 Yu Tian1,2
Yu Tian1,2 Xiaochuan Wu
Xiaochuan Wu Liang Zhou
Liang Zhou
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet. , 14 November 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.934829
Purpose: The study aimed to report a rare case of a patient with Alport syndrome, which was manifested as unilateral non-infectious uveitis after bilateral cataract surgery.
Methods: A case report.
Results: A 2-year-old boy was diagnosed with unilateral panuveitis based on the clinical and multimodal imaging findings. Intraocular fluid samples for metagenomic next-generation sequencing (mNGS) and microbial culture were negative. However, urine tests found proteinuria and microscopic hematuria. Pathologic findings of the kidney revealed a thickened membrane, and a diagnosis of Alport syndrome was considered. Gene analysis found deletions in exon 1 of COL4A5 and exons 1 and 2 of COL4A6. The uveitis resolved gradually, following the administration of oral steroids.
Conclusion: Uveitis may be an ocular manifestation of Alport syndrome.
Alport syndrome (AS) is a hereditary disease that involves the basement membrane of the glomerulus, cochlea, lens capsule, and retina. The ocular manifestations reported include anterior lenticonus, “dot and fleck” retinopathy, temporal macular thinning, and vitelliform maculopathy (Savige and ColvilleOpinion, 2009). In this report, we present a case of Alport syndrome manifested as unilateral non-infectious uveitis, which was successfully treated with steroids.
A 2-year-old boy with redness and hazy vitreous in the right eye was referred to our clinic. The patient underwent bilateral cataract surgeries 1 year ago. A review of the surgical recording and ultrasound revealed previous total cataracts, as well as whitening of nuclei in the cortex of both eyes. One month ago, in the cataract clinic, his mother reported redness and blurred vision in his right eye, and endophthalmitis was suspected.
Ophthalmic examination under anesthesia revealed normal intraocular pressure in both eyes (11 mmHg OD and 12 mmHg OS). The right eye had combined ciliary and conjunctival congestion, generally clear cornea, absent lens, and opacified vitreous. Ultrasonography demonstrated multiple hyperechogenic dots in the anterior chamber and vitreous opacity, indicating inflammation (Figures 1A,B). Wild-field fundus photography revealed a congested optic nerve head through the opacified vitreous (Figure 1C). Fluorescein angiography showed the retinal capillaries and hyperfluorescent optic disc leakage at the venous phase (Figure 1D). A single dose of combined vancomycin (1 mg/0.1 ml), ceftazidime (2.2 mg/0.1 ml), and dexamethasone (0.5 mg/0.1 ml) was injected consequently into the vitreous cavity. The aqueous fluid was drawn and analyzed via metagenomic next-generation sequencing (mNGS). Based on a published protocol, this technology can detect 20,343 different types of microbes including bacteria, viruses, fungi, parasites, and other pathogens (Beijing Zhide Medical Laboratory Co., Ltd., an Illumina NextSeq platform) (Qian et al., 2022). Briefly, the intraocular fluid sample was extracted and purified using a TIANamp micro DNA kit (TIANGEN, Beijing, P.R.C). Qualified DNA was applied to construct DNA libraries using the QIASeq™ Ultralow Input Library Kit (QIAGEN, Hilden, Germany). Qualified libraries were sequenced on a NextSeq 550 platform (Illumina, San Diego, United States). After obtaining the sequencing data, high-quality data were generated after filtering out the adapter, low-quality, low-complexity, and shorter reads. Next, human reads were removed by mapping the reads to the human reference genome by SNAP software. The remaining data were aligned to the microbial genome database using Burrows–Wheeler alignment. However, the result was negative. Inflammatory factor analysis of the aqueous fluid found elevated levels of VEGF at 97.7 pg/ml (normal range 1–40), basic fibroblast growth factor (BFGF) of 4.9 pg/ml (normal range <1.0), interleukin (IL)-6 of 7524.5 pg/ml (normal range 1.0–50.0), vascular adhesion molecule (VCAM) of 11066.9 pg/ml (normal range 200–1,000), and IL-8 of 3023.1 pg/ml (normal range 0–20.0). Finding no obvious cause for the vitreous inflammation, a vitreous sample was collected via vitrectomy. The sample was cultured for bacteria and fungi, and T-SPOT and procalcitonin tests were conducted to rule out tuberculosis and sepsis. All the results came out negative.
