 Tatsuaki Kurosaki
Tatsuaki Kurosaki Tetsuo Ashizawa
Tetsuo Ashizawa- 1Department of Biochemistry and Biophysics, School of Medicine and Dentistry, University of Rochester, Rochester, NY, United States
- 2Center for RNA Biology, University of Rochester, Rochester, NY, United States
- 3Stanley H. Appel Department of Neurology, Houston Methodist Research Institute and Weil Cornell Medical College at Houston Methodist Houston, TX, United States
Spinocerebellar ataxia type 10 (SCA10) is characterized by progressive cerebellar neurodegeneration and, in many patients, epilepsy. This disease mainly occurs in individuals with Indigenous American or East Asian ancestry, with strong evidence supporting a founder effect. The mutation causing SCA10 is a large expansion in an ATTCT pentanucleotide repeat in intron 9 of the ATXN10 gene. The ATTCT repeat is highly unstable, expanding to 280–4,500 repeats in affected patients compared with the 9–32 repeats in normal individuals, one of the largest repeat expansions causing neurological disorders identified to date. However, the underlying molecular basis of how this huge repeat expansion evolves and contributes to the SCA10 phenotype remains largely unknown. Recent progress in next-generation DNA sequencing technologies has established that the SCA10 repeat sequence has a highly heterogeneous structure. Here we summarize what is known about the structure and origin of SCA10 repeats, discuss the potential contribution of variant repeats to the SCA10 disease phenotype, and explore how this information can be exploited for therapeutic benefit.
Introduction
Non-coding microsatellite repeat expansions are responsible for a wide range of dominantly and recessively inherited autosomal or X-linked human disorders (summarized in Table 1). Compared with microsatellite repeats in coding regions, non-coding microsatellite repeats tend to be more unstable, resulting in massive repeat expansions of hundreds to thousands of repeats (Table 1). However, the disease mechanisms related to these repeats in non-coding regions remain largely uncharacterized.
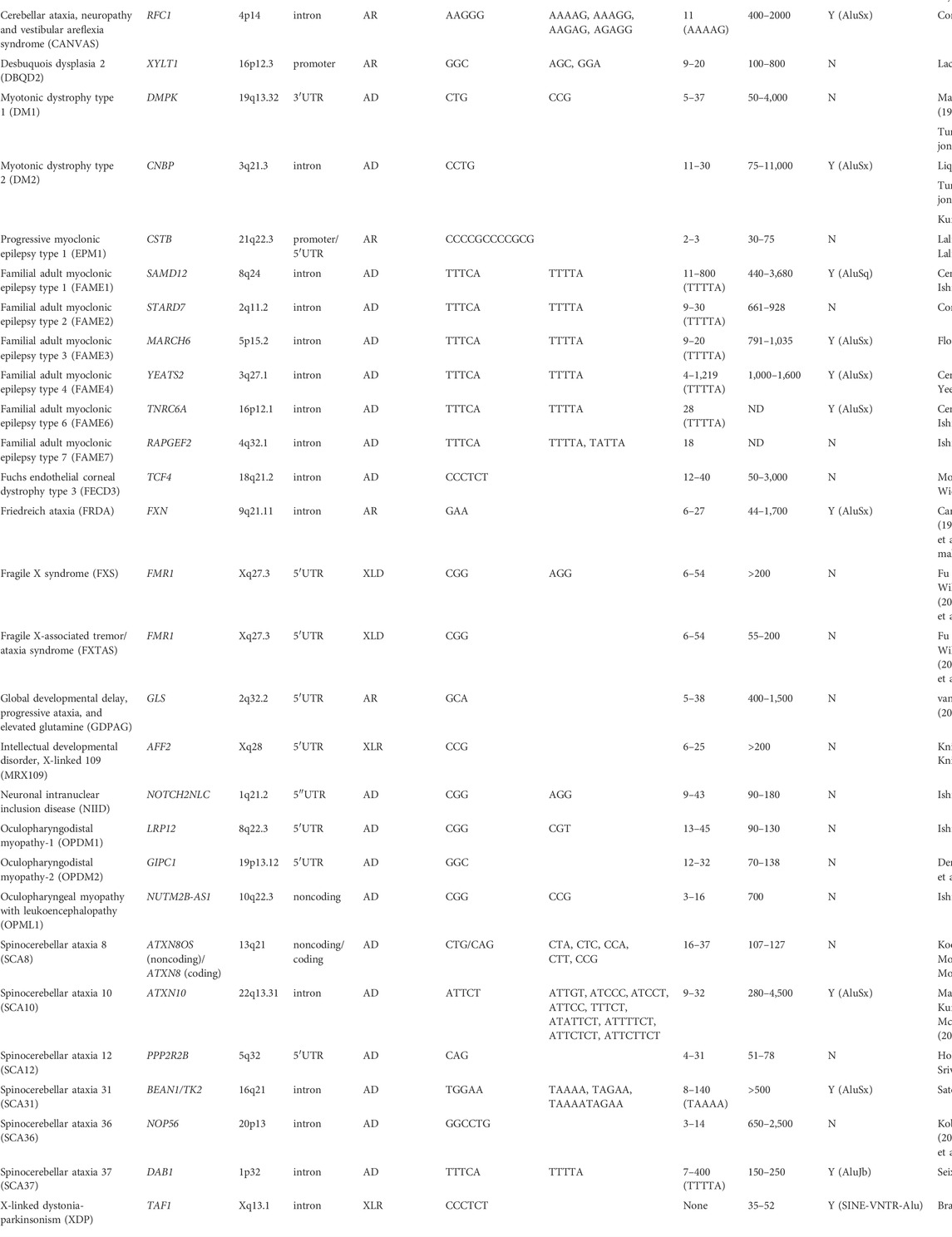
TABLE 1. Noncoding microsatellite repeat expansion diseases.
Spinocerebellar ataxia type 10 (SCA10) is an autosomal dominant neurodegenerative disorder that presents clinically with progressive cerebellar ataxia variably associated with epilepsy (Matsuura et al., 1999; Matsuura et al., 2000; Lin and Ashizawa, 2003; Lin and Ashizawa, 2005). SCA10 was the first human genetic disorder discovered to be caused by an expanded intronic pentanucleotide (ATTCT) repeat in intron 9 of the ATXN10 gene on chromosome 22q13.3 (Figure 1) (Matsuura et al., 1999; Matsuura et al., 2000). Normal individuals usually have 9–32 ATTCT repeats, but SCA10 patients can have up to 4,500 (∼22.5 kb) repeats (Matsuura et al., 2000; McFarland et al., 2013). Since the first discovery of the SCA10 mutation, many diseases have subsequently been reported to be caused by expanded intronic pentanucleotide repeats, including SCA31, SCA37, familial adult myoclonic epilepsy (FAME), and cerebellar ataxia, neuropathy and vestibular areflexia syndrome (CANVAS). Nevertheless, the SCA10 repeat is one of the largest expansions reported to date in microsatellite repeat expansion disorders (Table 1).
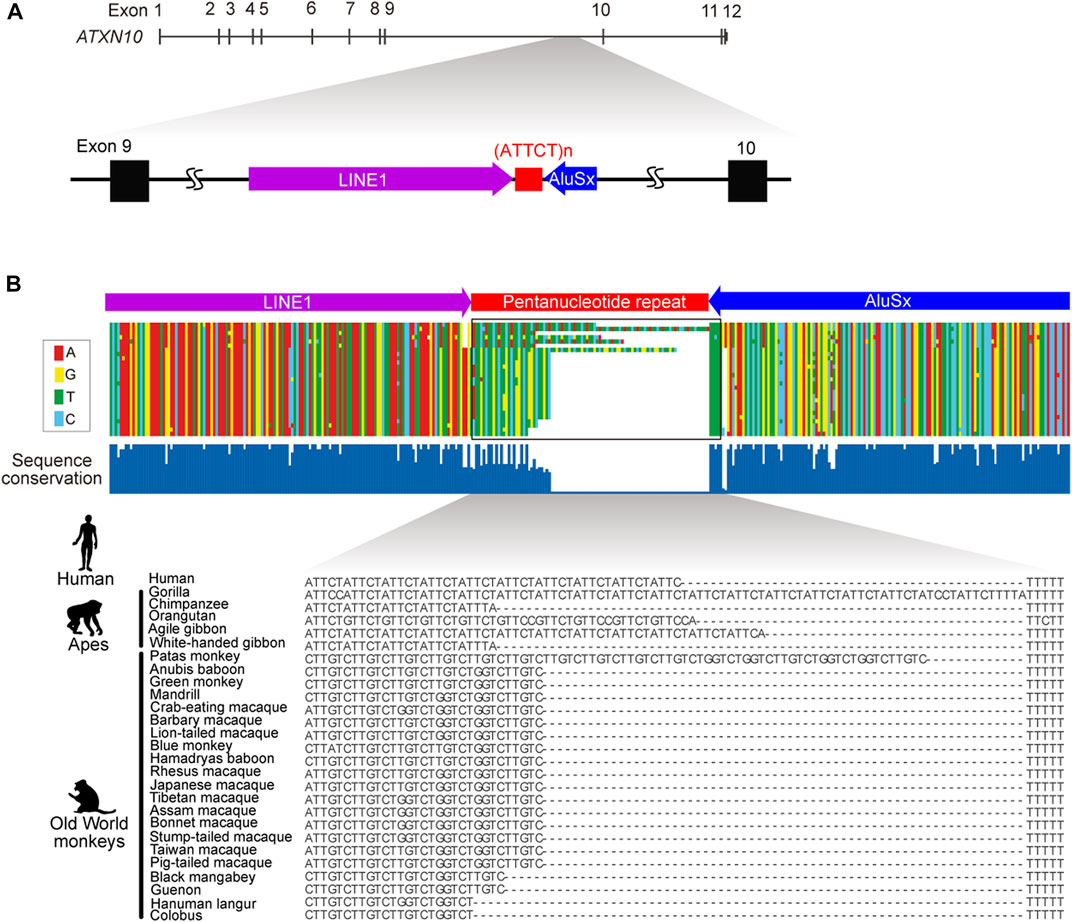
FIGURE 1. Genomic structure of ATXN10 pentanucleotide repeats. (A) The genomic location of the ATTCT pentanucleotide repeat between transposable LINE1 and the AluSx element in human ATXN10 intron 9. (B) Multiple alignments comparing ATXN10 pentanucleotide repeats in primate species using sequence data from Kurosaki et al., 2009.
