang, X., et al. (2010). Dysosteosclerosis Presents as an "Osteoclast-Poor" Form of Osteopetrosis: Comprehensive Investigation of a 3-Year-Old Girl and Literature Review. J. Bone Min. Res. 25, 2527–2539. doi:10.1002/jbmr.131
Xu, Y., Yu, X., and Huang, M. (2021). A Novel Mutation in TNFRSF11A Gene Causes Pediatric Osteopetrosis: Case Report. BMC Surg. 21, 269. doi:10.1186/s12893-021-01266-4
Xue, J.-Y., Ikegawa, S., and Guo, L. (2021). Genetic Disorders Associated with the RANKL/OPG/RANK Pathway. J. Bone Min. Metab. 39, 45–53. doi:10.1007/s00774-020-01148-4
Xue, J.-Y., Simsek-Kiper, P. O., Utine, G. E., Yan, L., Wang, Z., Taskiran, E. Z., et al. (2021). Expanding the Phenotypic Spectrum of TNFRSF11A-Associated Dysosteosclerosis: a Case with Intracranial Extramedullary Hematopoiesis. J. Hum. Genet. 66, 607–611. doi:10.1038/s10038-020-00891-w
Xue, J. Y., Wang, Z., Smithson, S. F., Burren, C. P., Matsumoto, N., Nishimura, G., et al. (2020). The Third Case of TNFRSF11A-Associated Dysosteosclerosis with a Mutation Producing Elongating Proteins. J. Hum. Genet. 9, 1–7. doi:10.1038/s10038-020-00831-8
Keywords: TNFRSF11A/TNR11/RANK, dysosteosclerosis, sclerosing bone dysplasia, osteopetrosis, mutation
Citation: Kırkgöz T, Özkan B, Hazan F, Acar S, Nalbantoğlu Ö, Özkaya B, Kulalı MA, Gürsoy S, Ikegawa S and Guo L (2022) A Null Mutation of TNFRSF11A Causes Dysosteosclerosis, Not Osteopetrosis. Front. Genet. 13:938814. doi: 10.3389/fgene.2022.938814
Received: 08 May 2022; Accepted: 06 June 2022;
Published: 24 June 2022.
Edited by:
Cristina Sobacchi, National Research Council (CNR), ItalyReviewed by:
Mario Abinun, Newcastle University, United KingdomCopyright © 2022 Kırkgöz, Özkan, Hazan, Acar, Nalbantoğlu, Özkaya, Kulalı, Gürsoy, Ikegawa and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sezer Acar, ZHIuYWNhcnNlemVyQGdtYWlsLmNvbQ==; Long Guo, bG9uZ2d1bzYwMUBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.