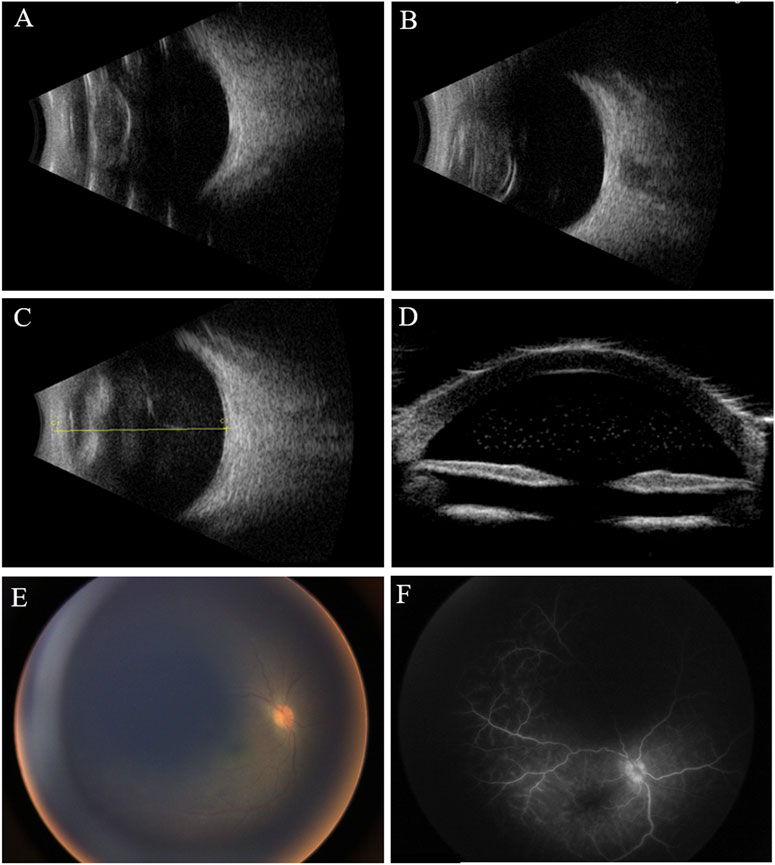
FIGURE 1. Clinical examination of the eyes at different times. (A,B) B-scan ultrasound shows opacified but otherwise normal-shaped lenses in both eyes before cataract surgeries. (C) B-scan ultrasound shows opacified vitreous 1 year after the cataract surgery; (D) ultrasound biomicroscopy (UBM) shows hyperechogenic dots in the anterior chamber; (E) fundus photography reveals hazy vitreous and congested optic nerve head; (F) venous phase of fluorescein angiography shows leakage of retinal capillaries and a hyperfluorescent optic nerve head.
The patient was generally well-developed with no obvious abnormality on systemic examination. The family history was not remarkable. A systemic evaluation was performed when a diagnosis of unilateral non-infectious uveitis was considered. The immunological and rheumatological evaluation was not contributory. However, urinalysis revealed proteinuria and microscopic hematuria. The patient was then transferred to the pediatric department for further diagnosis. Electron microscopic analysis of a renal biopsy showed mesangial proliferation and thickening of the glomerular basement membrane (Figures 2A,B). Based on the pathology, Alport disease was suspected. The patient was subjected to whole-exome sequencing (WES) using the HiSeq X Ten sequencing platform (Illumina, San Diego, California, United States) and by MyGenostics, as described previously (Yang et al., 2022). A deletion in the X chromosome (Xq22.3: NC_000023.11:g.(107681122_107683515)del) encompassing exon 1 of COL4A5 and exons 1 and 2 of COL4A6 (Figure 2C) was found, which was confirmed by multiplex ligation-dependent probe amplification (MLPA). No hearing abnormalities were detected by audiometry. The patient exhibited no signs of dysphagia; however, close follow-ups are advised for this young patient. The child’s mother had neither microscopic hematuria nor ocular manifestation. A family pedigree chart of the patient is shown in Figure 3.