The length of the expanded ATTCT repeat is highly unstable, especially during paternal transmission, and shows a variable degree of somatic and germline instability (Matsuura et al., 2004). Disease onset is usually in early adulthood, although initial symptoms can occur in teenagers and the elderly (Grewal et al., 2002; Bushara et al., 2013). While some families show conspicuous anticipation, others do not, suggesting that the genetic mechanisms underlying SCA10 are complex (Matsuura et al., 2000; Rasmussen et al., 2001; Grewal et al., 2002; Teive et al., 2004).
SCA10 can be diagnosed by southern blotting, repeat-primed PCR, or long-range PCR, which detect the repeat expansion (Matsuura et al., 2000; Matsuura et al., 2004; Matsuura et al., 2006; Kurosaki et al., 2008). However, due to technical limitations inherent in Sanger sequencing for reading long repetitive sequences (>∼1 kb), the sequence structure of SCA10 repeats was uncertain for a long time. However, recent progress in repeat-primed PCR coupled with pulse-field capillary electrophoresis or long-read sequencing technology, e.g., single-molecule real-time (SMRT) sequencing, has enabled the definition of the entire SCA10 repeat expansion sequence structure, providing new insights into the repetitive sequence and associated disease phenotypes (Mcfarland et al., 2015; Schüle et al., 2017; Hashem et al., 2020). In this review, we discuss recent progress in SCA10 research, focusing on its molecular genetics, sequence structure, and related disease mechanisms. In doing so, we explore the potential contribution of variant repeats to the SCA10 disease phenotype and explore how this information can be exploited for therapeutic benefit.
The clinical features of SCA10
Patients with SCA10 are characterized by the core clinical phenotype of progressive cerebellar ataxia, and although epilepsy is frequently observed, its occurrence is more variable within and between families than ataxia (Matsuura et al., 1999; Zu et al., 1999; Matsuura et al., 2000; Grewal et al., 2002; Seixas et al., 2005; Raskin et al., 2007; Teive et al., 2010; de Castilhos et al., 2014; Schüle et al., 2017; Domingues et al., 2019; Domingues et al., 2019; Nascimento et al., 2019; Ramirez-Garcia et al., 2022) (Table 2). SCA10 has been reported in Mexican, Brazilian, Colombian, Argentinian, Peruvian, Bolivian, or Venezuelan families with Indigenous American ancestry (Matsuura et al., 2000; Rasmussen et al., 2001; Alonso et al., 2007; Gatto et al., 2007; Gatto et al., 2007; Raskin et al., 2007; Roxburgh et al., 2013; Leonardi et al., 2014; Baizabal-Carvallo et al., 2015; Paradisi et al., 2015; Domingues et al., 2019; Ramirez-Garcia et al., 2022) and in Chinese and Japanese families (Wang et al., 2015; Naito et al., 2017; Mao et al., 2022). A majority (∼68%) of SCA10 patients exhibit pure cerebellar ataxia but, highlighting the clinical heterogeneity, only ∼5%–7% have epilepsy in Brazilian populations from the Parana and Santa Catarina states (Domingues et al., 2019; Domingues et al., 2019) but ∼65% of patients from other regions of Brazil develop seizures (de Castilhos et al., 2014). Similarly, epilepsy frequency in Mexican families ranges anywhere from 20% to 80% (Matsuura et al., 1999; Zu et al., 1999; Rasmussen et al., 2001; Grewal et al., 2002; Teive et al., 2004; Teive et al., 2010; Schüle et al., 2017). While SCA10 is usually diagnosed in patients aged 14–48 years (Table 2), one patient from Minnesota developed SCA10 ataxia at 83 years of age (Bushara et al., 2013). Of note, SCA10 patients with epilepsy tend to be younger (24 ± 16 years) than patients without epilepsy (35 ± 9 years) (Domingues et al., 2019). In addition to cerebellar ataxia and epilepsy, patients in some families also exhibit cognitive impairment and peripheral neuropathy (Table 2). Indeed, a recent magnetic resonance imaging (MRI) study of Mexican SCA10 patients has revealed neurodegeneration not only in the cerebellum but also in other brain regions, including the brainstem, thalamus, and putamen (Hernandez-Castillo et al., 2019).

TABLE 2. The clinical features of SCA10.
The most common signs and symptoms of cerebellar dysfunction in SCA10 patients are gait ataxia, dysarthria, and nystagmus (Table 2). The Scale for Assessment and Rating of Ataxia (SARA), a semi-quantitative instrument to assess impairment from ataxia, has been validated and is correlated with quality of life in SCA patients (Schmitz-Hübsch et al., 2006). The SARA score correlates positively with disease duration in SCA10 patients (r = 0.89, p < 0.0001) (Zonta et al., 2022). The disease progression rate, calculated as the SARA score divided by total disease duration in years, is slower in SCA10 than in other SCAs (e.g., SCA10 = 0.84; SCA2 = 1.16; SCA3 = 1.53) (Tensini et al., 2017; Zonta et al., 2022).
Geographic distribution and origin of the SCA10 repeat expansion
SCA10 is mainly reported in individuals from Latin American countries such as Mexico (Matsuura et al., 2000; Matsuura et al., 2002; Rasmussen et al., 2001; Alonso et al., 2007), Brazil (Raskin et al., 2007; Teive et al., 2010; Domingues et al., 2019; Nascimento et al., 2019), Peru (Leonardi et al., 2014), Bolivia (Baizabal-Carvallo et al., 2015), Venezuela (Teive et al., 2010; Paradisi et al., 2015), Colombia (Roxburgh et al., 2013), or Argentina (Gatto et al., 2007; Teive et al., 2010) but not in European, African, South Asian, or Oceanic countries. The geographic distribution strongly supports a founder effect in the SCA10 allele. The subsequent identification of one SCA10 patient with Sioux Indigenous American ancestry and no Hispanic or Latino heritage solidified the hypothesis that SCA10 originates from the Indigenous American population (Bushara et al., 2013). Haplotype analyses of SCA10 patients in Latin America showed a common haplotype of six polymorphic loci, i.e., four SNPs (rs5764850-C/A, rs72556348-G/A, rs72556349-G/A, rs72556350-C/T) and two dinucleotide repeats (D22S1140 and D22S1153). Strikingly, SCA10 families typically share the common or closely related haplotype, further strengthening the evidence that the SCA10 repeat originally emerged in Indigenous Americans migrating throughout North and South America around 7,000–15,000 years ago (Almeida et al., 2009; Bushara et al., 2013; Rodríguez-labrada et al., 2020). More recently, SCA10 families have also been reported in China (Wang et al., 2015; Mao et al., 2022) and Japan (Naito et al., 2017). Haplotype analyses of these individuals have revealed that the haplotypes (rs5764850-C, rs72556348-G, rs72556349-G, rs72556350-C) common in North and South American populations are shared by Chinese and Japanese SCA10 patients (Naito et al., 2017; Mao et al., 2022), suggesting that the SCA10 mutation initially emerged at an earlier time point in Indigenous Americans before migration from East Asia to North America.
