 Lin Cheng1,2,3†Fan Yang4†Xinlin Chen4Jiawei Kang1,2,3Jiafu Li1,2,3
Lin Cheng1,2,3†Fan Yang4†Xinlin Chen4Jiawei Kang1,2,3Jiafu Li1,2,3 Yuanzhen Zhang1,2,3Juan Liu1,2,3Jin Li2,3,5Jianhong Ma1,2,3*
Yuanzhen Zhang1,2,3Juan Liu1,2,3Jin Li2,3,5Jianhong Ma1,2,3* Jie Duan1,2,3*
Jie Duan1,2,3*- 1Department of Obstetrics and Gynecology, Zhongnan Hospital of Wuhan University, Wuhan, China
- 2Hubei Clinical Research Center for Prenatal Diagnosis and Birth Health, Wuhan, China
- 3Wuhan Clinical Research Center for Reproductive Science and Birth Health, Wuhan, China
- 4Department of Ultrasound Imaging, Hubei Maternal and Child Health Hospital, Wuhan, China
- 5Department of Laboratory Medicine, Zhongnan Hospital of Wuhan University, Wuhan, China
Matrix metalloproteinase 9 (MMP9) is an important member of the matrix metalloproteinase family and plays a key role in balancing extracellular matrix proteins. Studies have shown that the homozygous mutations in MMP9 can lead to metaphyseal anadysplasia type 2 (MANDP2, OMIM#613073). The clinical phenotype of this disease is limited and there were only five reported cases of MANDP2 associated with homozygous MMP9 mutations from three families. In this study, we described a case of a fetus with skeletal system malformation. The main clinical manifestations include the short bilateral femur, absence of right fibula, and curved ipsilateral tibia with short length. Importantly, two novel compound heterozygous variants of the MMP9 gene (NM_004,994.3: c.151C > T and c.929del) were found through the trio whole exome sequencing and Sanger sequencing. This is the first report that identified the compound heterozygous variants of the MMP9 gene associated with metaphyseal dysplasia type 2.
Introduction
Matrix metalloproteinase nine gene [MMP9, Online Mendelian Inheritance in Man (OMIM) 120,361] is located on q11.1-q13.1 of chromosome 20, which contains 13 exons and 12 introns (Huhtala et al., 1990; Huhtala et al., 1991). The gene-encoded protein belongs to the matrix metalloprotein family. The main function of MMP9 is to degrade and reshape the dynamic balance of the extracellular matrix. The study by Shinoda et al. demonstrated that the MMP9 gene plays an essential role in bone development (Shinoda et al., 2008). The mutations of the MMP9 gene have been reported to be linked with the cause of the metaphyseal anadysplasia type 2 (MANDP2) (Lausch et al., 2009). MANDP2 (OMIM: 613,073, also known as Maroteaux type) is a rare autosomal recessive disorder with the characteristic of short legs, short neck of femur, widening of the epiphysis, irregular epiphysis, and bent legs, which was first reported by Le Merrer M (LeMerrer and Maroteaux, 1998). Despite this fact, the studies that reported the association between the homozygous mutation of the MMP9 gene and MANDP2 are very limited (Lausch et al., 2009; Sharony et al., 2017; Bonilla-Fornés et al., 2021). As an indication, the impact of compound heterozygous MMP9 gene on MANDP2 was unknown previously. This work studied a Chinese fetus with MANDP2, presenting with the short femurs, absence of right fibula with short and curved right tibia, and we detected novel compound heterozygous variants in the MMP9 gene using trio whole exome sequencing (WES) and Sanger sequencing.
Methods
Study Participants
The study participators included the aborted fetus (the proband), her parents, and the lineages, all of whom provided informed consent for participation in the study. The study was approved by the institutional review board at Zhongnan Hospital of Wuhan University.