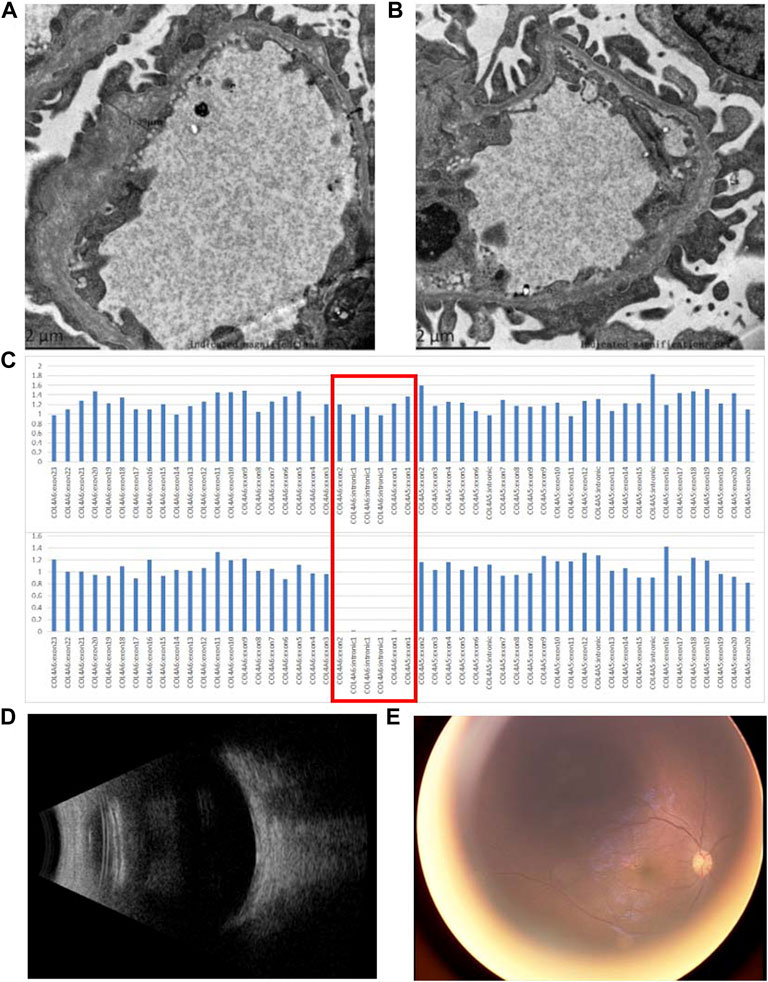
FIGURE 2. Pathological and genetic findings of the patient and the ocular findings after the treatment with steroids. (A,B) Electron microscopic analysis of a renal biopsy shows mesangial proliferation and thickening of the glomerular basement membrane (asterisks); (C) WES revealed that a deletion encompassing exon 1 of COL4A5 and exons 1 and 2 of COL4A6 was found in the patient. The red box indicates exon 1 of COL4A5, intron 1, and exons 1 and 2 of COL4A6. The top panel and bottom panels show the copy number of COL4A5 and COL4A6 from a normal individual and the patient, respectively; (D) B-scan ultrasound reveals a clear vitreous 1 month after treatment with steroids; (E) fundus photography shows resolved vitreous opacity and a clear optic nerve head.