Evolutionary origin of the ATXN10 ATTCT repeat
In normal individuals, ATXN10 intron 9 repeats are typically uninterrupted repetitive ATTCT units. By contrast, repeat interruptions are a common feature in the orthologous region in other higher primates (Figure 1B) (Kurosaki et al., 2009). Comparative analysis of primate genomes has shown that the pentanucleotide repeat locates at the 3′-end of the Alu element in humans, apes, and Old World monkeys but is entirely absent in prosimians, New World monkeys, and other primate species (Figure 1B). The pentanucleotide repeats originally arose from the poly(A) stretch of the Alu element in conjunction with the RNA polymerase III TTTT terminator sequence in the opposite direction of the ATXN10 gene around ∼50 million years ago (Kurosaki et al., 2009).
The Alu element is a primate-specific non-coding transposable element (TE). There are approximately one million copies of the Alu element in the human genome, representing ∼11% of the entire genome (Lander et al., 2001). Active Alu elements transcribed by RNA polymerase Ⅲ are sometimes retrotransposed into the human genome to cause several human diseases by disrupting coding sequences or splicing signals (Deininger, 2011). Thus, Alu elements generally suffer from purifying selection to inactivate the transposing activity by rapidly shortening and accumulating mutations in the poly(A) stretch (Roy-Engel et al., 2002; Comeaux et al., 2009). This heterogeneous poly(A) stretch then becomes a source of microsatellite repeats (Roy-Engel et al., 2002; Comeaux et al., 2009).
It is conspicuous that disease-causing microsatellite repeats are frequently observed in the vicinity of Alu elements, e.g., the GAA motif in Friedreich ataxia (Justice et al., 2001), the CCTG motif in myotonic dystrophy type 2 (DM2) (Kurosaki et al., 2012), the TGGAA/TAAAA motif in SCA31 (Sato et al., 2009), the TTTCA/TTTTA motif in SCA37 (Seixas et al., 2017), the CCCTCT motif in X-linked dystonia-parkinsonism (XDP) (Bragg et al., 2017), the ATTTC/ATTTT repeat motif in FAME1, FAME3, FAME4, and FAME6 (Ishiura et al., 2018; Florian et al., 2019; Yeetong et al., 2019), and AAGGG/AAAAG/AAAGG/AAGAG/AGAGG motifs in CANVAS (Cortese et al., 2019) (Table 1). Abundant Alu elements are supposed to lead to large genomic rearrangements during DNA replication through segmental duplication or Alu-Alu-mediated recombination (Bailey et al., 2003; Hedges and Deininger, 2007). Although TE-mediated genomic instability is potentially involved in the microsatellite repeat instability in these neurological disorders, further mechanistic studies are warranted to establish the exact pathobiology.
Heterogeneity of SCA10 pentanucleotide repeats and associations with disease phenotype
The origin of the mutant SCA10 repeat is unknown, and there are only limited data on the molecular basis of the instability of this repeat. SCA10 repeat expansions are beyond the limits of analysis by conventional Sanger sequencing. However, recent characterization of the complex pattern of ATTCT repeat sequences in normal individuals and some SCA10 families has provided new avenues for understanding the genetic basis and molecular mechanisms underlying SCA10 (Matsuura et al., 2006; Mcfarland et al., 2014; Mcfarland et al., 2015).
In normal individuals, the repeat shows length polymorphism of 9–32 tandem ATTCT units, although some large normal alleles (≥17 repeats) have TTTCT or TTTCT-ATTGT insertions in the 3′ end of the repeat (Matsuura et al., 2006). Initial studies suggested that highly interrupted intermediate alleles of 280 and 850 repeats have reduced penetrance, whereas alleles larger than this range were thought to be fully penetrant (Matsuura et al., 2006; Raskin et al., 2007). However, recent evidence indicates that SCA10 repeat expansions containing ATCCT interruptions lead to contractions during paternal transmission, with no correlation between repeat size and age of onset (McFarland et al., 2013; Mcfarland et al., 2014), contradicting the classical rule of genetic anticipation in repeat expansion diseases. Furthermore, data indicate that ATCCT interruptions appear to be a significant risk factor for an epileptic phenotype in SCA10 (McFarland et al., 2013; Mcfarland et al., 2014). By contrast, pure ATTCT expansion is sometimes associated with parkinsonism (Schüle et al., 2017). These observations suggest that the mechanisms of disease associated with the repeat structure in SCA10 are complex.
Recent progress in next-generation DNA sequencing technology, especially single-molecule real-time (SMRT) sequencing permitting exceptionally long read lengths, has made it possible to determine the entire SCA10 expansion sequence. SCA10 expansions are frequently interrupted by ATCCT ATCCC, ATTCC, ATTTCT, ATATTCT, ATTCTTCT, or ATTCTTCT (Table 1) (Mcfarland et al., 2015; Landrian et al., 2017). Given that certain types of repeat expansion, which typically consist of the TTTCA motif but not the TTTTA motif, accompany the disease phenotype in SCA37, FAME1, FAME2, FAME3, FAME4, FAME6, and FAME7 (Table 1), variant repeats in SCA10 may differentially contribute to the disease phenotype.
At the molecular level, short tandem repeats tend to change the repeat length by forming stable hairpin structures to induce misalignment of DNA strands during DNA replication, which is sometimes prone to expansion (Pearson et al., 2005). Repeat interruptions are generally thought to function as a repeat stabilizing factor by disrupting the long hairpin structure to reduce replication slippage and protect the repeats from expansion (Richards, 2001; Chintalaphani et al., 2021). Additionally, repeat interruptions modulate disease penetrance and severity, which are widely reported in several repeat expansion disorders such as DM1, SCA2, SCA8, and Huntington’s disease (Moseley et al., 2000; Sobczak and Krzyzosiak, 2005; Moseley et al., 2006; Braida et al., 2010; Wright et al., 2019). However, how these repeat interruptions drive the disease phenotype remains poorly understood.
SCA10 repeat interruptions such as ATTTTCT and ATATTCT appear to strengthen assembly with hyperacetylated histones (Hagerman et al., 2009). Since histone acetylation is critical for chromatin disassembly and transcriptional activation (Verdin and Ott, 2015), SCA10 repeat interruptions may upregulate gene expression and induce the accumulation of expanded repeats relative to pure repeats, influencing the SCA10 disease phenotype (Figure 2A). Additionally, histone acetylation may also play a significant role in chromatin decompaction to promote DNA replication (Ruan et al., 2015). The long ATTCT repeat has been shown to function as a DNA unwinding element to induce aberrant DNA replication (Liu et al., 2007). Thus, repeat interruptions may also be involved in the DNA replication process (Figure 2A). However, further studies on the relationship between the interrupted structure of SCA10 repeats and the underlying molecular mechanisms are still needed.
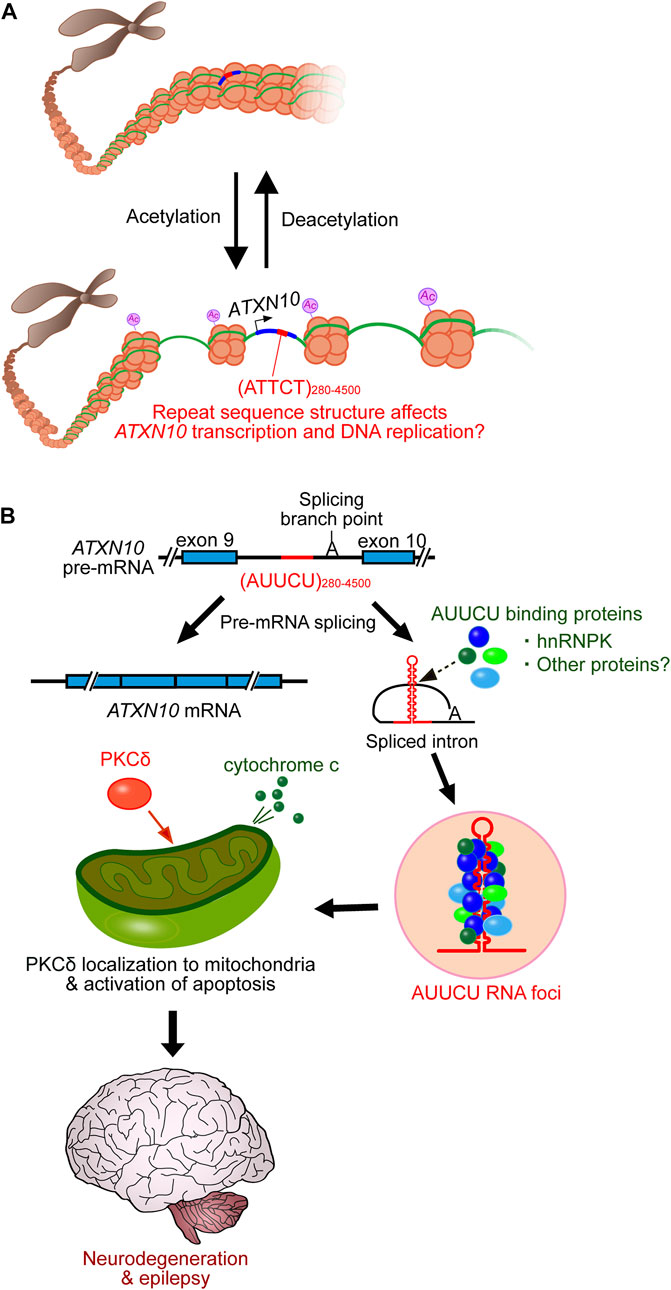
FIGURE 2. SCA10 disease model. (A) SCA10 ATTCT repeat expansion promotes histone acetylation, which subsequently enhances transcription and DNA replication, which may be affected by repeat interruptions. (B) Expanded AUUCU repeats trap hnRNPK in RNA accumulations (RNA foci). Dysfunction of hnRNPK induces PKCδ localization to mitochondria, which subsequently triggers cytochrome c release from mitochondria, activating apoptosis and eventually triggering neurodegeneration.