Genomic DNA Extraction
Genomic DNA was extracted from umbilical cord blood of the proband and peripheral blood of the family members separately, using a Qiagen DNA Blood Midi/Mini kit (Qiagen GmbH, Hilden, Germany, 69,506). NanoDrop spectrophotometer and agarose gel electrophoresis were employed in determining the purity and yield of DNA products.
Copy Number Variation Sequencing
Genomic DNA was firstly fragmented. DNA libraries constructed by end filling, adapter ligation, and PCR amplification were subjected to massively parallel sequencing on the NextSeq 500 platform (Illumina, San Diego, CA). The sample sequences were screened using the hg19 genomic sequence as reference. Identified and mapped CNVs were investigated in publicly available databases, including Decipher (https://www.deciphergenomics.org), Database of Genomic Variants (DGV) (MacDonald et al., 2014), 1,000 genomes (1000G, http://www.1000genomes.org/), and OMIM (Hamosh et al., 2002), and their pathogenicity was assessed according to the guidelines outlined by the American College of Medical Genetics (ACMG) for interpretation of sequence variants. Variants were classified into five categories: pathogenic, likely pathogenic, variants of uncertain significance (VUS), likely benign, and benign.
Whole Exome Sequencing
Genomic DNA was interrupted to 200bp around by fragmentation enzymes. The DNA fragments were end-repaired by adding an A base at the 3′ end. Followingly, the DNA fragments were ligated with barcoded sequencing adaptors and hybridized by Berry’s NanoWES Human Exome V1.0 (Berry Genomics, Beijing, China) according to the manufacturer’s standard operating procedure. The hybrid products after elution and collection were subjected to PCR (polymerase chain reaction) amplification and the purification and subsequently were ready for sequencing. Novaseq6000 platform (Illumina, San Diego, United States), with 150 bp pair-end sequencing mode, was used for sequencing the genomic DNA samples of the proband and the participated family members. Raw image files were processed using CASAVA pipeline v1.82 (Illumina, San Diego, United States) for base calling and raw data generation. The sequencing reads were aligned to the human reference genome (hg38/GRCh38) using the Burrows-Wheeler Aligner (Li and Durbin, 2009; Li and Durbin, 2010) tool with default parameters and PCR duplicates were removed by using Picard v1.57 (http://picard.sourceforge.net/). Verita Trekker® Variants Detection System by Berry Genomics and the third-party software GATK (https://software.broadinstitute.org/gatk/) with default parameters was employed for detecting any variant. Variant annotation and interpretation were conducted by ANNOVAR (Wang et al., 2010) and the Enliven® Variants Annotation Interpretation System authorized by Berry Genomics. The variants were classified into the same five categories mentioned earlier (Richards et al., 2015).
Sanger Sequencing
Primer Blast was used to design the forward and reverse primers for MMP9 variant sites (NM_004,994.3: c.151C > T and c.929del) (Table 1). The bidirectional sequencing was performed by Sanger sequencing after PCR. The sequencing results included all the target exon sequences, which are at least 30 bp flanking sequences. The purified sequencing results were bidirectionally aligned with the sequences published by Ensembl genome browser 90.

TABLE 1. Forward and reverse primers for MMP9 gene variant sites.
Protein Structure Modeling
We performed the prediction with the online server, SWISS-MODEL (http://swissmodel.expasy.org/), to construct the three-dimensional structure of the MMP9 protein.
Results
Clinical Phenotype
The participating couple is non-consanguineous and healthy. The pregnant woman was 31 years old. The abnormal development of the right leg with the aberrant right subclavian artery (ARSA) was detected in the fetus during 1st-trimester routine scans. One week later, a tertiary ultrasound revealed that the fetus had a single long bone echo on the right lower leg with a length of 0.6 cm, and the connection position between calf and footplate was abnormal (Figure 1AB). An ultrasound at 14+4 weeks of gestation showed a short femur (1.1 cm, −2.55 SD according to Hadlock curve) and detected a slightly curved long bone with a single echo on the right lower leg (length at 0.83 cm). Due to the fetal skeletal malformation, induced abortion was decided and performed at 15+4 weeks of gestation at the request of the couple. Meanwhile, the umbilical cord blood of proband and the peripheral blood of parents were collected for the trio whole-exome sequencing (trio WES). The copy number variant sequencing (CNV-seq) was employed to exclude the CNVs of the proband. Autopsy examination confirmed that the right fibula of the proband was absent, and the X-ray image was shown in Figure 1C.