Molecular mechanisms of SCA10: Toxic RNA-mediated gain-of-function
ATXN10 mRNAs are abundantly expressed in the juvenile (10-day-old) and adult (4-month-old) mouse (Matsuura et al., 2000) and human brain as well as the heart, skeletal muscle, and kidney (Wakamiya et al., 2006). Ataxin 10 appears to play an essential role in neurite genesis (Waragai et al., 2006) and cerebellar neuron survival (März et al., 2004). However, there is no aberrant expression of ATXN10 (including splicing abnormalities) or of its flanking genes FBLN1 and PPARA in SCA10 lymphoblasts, fibroblasts, and myoblasts (Wakamiya et al., 2006). Moreover, while Atxn10 knockout mice show embryonic lethality, heterozygous (Atxn10+/−) mice have no motor phenotype (Wakamiya et al., 2006). In addition, individuals harboring a translocation of chromosome 2p25.3 into intron 2 of ATXN10 (22q13.31) show no SCA10-like symptoms, indicating that ATXN10 haploinsufficiency does not cause the disease (Keren et al., 2010). Thus, a gain or loss of ATXN10 function is unlikely to be the main pathogenic mechanism in SCA10.
Similar to other non-coding repeat expansion disorders, SCA10 is proposed to be caused by a toxic RNA-mediated gain-of-function mechanism (White et al., 2010; White et al., 2012). The AUUCU expansions form RNA accumulations, detected as RNA foci in the nucleus and cytoplasm of SCA10 fibroblasts and lymphoblasts (White et al., 2010). The RNA foci trap heterogeneous nuclear ribonucleoprotein K (hnRNPK) and compromise its function. This dysfunction in hnRNPK induces translocation of protein kinase C (PKC)δ to mitochondria, which subsequently induces an apoptotic pathway by releasing cytochrome c and activating caspase 3 in SCA10 cells (Figure 2B) (White et al., 2010; White et al., 2012). A transgenic mouse model of SCA10 expressing 500 ATTCT repeats within the 3′UTR of the LacZ gene, driven by the prion (Prnp) promoter (White et al., 2012), shares many phenotypic similarities with SCA10 patients, including irregular gait, increased seizure susceptibility, and neuronal loss in the cerebral cortex and hippocampus. However, this mouse does not recapitulate the cerebellar degeneration typically seen in SCA10 in humans (White et al., 2012). Thus, efforts are underway to further model the SCA10 disease phenotype, for instance, by introducing ATTCT repeats with or without repeat interruptions into the intronic region of a gene using either a pan-neuronal neuronal enolase (Eno2) promoter or a Purkinje cell-specific Purkinje cell protein-2 (Pcp2) promoter to express more expanded repeats in the cerebellum and brainstem (McFarland and Ashizawa, 2012). Furthermore, it remains unclear whether other RNA-binding proteins play a role in SCA10 pathogenesis to cause the variable SCA10 phenotype.
An alternative pathogenetic mechanism for SCA10 is bidirectional transcription producing toxic antisense transcripts or repeat-associated non-ATG (RAN) translation, as observed in DM1, DM2, fragile X-associated tremor/ataxia syndrome (FXTAS), C9orf72 amyotrophic lateral sclerosis and frontotemporal dementia ALS/FTD, and SCA8 (Zu et al., 2011; Ash et al., 2013; Todd et al., 2013; Zu et al., 2013; Zu et al., 2017). Translation of SCA10 AUUCU repeats would produce Ile-Leu-Phe-Tyr-Ser (ILFYS) pentapeptide repeats. However, there has yet to be a study of these pentapeptides in SCA10 cells and SCA10 mice, and a thorough molecular analysis is still required.
Moving towards effective SCA10 therapeutics
The molecular basis of the unstable repeat expansion and the underlying disease mechanism in SCA10 patients are still poorly characterized, hampering efforts to develop effective therapeutics for affected individuals. The AUUCU repeat forms an unusual hairpin structure in vitro via the hydrogen bonds formed between A-U and U-U base pairs (Handa et al., 2005). The small molecule dimeric compound 2AU-2 selectively binds to A-U base pairs to disrupt RNA folding and AUUCU accumulations in SCA10 fibroblasts (Yang et al., 2016). Furthermore, 2AU-2 treatment effectively reduced PKCδ localization to mitochondria and reduced apoptosis (Yang et al., 2016). Remarkably, 2AU-2 treatment neither reduced the abundance of normal ATXN10 transcripts nor triggered apoptosis in healthy fibroblasts (Yang et al., 2016). Thus, the bioactivity of 2AU-2, which is applicable not only to SCA10 patients but also to other AU-rich repeat expansion disorders, needs further evaluation in relevant neuronal models.
In addition to small molecules, other RNA silencing therapies could be applicable to SCA10. RNA silencing therapies include antisense oligonucleotides (ASOs) (Mceachin et al., 2020; Schwartz et al., 2021), artificial microRNAs (miRNAs) (Martier et al., 2019), CRISPR-Cas9 system approaches (Batra et al., 2017; Zhang et al., 2020), and DNAzymes (Zhang et al., 2021). SCA10 is suited to these therapeutic strategies because 1) the repeat is in the intron of both normal and expanded alleles and is not translated, so the repeat can be silenced whilst minimizing adverse consequences; 2) the silencing of this expanded pentanucleotide repeat would suppress all downstream pathogenic pathways; 3) no opposite strand transcripts containing the repeat have been detected at the SCA10 locus; and 4) preferential targeting of the mutant RNA may be feasible with therapeutics that directly engage the repeat units because of the unique and large size of mutant repeat alleles. Further mechanistic studies are likely to facilitate the development of effective therapeutics for SCA10.
Conclusion
SCA10 was the first human genetic disorder discovered to be caused by an expanded intronic pentanucleotide repeat. Since the first discovery of the SCA10 mutation, many diseases have subsequently been reported to be caused by expanded intronic pentanucleotide repeats, including SCA31, SCA37, FAME, and CANVAS. Therefore, elucidating the molecular basis of SCA10 will provide more generalizable insights into the disease mechanisms underpinning similar intronic repeat expansion disorders. Furthermore, in the absence of reliable biomarkers and the variable disease onset, this molecular knowledge should provide new avenues for the development of biomarkers so that affected individuals can receive early interventions and support.
While there is currently no therapy for SCA10, advances in sequencing technology, disease models, our understanding of the genetic, transcriptomic, and proteomic features of the consequences of expanded SCA10 repeats, and developments in nucleic acid-based therapies are likely to contribute to the development of a clinically translatable strategy to detect and treat patients with SCA10 and other similar neuronal disorders.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work was supported by grants from the NIH (R01NS115002, R01NS083564, R01NS041547) to TA. and a URMC Schmitt Program on Integrative Neuroscience (SPIN) research grant to TK.
Acknowledgments
We thank Charles Thornton for critical comments on this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Al-mahdawi, S., Ging, H., Bayot, A., Cavalcanti, F., Cognata, V., Giunti, P., et al. (2018). Large interruptions of GAA repeat expansion mutations in Friedreich ataxia are very rare. Front. Cell. Neurosci. 12, 443. doi:10.3389/fncel.2018.00443
Almeida, T., Alonso, I., Martins, S., Ramos, E. M., Azevedo, L., Ohno, K., et al. (2009). Ancestral origin of the ATTCT repeat expansion in spinocerebellar ataxia type 10 (SCA10). PLoS One 4, e4553. doi:10.1371/journal.pone.0004553
Alonso, E., Martı, L., Ochoa, A., Yescas, P., Gutie, R., White, M., et al. (2007). Distinct distribution of autosomal dominant spinocerebellar ataxia in the Mexican population. Mov. Disord. 22, 1050–1053. doi:10.1002/mds.21470
Ash, P. E. A., Bieniek, K. F., Gendron, T. F., Caulfield, T., Lin, W. L., DeJesus-Hernandez, M., et al. (2013). Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to C9FTD/ALS. Neuron 77, 639–646. doi:10.1016/j.neuron.2013.02.004
Bailey, J. A., Liu, G., and Eichler, E. E. (2003). An Alu transposition model for the origin and expansion of human segmental duplications. Am. J. Hum. Genet. 73, 823–834. doi:10.1086/378594
Baizabal-Carvallo, J. F., Xia, G., Botros, P., Laguna, J., Ashizawa, T., and Jankovic, J. (2015). Bolivian kindred with combined spinocerebellar ataxia types 2 and 10. Acta Neurol. Scand. 132, 139–142. doi:10.1111/ane.12371
Batra, R., Nelles, D. A., Pirie, E., Corbett, K. D., Swanson, M. S., Yeo, G. W., et al. (2017). Elimination of toxic microsatellite repeat expansion RNA by RNA-targeting Cas9. Cell 170, 899–912. doi:10.1016/j.cell.2017.07.010
Bragg, D. C., Mangkalaphiban, K., Vaine, C. A., Kulkarni, N. J., Shin, D., Yadav, R., et al. (2017). Disease onset in X-linked dystonia-parkinsonism correlates with expansion of a hexameric repeat within an SVA retrotransposon in TAF1. Proc. Natl. Acad. Sci. U. S. A. 114, E11020–E11028. doi:10.1073/pnas.1712526114
Braida, C., Stefanatos, R. K. A., Adam, B., Mahajan, N., Smeets, H. J. M., Niel, F., et al. (2010). Variant CCG and GGC repeats within the CTG expansion dramatically modify mutational dynamics and likely contribute toward unusual symptoms in some myotonic dystrophy type 1 patients. Hum. Mol. Genet. 19, 1399–1412. doi:10.1093/hmg/ddq015
Bushara, K., Bower, M., Liu, J., Mcfarland, K. N., Landrian, I., Hutter, D., et al. (2013). Expansion of the spinocerebellar ataxia type 10 (SCA10) repeat in a patient with Sioux native American ancestry. PLoS One 8, e81342–11. doi:10.1371/journal.pone.0081342
Campuzano, V., Montermini, L., Moltò, M. D., Pianese, L., Cossée, M., Cavalcanti, F., et al. (1996). Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 80271, 1423–1427. doi:10.1126/science.271.5254.1423
Cen, Z., Jiang, Z., Chen, Y., Zheng, X., Xie, F., Yang, X., et al. (2018). Intronic pentanucleotide TTTCA repeat insertion in the SAMD12 gene causes familial cortical myoclonic tremor with epilepsy type 1. Brain 141, 2280–2288. doi:10.1093/brain/awy160
Corbett, M. A., Kroes, T., Veneziano, L., Bennett, M. F., Florian, R., Schneider, A. L., et al. (2019). Intronic ATTTC repeat expansions in STARD7 in familial adult myoclonic epilepsy linked to chromosome 2. Nat. Commun. 10, 4920. doi:10.1038/s41467-019-12671-y
Chintalaphani, S. R., Pineda, S. S., Deveson, I. W., and Kumar, K. R. (2021). An update on the neurological short tandem repeat expansion disorders and the emergence of long - read sequencing diagnostics. Acta Neuropathol. Commun. 9, 98. doi:10.1186/s40478-021-01201-x
Comeaux, M. S., Roy-Engel, A. M., Hedges, D. J., and Deininger, P. L. (2009). Diverse cis factors controlling Alu retrotransposition: What causes Alu elements to die? Genome Res. 19, 545–555. doi:10.1101/gr.089789.108
Cortese, A., Simone, R., Sullivan, R., Vandrovcova, J., Tariq, H., Yau, W. Y., et al. (2019). Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat. Genet. 51, 649–658. doi:10.1038/s41588-019-0372-4
de Castilhos, R. M., Furtado, G. V., Gheno, T. C., Schaeffer, P., Russo, A., Barsottini, O., et al. (2014). Spinocerebellar ataxias in Brazil — Frequencies and modulating effects of related genes. Cerebellum 13, 17–28. doi:10.1007/s12311-013-0510-y
Deininger, P. (2011). Alu elements: Know the SINEs. Genome Biol. 12, 236–312. doi:10.1186/gb-2011-12-12-236
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi:10.1016/j.neuron.2011.09.011
Deng, J., Yu, J., Li, P., Luan, X., Cao, L., Zhao, J., et al. (2020). Expansion of GGC repeat in GIPC1 is associated with oculopharyngodistal myopathy. Am. J. Hum. Genet. 106, 793–804. doi:10.1016/j.ajhg.2020.04.011
Domingues, B. M. D., Nascimento, F. A., Meira, A. T., Moro, A., Raskin, S., Ashizawa, T., et al. (2019). Clinical and genetic evaluation of spinocerebellar ataxia type 10 in 16 Brazilian families. Cerebellum 18, 849–854. doi:10.1007/s12311-019-01064-y
Florian, R. T., Kraft, F., Leitao, E., Kaya, S., Klebe, S., Magnin, E., et al. (2019). Unstable TTTTA/TTTCA expansions in MARCH6 are associated with familial adult myoclonic epilepsy type 3. Nat. Commun. 10, 4919. doi:10.1038/s41467-019-12763-9
Fu, Y. H., Kuhl, D. P. A., Pizzuti, A., Pieretti, M., Sutcliffe, J. S., Richards, S., et al. (1991). Variation of the CGG repeat at the fragile X site results in genetic instability: Resolution of the Sherman paradox. Cell 67, 1047–1058. doi:10.1016/0092-8674(91)90283-5
García-Murias, M., Quintáns, B., Arias, M., Seixas, A. I., Cacheiro, P., Tarrío, R., et al. (2012). Costa da Morte” ataxia is spinocerebellar ataxia 36: Clinical and genetic characterization. Brain 135, 1423–1435. doi:10.1093/brain/aws069
Gatto, E. M., Gao, R., White, M. C., Uribe Roca, M. C., Etcheverry, J. L., Persi, G., et al. (2007). Ethnic origin and extrapyramidal signs in an Argentinean spinocerebellar ataxia type 10 family. Neurology 69, 216–218. doi:10.1212/01.wnl.0000265596.72492.89
Gijselinck, I., Van Mossevelde, S., Van Der Zee, J., Sieben, A., Engelborghs, S., De Bleecker, J., et al. (2016). The C9orf72 repeat size correlates with onset age of disease, DNA methylation and transcriptional downregulation of the promoter. Mol. Psychiatry 21, 1112–1124. doi:10.1038/mp.2015.159
Grewal, R. P., Achari, M., Matsuura, T., Durazo, A., Tayag, E., Zu, L., et al. (2002). Clinical features and ATTCT repeat expansion in spinocerebellar ataxia type 10. Arch. Neurol. 59, 1285–1290. doi:10.1001/archneur.59.8.1285
Hagerman, K., Ruan, H., Edamura, K. N., Matsuura, T., Pearson, C. E., and Wang, Y.-H. (2009). The ATTCT repeats of spinocerebellar ataxia type 10 display strong nucleosome assembly which is enhanced by repeat interruptions. Gene 434, 29–34. doi:10.1016/j.gene.2008.12.011
Hagerman, R. J., Berry-Kravis, E., Hazlett, H. C., Bailey, D. B., Moine, H., Kooy, R. F., et al. (2017). Fragile X syndrome. Nat. Rev. Dis. Prim. 3, 17065. doi:10.1038/nrdp.2017.65
Handa, V., Yeh, H. J. C., McPhie, P., and Usdin, K. (2005). The AUUCU repeats responsible for spinocerebellar ataxia type 10 form unusual RNA hairpins. J. Biol. Chem. 280, 29340–29345. doi:10.1074/jbc.M503495200
Hashem, V., Tiwari, A., Bewick, B., Teive, H. A. G., Moscovich, M., Schüele, B., et al. (2020). Pulse-Field capillary electrophoresis of repeat-primed PCR amplicons for analysis of large repeats in Spinocerebellar Ataxia Type 10. PLoS One 15, e0228789. doi:10.1371/journal.pone.0228789
Hedges, D. J., and Deininger, P. L. (2007). Inviting instability: Transposable elements, double-strand breaks, and the maintenance of genome integrity. Mutat. Res. 616, 46–59. doi:10.1016/j.mrfmmm.2006.11.021
Hernandez-Castillo, C. R., Diaz, R., Vaca-Palomares, I., Torres, D. L., Chirino, A., Campos-Romo, A., et al. (2019). Extensive cerebellar and thalamic degeneration in spinocerebellar ataxia type 10. Park. Relat. Disord. 66, 182–188. doi:10.1016/j.parkreldis.2019.08.011
Holmes, S. E., O’Hearn, E. E., Mclnnis, M. G., Gorelick-Feldman, D. A., Kleiderlein, J. J., Callahan, C., et al. (1999). Expansion of a novel CAG trinucleotide repeat in the 5’ region of PPP2R2B is associated with SCA12. Nat. Genet. 23, 391–392. doi:10.1038/70493
Ishiura, H., Koichiro, D., Jun, M., Jun, Y., Kawabe Matsukawa, M., Fujiyama, A., et al. (2018). Expansions of intronic TTTCA and TTTTA repeats in benign adult familial myoclonic epilepsy. Nat. Genet. 12, 581–590. doi:10.1038/s41588-018-0067-2
Ishiura, H., Shibata, S., Yoshimura, J., Suzuki, Y., Qu, W., Doi, K., et al. (2019). Noncoding CGG repeat expansions in neuronal intranuclear inclusion disease, oculopharyngodistal myopathy and an overlapping disease. Nat. Genet. 51, 1222–1232. doi:10.1038/s41588-019-0458-z
Justice, C. M., Den, Z., Nguyen, S. V., Stoneking, M., Deininger, P. L., Batzer, M. A., et al. (2001). Phylogenetic analysis of the Friedreich ataxia GAA trinucleotide repeat. J. Mol. Evol. 52, 232–238. doi:10.1007/s002390010151
Keren, B., Jacquette, A., Depienne, C., Leite, P., Durr, A., Carpentier, W., et al. (2010). Evidence against haploinsuffiency of human ataxin 10 as a cause of spinocerebellar ataxia type 10. Neurogenetics 11, 273–274. doi:10.1007/s10048-009-0227-8
Knight, S. J. L., Flannery, A. V., Hirst, M. C., Campbell, L., Christodoulou, Z., Phelps, S. R., et al. (1993). Trinucleotide repeat amplification and hypermethylation of a CpG island in FRAXE mental retardation. Cell 74, 127–134. doi:10.1016/0092-8674(93)90300-f
Knight, S. J. L., Voelckel, M. A., Hirst, M. C., Flannery, A. V., Moncla, A., and Davies, K. E. (1994). Triplet repeat expansion at the FRAXE locus and X-linked mild mental handicap. Am. J. Hum. Genet. 55, 81–86.