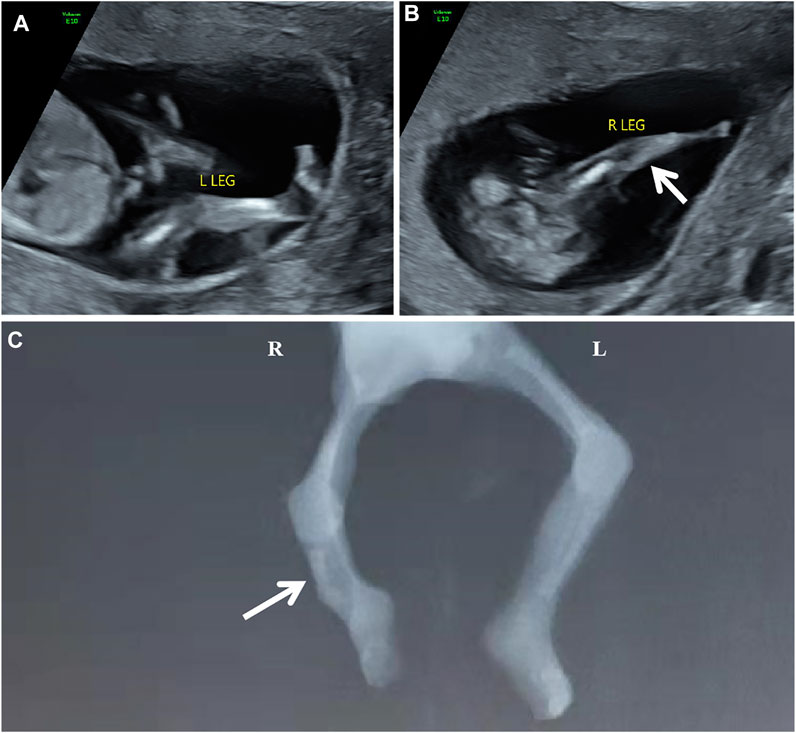
FIGURE 1. The scan images of the proband [(A,B) the ultrasonography at 13+6 weeks of gestation showed abnormal development of the right calf. (C) the X-ray of the proband showed the absence of the right fibula with the bent tibia.]
Laboratory Findings
The trio WES detected two compound heterozygous variants in exon two and exon six of the MMP9 gene in the proband (NM_004,994.3: c.151C > T and c.929del), with the findings that c.151C > T was identified as a paternal missense variant and c.929del was determined as a maternal frameshift variant. The analysis of MMP9 gene variation was extended in the other family members using Sanger sequencing. These detected variants were found in multiple family members of the proband (cases I-1, II-2, II-3, and III-1 with c.151C > T, while cases I-3, II-4, and II-5 with c.929del, no blood samples obtained in cases I-4 and II-1 for personal reason.) The pedigree-chart-based Sanger sequencing results were presented in Figure 2. However, neither chromosomal aneuploidy nor pathogenic CNVs above 100bp were found using the CNV-seq.