Kobayashi, H., Abe, K., Matsuura, T., Ikeda, Y., Hitomi, T., Akechi, Y., et al. (2011). Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am. J. Hum. Genet. 89, 121–130. doi:10.1016/j.ajhg.2011.05.015
Koob, M. D., Moseley, M. L., Schut, L. J., Benzow, K. A., Bird, T. D., Day, J. W., et al. (1999). An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8). Nat. Genet. 21, 379–384. doi:10.1038/7710
Kurosaki, T., Matsuura, T., Ohno, K., and Ueda, S. (2009). Alu-mediated acquisition of unstable ATTCT pentanucleotide repeats in the human ATXN10 gene. Mol. Biol. Evol. 26, 2573–2579. doi:10.1093/molbev/msp172
Kurosaki, T., Matsuura, T., Ohno, K., and Ueda, S. (2008). Long-range PCR for the diagnosis of spinocerebellar ataxia type 10. Neurogenetics 9, 151–152. doi:10.1007/s10048-007-0117-x
Kurosaki, T., Ueda, S., Ishida, T., Abe, K., Ohno, K., and Matsuura, T. (2012). The unstable CCTG repeat responsible for myotonic dystrophy type 2 originates from an AluSx element insertion into an early primate genome. PLoS One 7, e38379. doi:10.1371/journal.pone.0038379
Lacroix, A. J., Stabley, D., Sahraoui, R., Adam, M. P., Mehaffey, M., Kernan, K., et al. (2019). GGC repeat expansion and exon 1 methylation of XYLT1 is a common pathogenic variant in Baratela-Scott syndrome. Am. J. Hum. Genet. 104, 35–44. doi:10.1016/j.ajhg.2018.11.005
Lalioti, M. D., Scott, H. S., Buresit, C., Rossier, C., Bottani, A., Morris, M. A., et al. (1997). Dodecamer repeat expansion in cystatin B gene in progressive myoclonus epilepsy. Nature 386, 847–851. doi:10.1038/386847a0
Lalioti, M. D., Scott, H. S., Genton, P., Grid, D., Rabet, A. M., Ibrahim, S., et al. (1998). A PCR amplification method reveals instability of the dodecamer repeat in progressive myoclonus epilepsy (EPM1) and no correlation between the size of the repeat and age at onset. Am. J. Hum. Genet. 62, 842–847. doi:10.1086/301798
Lander, E. S., Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J., et al. (2001). Initial sequencing and analysis of the human genome. Nature 409, 860–921. doi:10.1038/35057062
Landrian, I., Mcfarland, K. N., Liu, J., Mulligan, C. J., Rasmussen, A., and Ashizawa, T. (2017). Inheritance patterns of ATCCT repeat interruptions in spinocerebellar ataxia type 10 (SCA10) expansions. PLoS One 12, e0175958. doi:10.1371/journal.pone.0175958
Leonardi, L., Marcotulli, C., Mcfarland, K. N., Tessa, A., Difabio, R., Santorelli, F. M., et al. (2014). Spinocerebellar ataxia type 10 in Peru : The missing link in the amerindian origin of the disease. J. Neurol. 57, 1691–1694. doi:10.1007/s00415-014-7394-8
Lin, X., and Ashizawa, T. (2005). Recent progress in spinocerebellar ataxia type 10 (SCA10). Cerebellum 4, 37–42. doi:10.1080/14734220510007897
Lin, X., and Ashizawa, T. (2003). SCA10 and ATTCT repeat expansion : Clinical features and molecular aspects. Cytogenet. Genome Res. 188, 184–188. doi:10.1159/000072853
Liquori, C. L., Ricker, K., Moseley, M. L., Jacobsen, J. F., Kress, W., Naylor, S. L., et al. (2001). Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 293, 864–867. doi:10.1126/science.1062125
Liu, G., Bissler, J. J., Sinden, R. R., and Leffak, M. (2007). Unstable spinocerebellar ataxia type 10 (ATTCT)(AGAAT) repeats are associated with aberrant replication at the ATX10 locus and replication origin-dependent expansion at an ectopic site in human cells. Mol. Cell. Biol. 27, 7828–7838. doi:10.1128/MCB.01276-07
Mahadevan, M., Tsilfidis, C., Sabourin, L., Shutler, G., Amemiya, C., Jansen, G., et al. (1992). Myotonic dystrophy mutation: An unstable CTG repeat in the 3’ untranslated region of the gene. Science 255, 1253–1255. doi:10.1126/science.1546325
Mao, C., Li, X., Su, Y., Luo, H., Fan, L., Zheng, H., et al. (2022). Spinocerebellar ataxia type 10 with atypical clinical manifestation in Han Chinese. Cerebellum. doi:10.1007/s12311-022-01405-4
Martier, R., Liefhebber, J. M., Miniarikova, J., Zon, T. V., Snapper, J., Kolder, I., et al. (2019). Artificial microRNAs targeting C9orf72 can reduce accumulation of intra-nuclear transcripts in ALS and FTD patients. Mol. Ther. Nucleic Acids 14, 593–608. doi:10.1016/j.omtn.2019.01.010
März, P., Probst, A., Lang, S., Schwager, M., Rose-John, S., Otten, U., et al. (2004). Ataxin-10, the spinocerebellar ataxia type 10 neurodegenerative disorder protein, is essential for survival of cerebellar neurons. J. Biol. Chem. 279, 35542–35550. doi:10.1074/jbc.M405865200
Matsuura, T., Achari, M., Khajavi, M., Bachinski, L. L., Zoghbi, H. Y., and Ashizawa, R. (1999). Mapping of the gene for a novel spinocerebellar ataxia with pure cerebellar signs and epilepsy. Ann. Neurol. 45, 407–411. doi:10.1002/1531-8249(199903)45:3<407::aid-ana21>3.0.co;2-d
Matsuura, T., Fang, P., Lin, X., Khajavi, M., Tsuji, K., Rasmussen, A., et al. (2004). Somatic and germline instability of the ATTCT repeat in spinocerebellar ataxia type 10. Am. J. Hum. Genet. 74, 1216–1224. doi:10.1086/421526
Matsuura, T., Fang, P., Pearson, C. E., Jayakar, P., Ashizawa, T., Roa, B. B., et al. (2006). Interruptions in the expanded ATTCT repeat of spinocerebellar ataxia type 10: Repeat purity as a disease modifier? Am. J. Hum. Genet. 78, 125–129. doi:10.1086/498654
Matsuura, T., Ranum, L. P. W., Volpini, V., Pandolfo, M., Sasaki, H., Tashiro, K., et al. (2002). Spinocerebellar ataxia type 10 is rare in populations other than Mexicans. Neurology 58, 983–984. doi:10.1212/WNL.58.6.983
Matsuura, T., Yamagata, T., Burgess, D. L., Rasmussen, A., Grewal, R. P., Watase, K., et al. (2000). Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat. Genet. 26, 191–194. doi:10.1038/79911
Mceachin, Z. T., Parameswaran, J., Raj, N., Bassell, G. J., and Jiang, J. (2020). RNA-mediated toxicity in C9orf72 ALS and FTD. Neurobiol. Dis. 145, 105055. doi:10.1016/j.nbd.2020.105055
McFarland, K. N., and Ashizawa, T. (2012). Transgenic models of spinocerebellar ataxia type 10: Modeling a repeat expansion disorder. Genes (Basel) 3, 481–491. doi:10.3390/genes3030481
McFarland, K. N., Liu, J., Landrian, I., Gao, R., Sarkar, P. S., Raskin, S., et al. (2013). Paradoxical effects of repeat interruptions on spinocerebellar ataxia type 10 expansions and repeat instability. Eur. J. Hum. Genet. 21, 1272–1276. doi:10.1038/ejhg.2013.32