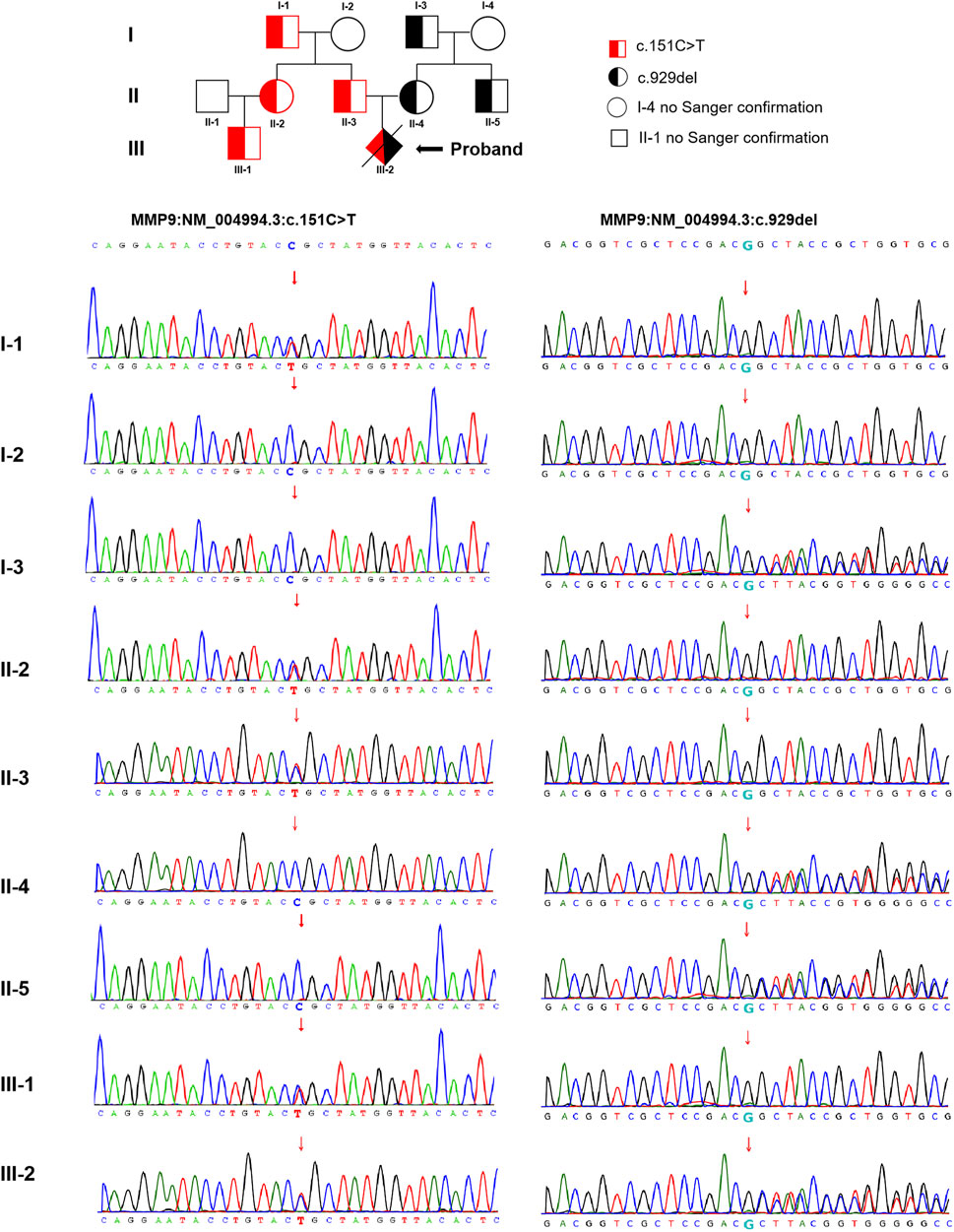
FIGURE 2. The pedigree-chart-based Sanger sequencing results of the proband (Cases I-1, II-2, II-3, and III-1 with c.151C > T, whereas cases I-3, II-4, and II-5 with c.929del.)
Protein Structure Prediction
The protein structures of the compound heterozygous variants of the MMP9 gene were predicted with high confidence. The predicted protein structures show a good Global Model Quality Estimation (GMQE) score of 0.67 and a Qualitative Model Energy Analysis (QMEAN) score of 0.69. The substitution of 51 Arg was identified in the structure prediction of the MMP9 protein. The c.929del variant caused the generation of a stop codon, which can lead to a truncated protein (Figure 3AB). The MMP9 protein prediction indicated that the p. R51C substitution could break the hydrogen bond (Figure 3CD).
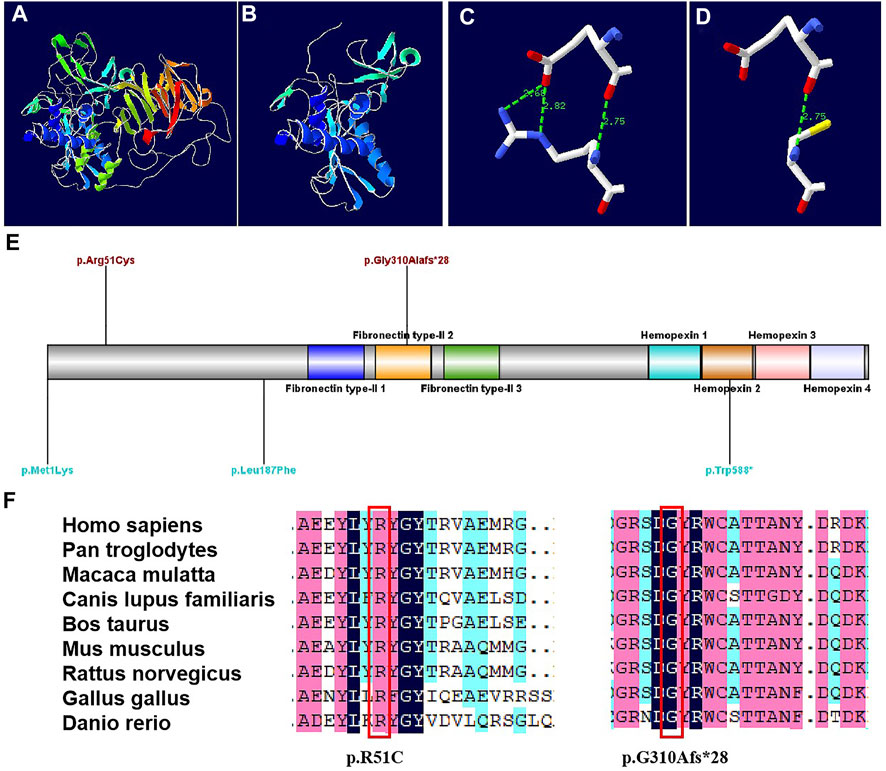
FIGURE 3. Models of the MMP9 protein, and conservation sites of the MMP9 amino acid sequences among species [(A). Three-dimensional structure of normal MMP9 protein. (B) Three-dimensional structure of MMP9 protein variant when a stop codon introduced at position 310.(C) Partial three-dimensional structure when a stop codon introduced at position 310. (D) Partial three-dimensional structure when Arg changed to Cys at position 51, where H bond breaks. (E) The variant sites previously reported are demonstrated in blue, while the variants sites reported in this case are in red. (F) The MMP9 amino acid sequences were highly conserved among species.]
Discussion
This study reported a proband showing a short femur, unilateral absence of fibula, and short curvature of the tibia. The results of trio-WES and Sanger sequencing identified two compound heterozygous variants of the MMP9 gene (NM_004,994.3: c.151C > T and c.929del).
The missense variant c.151C > T occurred at exon two of the MMP9 gene on chromosomal 20, in which arginine at 51 was replaced by cysteine. To date, this variant has not been found in the 1,000 Genomes (1000G, http://www.1000genomes.org/), and the frequency of this variant in Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org/) and Genome Aggregation Database (gnomAD, https://gnomad.broadinstitute.org/) are 9.52 × 10−5 and 9.6 × 10−4, respectively (PM2). This variant was a compound heterozygous with the c.929del (PM3). Besides, this variant was predicted of the domain by InterPro software (http://www.ebi.ac.uk/interpro/), and there is no benign variation in this functional domain (PP3).