Mcfarland, K. N., Liu, J., Landrian, I., Godiska, R., Shanker, S., Yu, F., et al. (2015). SMRT sequencing of long tandem nucleotide repeats in SCA10 reveals unique insight of repeat expansion structure. PLoS One 10, e0135906. doi:10.1371/journal.pone.0135906
Mcfarland, K. N., Liu, J., Landrian, I., Zeng, D., Raskin, S., Moscovich, M., et al. (2014). Repeat interruptions in spinocerebellar ataxia type 10 expansions are strongly associated with epileptic seizures. Neurogenetics 15, 59–64. doi:10.1007/s10048-013-0385-6
Montermini, L., Andermann, E., Labuda, M., Richter, A., Pandolfo, M., Cavalcanti, F., et al. (1997). The Friedreich ataxia GAA triplet repeat: Premutation and normal alleles. Hum. Mol. Genet. 6, 1261–1266. doi:10.1093/hmg/6.8.1261
Mootha, V. V., Gong, X., Ku, H., and Xing, C. (2014). Association and familial segregation of CTG18.1 trinucleotide repeat expansion of TCF4 gene in Fuchs’ endothelial corneal dystrophy. Invest. Ophthalmol. Vis. Sci. 55, 33–42. doi:10.1167/iovs.13-12611
Moseley, M. L., Schut, L. J., Bird, T. D., Koob, M. D., Day, J. W., and Ranum, L. P. W. (2000). SCA8 CTG repeat : En masse contractions in sperm and intergenerational sequence changes may play a role in reduced penetrance. Hum. Mol. Genet. 9, 2125–2130. doi:10.1093/hmg/9.14.2125
Moseley, M. L., Zu, T., Ikeda, Y., Gao, W., Mosemiller, A. K., Daughters, R. S., et al. (2006). Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat. Genet. 38, 758–769. doi:10.1038/ng1827
Naito, H., Takahashi, T., Kamada, M., Morino, H., Yoshino, H., Hattori, N., et al. (2017). First report of a Japanese family with spinocerebellar ataxia type 10: The second report from Asia after a report from China. PLoS One 12, e0177955. doi:10.1371/journal.pone.0177955
Nascimento, F. A., Rodrigues, V. O. R., Pelloso, F. C., Henrique, C., Camargo, F., Moro, A., et al. (2019). Spinocerebellar ataxias in Southern Brazil: Genotypic and phenotypic evaluation of 213 families. Clin. Neurol. Neurosurg. 184, 105427. doi:10.1016/j.clineuro.2019.105427
Paradisi, I., Ikonomu, V., and Arias, S. (2015). Spinocerebellar ataxias in Venezuela: Genetic epidemiology and their most likely ethnic descent. J. Hum. Genet. 61, 215–222. doi:10.1038/jhg.2015.131
Pearson, C. E., Edamura, K. N., and Cleary, J. D. (2005). Repeat instability: Mechanisms of dynamic mutations. Nat. Rev. Genet. 6, 729–742. doi:10.1038/nrg1689
Ramirez-Garcia, S. A., Sánchez-Corona, J., Volpini-Bertran, V., Moran-Moguel, M. C., Gutiérrez-Rubio, S. A., Castañeda-Cisneros, G., et al. (2022). A Female case of spinocerebellar ataxia type 10 with suicidal behavior and endocrinpathies associated with a massive expansion (ATTCT) of the gene ATXN10. Actas Esp. Psiquiatr. 50, 58–62.
Raskin, S., Ashizawa, T., Teive, H. a. G., Arruda, W. O., Fang, P., Gao, R., et al. (2007). Reduced penetrance in a Brazilian family with spinocerebellar ataxia type 10. Arch. Neurol. 64, 591–594. doi:10.1001/archneur.64.4.591
Rasmussen, A., Matsuura, T., Ruano, L., Yescas, P., Ochoa, A., Ashizawa, T., et al. (2001). Clinical and genetic analysis of four Mexican families with spinocerebellar ataxia type 10. Ann. Neurol. 50, 234–239. doi:10.1002/ana.1081
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi:10.1016/j.neuron.2011.09.010
Richards, R. I. (2001). Dynamic mutations : A decade of unstable expanded repeats in human genetic disease. Hum. Mol. Genet. 10, 2187–2194. doi:10.1093/hmg/10.20.2187
Rodríguez-Labrada, R., Martins, A. C., Magaña, J. J., Vazquez-mojena, Y., Medrano-montero, J., Fernandez-ruíz, J., et al. (2020). Founder effects of spinocerebellar ataxias in the American continents and the Caribbean. Cerebellum 19, 446–458. doi:10.1007/s12311-020-01109-7
Roxburgh, R. H., Smith, C. O., Lim, J. G., Bachman, D. F., Byrd, E., and Bird, T. D. (2013). The unique co-occurrence of spinocerebellar ataxia type 10 (SCA10) and Huntington disease. J. Neurol. Sci. 324, 176–178. doi:10.1016/j.jns.2012.09.030
Roy-Engel, A. M., Salem, A. H., Oyeniran, O. O., Deininger, L., Hedges, D. J., Kilroy, G. E., et al. (2002). Active Alu element “A-tails”: Size does matter. Genome Res. 12, 1333–1344. doi:10.1101/gr.384802
Ruan, K., Yamamoto, T. G., Asakawa, H., Chikashige, Y., Kimura, H., Masukata, H., et al. (2015). Histone H4 acetylation required for chromatin decompaction during DNA replication. Sci. Rep. 5, 12720. doi:10.1038/srep12720
Sato, N., Amino, T., Kobayashi, K., Asakawa, S., Ishiguro, T., Tsunemi, T., et al. (2009). Spinocerebellar ataxia type 31 is associated with ‘“Inserted”’ penta-nucleotide repeats containing (TGGAA)n. Am. J. Hum. Genet. 85, 544–557. doi:10.1016/j.ajhg.2009.09.019
Schmitz-Hübsch, T., Du Montcel, S. T., Baliko, L., Berciano, J., Boesch, S., Depondt, C., et al. (2006). Scale for the assessment and rating of ataxia: Development of a new clinical scale. Neurology 66, 1717–1720. doi:10.1212/01.wnl.0000219042.60538.92
Schüle, B., McFarland, K. N., Lee, K., Tsai, Y.-C., Nguyen, K.-D., Sun, C., et al. (2017). Parkinson’s disease associated with pure ATXN10 repeat expansion. NPJ Park. Dis. 3, 27. doi:10.1038/s41531-017-0029-x
Schwartz, J. L., Jones, K. L., Yeo, G. W., Schwartz, J. L., Jones, K. L., and Yeo, G. W. (2021). Repeat RNA expansion disorders of the nervous system: Post-transcriptional mechanisms and therapeutic strategies. Crit. Rev. Biochem. Mol. Biol. 56, 31–53. doi:10.1080/10409238.2020.1841726
Seixas, A. I., Loureiro, J. R., Costa, C., Ordóñez-Ugalde, A., Marcelino, H., Oliveira, C. L., et al. (2017). A pentanucleotide ATTTC repeat insertion in the non-coding region of DAB1, mapping to SCA37, causes spinocerebellar ataxia. Am. J. Hum. Genet. 101, 87–103. doi:10.1016/j.ajhg.2017.06.007
Seixas, A. I., Maurer, M. H., Lin, M., Callahan, C., Ahuja, A., Matsuura, T., et al. (2005). FXTAS, SCA10, and SCA17 in American patients with movement disorders. Am. J. Med. Genet. A 136, 87–89. doi:10.1002/ajmg.a.30761
Sobczak, K., and Krzyzosiak, W. J. (2005). CAG repeats containing CAA interruptions form branched hairpin structures in spinocerebellar ataxia type 2 transcripts. J. Biol. Chem. 280, 3898–3910. doi:10.1074/jbc.M409984200
Srivastava, A. K., Takkar, A., Garg, A., and Faruq, M. (2017). Clinical behaviour of spinocerebellar ataxia type 12 and intermediate length abnormal CAG repeats in PPP2R2B. Brain 140, 27–36. doi:10.1093/brain/aww269