The c.929del is a frameshift variant that occurs in exon six of the MMP9 on chromosomal 20. This frameshift variant was caused by introducing a stop codon at the following 28 amino acids, leading to the truncation of the protein by changing the open reading frame of the gene (PVS1). Nevertheless, the variant has not been reported in the 1000G, ExAC, or gnomAD databases (PM2). Uniprot analysis showed that Gly at position 310 was in the crimp domain of the MMP9 ion channel pore, and the loss of this amino acid indicated a decreased stability or rigidity of the crimp motif.
Generally, the findings demonstrated that the discovered variants were novel. Both amino acid sequences were highly conserved among species (Figure 3F). According to the guidelines of the American College of Medical Genetics and Genomics (ACMG), the missense variant c.151C > T was classified as a “variant of uncertain significance (VUS)”, whereas the frameshift variant c.929del was classified as “likely pathogenic (LP)” (Richards et al., 2015).
There are three MANDP2 reports determined with MMP9 gene homozygous mutation, which were shown in Table 2 and Figure 3E. In our study, we found the compound heterozygote variants of the MMP9 gene (NM_004,994.3: c.151C > T and c.929del) through trio WES, which were recognized as novel variants. Besides the shortening of fetal lower limb long bones, the unilateral fibula absence and tibial curvature were clinical phenotypes in the case. It is well known that vascularization is crucial for transforming cartilage scaffolds into the bone during fetal skeletal development. The osteoblasts invade cartilage with blood vessels and form the bone matrix (Watson and Adams, 2018). Studies have shown that the MMP family plays an important role in bone formation and growth (Paiva and Granjeiro, 2014; Paiva and Granjeiro, 2017). Among them, MMP9 is mainly secreted by chondrocytes, osteoclasts, and endothelial cells, and it can regulate the bioavailability of vascular endothelial growth factor A (VEGF-A). The fetal fibula deficiency in the case of our study could result from the compound heterozygous various of the MMP9 gene, which is associated with VEGF-A reduction and impaired angiogenesis during endochondral ossification, consequently affecting the formation of the fibula. The loss of the fibula could further cause the curvature of the tibia due to the missing support from the fibula. However, the assumption requires further investigation.
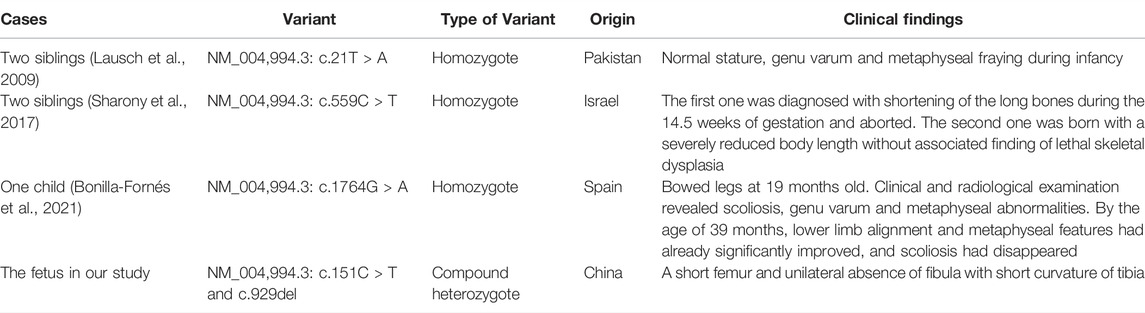
TABLE 2. Summary of clinical features and molecular findings in MANDP2 patients caused by MMP9 variants.
Aberrant right subclavian artery (ARSA) is also considered in the investigation. It occurs both as a variant of normal and in association with other cardiac malformations or chromosomal abnormalities. Other studies have reported the incidence of ARSA in the second trimester as about 35% in trisomy 21 fetuses and 1.4% in chromosomally normal fetuses (Borenstein et al., 2008; Svirsky et al., 2017). A multi-center study found increased risks of chromosomal aneuploidy and pathogenic copy number variations (pCNVs) of fetal ARSA with extracardiac abnormalities (Maya et al., 2017). The presence of an isolated ARSA is benign and is not associated with chromosomal abnormalities. In our case, neither chromosomal aneuploidy nor copy number variation was found in the proband, and ARSA-related gene variant was also absent in WES and CNV-seq. Therefore, we considered ARSA to be a normal anatomical variation in this study.
Conclusion
In summary, we have identified the novel compound heterozygous variants of the MMP9 gene (NM_004,994.3: c.151C > T and c.929del) in the fetus with MANDP2. The compound heterozygous variants could be associated with the absence of unilateral fibula and bent tibia. However, since the c.151C > T is a VUS variant, it is necessary to research the regulation of protein structure and function by this variant, which will not only be helpful to expand the spectrum of MMP9 gene variations and the clinical phenotype of MANDP2, but also provide guidance for prenatal diagnosis and clinical genetic consultation.
Data Availability Statement
The data presented in the study are deposited in the GenBank repository, accession numbers are SAMN29672838, SAMN29672839, and SAMN29672840.
Ethics Statement
Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
LC drafted the manuscript. FY, XC, JK, and JIL performed the clinical examination of the proband and its family. YZ and JUL followed up with the family. Jin L analyzed the WES and CNV-seq data. JM and JD reviewed the manuscript and critically revised the paper. All authors read and approved the final manuscript.
Funding
This work was funded by the National Key Research and Development Program of China (No. 2020YFA0803900).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We acknowledge Guowei Chen (Berry Genomics), who provided technical assistance.
References
Bonilla-Fornés, S., Galán-Ledesma, L., Pérez, P. M., Modamio-Høybjør, S., Carbonell-Pérez, J. M., Parrón-Pajares, M., et al. (2021). Early Clinical and Radiological Improvement in a Young Boy with Metaphyseal Anadysplasia Type 2. Eur. J. Med. Genet. 64 (10), 104307. doi:10.1016/j.ejmg.2021.104307
Borenstein, M., Cavoretto, P., Allan, L., Huggon, I., and Nicolaides, K. H. (2008). Aberrant Right Subclavian Artery at 11 + 0 to 13 + 6 Weeks of Gestation in Chromosomally Normal and Abnormal Fetuses. Ultrasound Obstet. Gynecol. 31 (1), 20–24. doi:10.1002/uog.5226
Hamosh, A., Scott, A. F., Amberger, J., Bocchini, C., Valle, D., and McKusick, V. A. (2002). Online Mendelian Inheritance in Man (OMIM), a Knowledgebase of Human Genes and Genetic Disorders. Nucleic Acids Res. 30 (1), 52–55. doi:10.1093/nar/30.1.52
Huhtala, P., Chow, L. T., and Tryggvason, K. (1990). Structure of the Human Type IV Collagenase Gene. J. Biol. Chem. 265 (19), 11077–11082. doi:10.1016/s0021-9258(19)38559-x
Huhtala, P., Tuuttila, A., Chow, L. T., Lohi, J., Keski-Oja, J., and Tryggvason, K. (1991). Complete Structure of the Human Gene for 92-kDa Type IV Collagenase. Divergent Regulation of Expression for the 92- and 72-kilodalton Enzyme Genes in HT-1080 Cells. J. Biol. Chem. 266 (25), 16485–16490. doi:10.1016/s0021-9258(18)55326-6