Teive, H. A. G., Munhoz, R. P., Raskin, S., Arruda, W. O., Paola, L., Werneck, L. C., et al. (2010). Spinocerebellar Ataxia Type 10: Frequency of epilepsy in a large sample of Brazilian patients. Mov. Disord. 25, 2875–2878. doi:10.1002/mds.23324
Teive, H. A. G., Roa, B. B., Raskin, S., Fang, P., Arruda, W. O., Neto, Y. C., et al. (2004). Clinical phenotype of Brazilian families with spinocerebellar ataxia 10. Neurology 63, 150963–151512. doi:10.1212/01.wnl.0000142109.62056.57
Tensini, F. S., Sato, M. T., Shiokawa, N., Ashizawa, T., and Teive, H. A. G. (2017). A comparative optical coherence tomography study of spinocerebellar ataxia types 3 and 10. Cerebellum 16, 797–801. doi:10.1007/s12311-017-0856-7
Todd, P. K., Oh, S. Y., Krans, A., He, F., Sellier, C., Frazer, M., et al. (2013). CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron 78, 440–455. doi:10.1016/j.neuron.2013.03.026
Turner, C., and Hilton-jones, D. (2010). The myotonic dystrophies: Diagnosis and management. J. Neurol. Neurosurg. Psychiatry 81, 358–367. doi:10.1136/jnnp.2008.158261
van Kuilenburg, A. B. P., Tarailo-Graovac, M., Richmond, P. A., Drögemöller, B. I., Pouladi, M. A., Leen, R., et al. (2019). Glutaminase deficiency caused by short tandem repeat expansion in GLS. N. Engl. J. Med. 380, 1433–1441. doi:10.1056/NEJMoa1806627
Verdin, E., and Ott, M. (2015). 50 years of protein acetylation: From gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 16, 258–264. doi:10.1038/nrm3931
Wakamiya, M., Matsuura, T., Liu, Y., Schuster, G. C., Gao, R., Xu, W., et al. (2006). The role of ataxin 10 in the pathogenesis of spinocerebellar ataxia type 10. Neurology 67, 607–613. doi:10.1212/01.wnl.0000231140.26253.eb
Wang, K., McFarland, K., Liu, J., Ms, M. D., Zeng, D., Landrian, I., et al. (2015). Spinocerebellar ataxia type 10 in Chinese Han. Neurol. Genet. 1, e26. doi:10.1212/NXG.0000000000000026
Waragai, M., Nagamitsu, S., Xu, W., Li, Y. J., Lin, X., and Ashizawa, T. (2006). Ataxin 10 induces neuritogenesis via interaction with G-protein beta2 subunit. J. Neurosci. Res. 83, 1170–1178. doi:10.1002/jnr.20807
White, M. C., Gao, R., Xu, W., Mandal, S. M., Lim, J. G., Hazra, T. K., et al. (2010). Inactivation of hnRNP K by expanded intronic AUUCU repeat induces apoptosis via translocation of PKCdelta to mitochondria in spinocerebellar ataxia 10. PLoS Genet. 6, e1000984. doi:10.1371/journal.pgen.1000984
White, M., Xia, G., Gao, R., Wakamiya, M., Sarkar, P. S., McFarland, K., et al. (2012). Transgenic mice with SCA10 pentanucleotide repeats show motor phenotype and susceptibility to seizure: A toxic RNA gain-of-function model. J. Neurosci. Res. 90, 706–714. doi:10.1002/jnr.22786
Wieben, E. D., Aleff, R. A., Rinkoski, T. A., Baratz, K. H., Basu, S., Id, S. V. P., et al. (2021). Comparison of TCF4 repeat expansion length in corneal endothelium and leukocytes of patients with Fuchs endothelial corneal dystrophy. PLoS One 4, e0260837. doi:10.1371/journal.pone.0260837
Willemsen, R., Levenga, J., and Oostra, B. A. (2013). CGG repeat in the FMR1 gene: Size matters. Clin. Genet. 80, 214–225. doi:10.1111/j.1399-0004.2011.01723.x
Wright, G. E. B., Collins, J. A., Kay, C., McDonald, C., Dolzhenko, E., Xia, Q., et al. (2019). Length of uninterrupted CAG, independent of polyglutamine size, results in increased somatic instability, hastening onset of Huntington disease. Am. J. Hum. Genet. 104, 1116–1126. doi:10.1016/j.ajhg.2019.04.007
Xi, J., Wang, X., Yue, D., Dou, T., Wu, Q., Lu, J., et al. (2021). 5’ UTR CGG repeat expansion in GIPC1 is associated with oculopharyngodistal myopathy. Brain. 144, 601–614. doi:10.1093/brain/awaa426
Yang, W.-Y., Gao, R., Southern, M., Sarkar, P. S., and Disney, M. D. (2016). Design of a bioactive small molecule that targets r(AUUCU) repeats in spinocerebellar ataxia 10. Nat. Commun. 7, 11647. doi:10.1038/ncomms11647
Yeetong, P., Pongpanich, M., Srichomthong, C., Assawapitaksakul, A., Shotelersuk, V., Tantirukdham, N., et al. (2019). TTTCA repeat insertions in an intron of YEATS2 in benign adult familial myoclonic epilepsy type 4. Brain 142, 3360–3366. doi:10.1093/brain/awz267
Zhang, N., Bewick, B., Schultz, J., Tiwari, A., Krencik, R., Zhang, A., et al. (2021). DNAzyme cleavage of CAG repeat RNA in polyglutamine diseases. Neurotherapeutics 18, 1710–1728. doi:10.1007/s13311-021-01075-w
Zhang, N., Bewick, B., Xia, G., Furling, D., and Ashizawa, T. (2020). A CRISPR-Cas13a based strategy that tracks and degrades toxic RNA in myotonic dystrophy type 1. Front. Genet. 11, 594576–594613. doi:10.3389/fgene.2020.594576
Zonta, M. B., Teive, H. A. G., Camargo, C. H. F., Meira, A. T., Neto, F. D. N. L., Tensini, F. S., et al. (2022). Comparing loss of balance and functional capacity among patients with SCA2, SCA3 and SCA10. Clin. Neurlogy Neurosurg. 214, 107150. doi:10.1016/j.clineuro.2022.107150
Zu, L., Figueroa, K. P., Grewal, R., and Pulst, S. M. (1999). Mapping of a new autosomal dominant spinocerebellar ataxia to chromosome 22. Am. J. Hum. Genet. 64, 594–599. doi:10.1086/302247
Zu, T., Cleary, J. D., Liu, Y., Bañez-Coronel, M., Bubenik, J. L., Ayhan, F., et al. (2017). RAN translation regulated by muscleblind proteins in myotonic dystrophy type 2. Neuron 95, 1292–1305. e5. doi:10.1016/j.neuron.2017.08.039
Zu, T., Gibbens, B., Doty, N. S., Gomes-Pereira, M., Huguet, A., Stone, M. D., et al. (2011). Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. U. S. A. 108, 260–265. doi:10.1073/pnas.1013343108
Keywords: spinocerebellar ataxia type 10, intronic repeat expansion, pentanucleotide repeat, repeat interruption, RNA-gain-of-function mechanism
Citation: Kurosaki T and Ashizawa T (2022) The genetic and molecular features of the intronic pentanucleotide repeat expansion in spinocerebellar ataxia type 10. Front. Genet. 13:936869. doi: 10.3389/fgene.2022.936869
Received: 05 May 2022; Accepted: 25 August 2022;
Published: 15 September 2022.
Edited by:
John Douglas Cleary, State University of New York at Albany, United StatesReviewed by:
Jose Laffita Mesa, Karolinska Institutet (KI), SwedenMarija Cvetanovic, University of Minnesota Twin Cities, United States
Hannah Shorrock, University at Albany, United States
Copyright © 2022 Kurosaki and Ashizawa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tatsuaki Kurosaki, dGF0c3Vha2lfa3Vyb3Nha2lAdXJtYy5yb2NoZXN0ZXIuZWR1; Tetsuo Ashizawa, dGFzaGl6YXdhQGhvdXN0b25tZXRob2Rpc3Qub3Jn