Lausch, E., Keppler, R., Hilbert, K., Cormier-Daire, V., Nikkel, S., Nishimura, G., et al. (2009). Mutations in MMP9 and MMP13 Determine the Mode of Inheritance and the Clinical Spectrum of Metaphyseal Anadysplasia. Am. J. Hum. Genet. 85 (2), 168–178. doi:10.1016/j.ajhg.2009.06.014
LeMerrer, M., and Maroteaux, P. (1998). Metaphyseal Anadysplasia Type II: a New Regressive Metaphyseal Dysplasia. Pediatr. Radiol. 28 (10), 771–775. doi:10.1007/s002470050463
Li, H., and Durbin, R. (2010). Fast and Accurate Long-Read Alignment with Burrows-Wheeler Transform. Bioinformatics 26 (5), 589–595. doi:10.1093/bioinformatics/btp698
Li, H., and Durbin, R. (2009). Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 25 (14), 1754–1760. doi:10.1093/bioinformatics/btp324
MacDonald, J. R., Ziman, R., Yuen, R. K. C., Feuk, L., and Scherer, S. W. (2014). The Database of Genomic Variants: a Curated Collection of Structural Variation in the Human Genome. Nucl. Acids Res. 42, D986–D992. doi:10.1093/nar/gkt958
Maya, I., Kahana, S., Yeshaya, J., Tenne, T., Yacobson, S., Agmon‐Fishman, I., et al. (2017). Chromosomal Microarray Analysis in Fetuses with Aberrant Right Subclavian Artery. Ultrasound Obstet. Gynecol. 49 (3), 337–341. doi:10.1002/uog.15935
Paiva, K. B. S., and Granjeiro, J. M. (2014). Bone Tissue Remodeling and Development: Focus on Matrix Metalloproteinase Functions. Archives Biochem. Biophysics 561, 74–87. doi:10.1016/j.abb.2014.07.034
Paiva, K. B. S., and Granjeiro, J. M. (2017). Matrix Metalloproteinases in Bone Resorption, Remodeling, and Repair. Prog. Mol. Biol. Transl. Sci. 148, 203–303. doi:10.1016/bs.pmbts.2017.05.001
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Sharony, R., Borochowitz, Z., Cohen, L., Shtorch-Asor, A., Rosenfeld, R., Modai, S., et al. (2017). Prenatal Course of Metaphyseal Anadysplasia Associated with Homozygous Mutation in MMP9 Identified by Exome Sequencing. Clin. Genet. 92 (6), 645–648. doi:10.1111/cge.13020
Shinoda, Y., Ogata, N., Higashikawa, A., Manabe, I., Shindo, T., Yamada, T., et al. (2008). Krüppel-like Factor 5 Causes Cartilage Degradation through Transactivation of Matrix Metalloproteinase 9. J. Biol. Chem. 283 (36), 24682–24689. doi:10.1074/jbc.M709857200
Svirsky, R., Reches, A., Brabbing-Goldstein, D., Bar-Shira, A., and Yaron, Y. (2017). Association of Aberrant Right Subclavian Artery with Abnormal Karyotype and Microarray Results. Prenat. Diagn. 37 (8), 808–811. doi:10.1002/pd.5092
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 38 (16), e164. doi:10.1093/nar/gkq603
Keywords: matrix metalloproteinase 9 gene, absence of fibula, short femur length, prenatal diagnosis, metaphyseal anadysplasia type 2
Citation: Cheng L, Yang F, Chen X, Kang J, Li J, Zhang Y, Liu J, Li J, Ma J and Duan J (2022) Identification of Novel Compound Heterozygous Variants of MMP9 in Fetus With Metaphyseal Anadysplasia Type 2. Front. Genet. 13:938457. doi: 10.3389/fgene.2022.938457
Received: 07 May 2022; Accepted: 14 June 2022;
Published: 12 August 2022.
Edited by:
Stephen J. Bush, University of Oxford, United KingdomReviewed by:
Hong Luo, Central South University, ChinaPatricia Canto, Universidad Nacional Autónoma de México, Mexico
Copyright © 2022 Cheng, Yang, Chen, Kang, Li, Zhang, Liu, Li, Ma and Duan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianhong Ma, bWFqaWFuaDIwMDVAMTYzLmNvbQ==; Jie Duan, amllZHVhbjEzMUBob3RtYWlsLmNvbQ==
†These authors have contributed equally to this work and share first authorship.