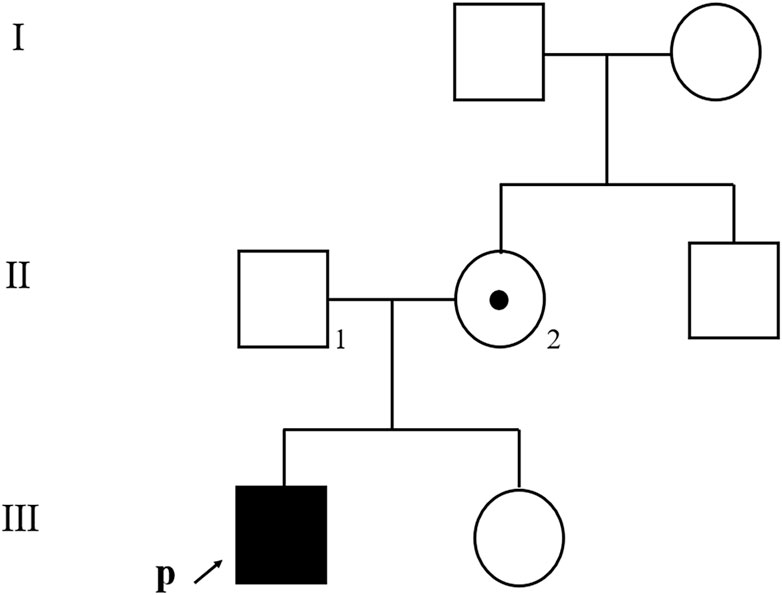
FIGURE 3. Family pedigree chart of the patient. An affected family member is indicated with filled symbols; unaffected relatives are indicated by open symbols; a heterozygous carrier is indicated with a dot in the middle of the symbol. Arrows indicate the proband. Numbers are allotted to the family members whose DNA samples were used in this study.
Oral prednisone was prescribed for uveitis, starting at 15 mg/day and was gradually tampered. One month after steroid treatment, inflammation in the vitreous cavity resolved. Congestion of the optic nerve head was resolved, as shown in fundus photography (Figure 2D). However, proteinuria and hematuria still existed.
Alport syndrome was first reported in a family with both nephritis and deafness by Arthur C. Alport in 1927 (Alport, 1927). The diagnosis of Alport syndrome was based on a combination of characteristic clinical findings (hematuria, lenticonus, retinopathy, etc.), diffuse GBM lamellation in pathological findings, and genetic confirmation of associated mutations (Savige et al., 2013; Kashtan, 2021). Variants of COL4A5 (X-linked dominant inheritance pattern), including deletion, duplication, substitution, and splicing mutation, account for about 80% of patients with Alport syndrome (Martin et al., 1998). Autosomal recessive and dominant inheritance with COL4A3 or COL4A4 mutations accounts for a lesser portion (Martin et al., 1998). Alport patients with COL4A5 variants are more likely to suffer from proteinuria, focal segmental glomerulosclerosis, and kidney failure than the COL4A3 and COL4A4 variants, especially proteinuria which develops at a young age in male patients (Savige and Harraka, 2021). Large deletions, rearrangements, and nonsense mutations of COL4A5 were reported to be associated with early-onset renal and ocular manifestations (Savige and ColvilleOpinion, 2009). In previous studies, contiguous gene deletions in COL4A5 and COL4A6 were reported to be responsible for Alport syndrome–diffuse leiomyomatosis (AS-DL) (Mothes et al., 2002; Murata et al., 2016; Zhou et al., 2021). However, COL4A6 gene mutation alone was not contributable to Alport syndrome or diffuse leiomyomatosis (Murata et al., 2016). The upregulation of IRS4, a neighboring gene of COL4A5, was considered to be important in the development of leiomyomatosis in patients with AS-DL (Thielen et al., 2003; Mehine et al., 2013). In our case, we consider that the contiguous gene deletions in COL4A6 and COL4A5 are the disease-associated mutations that contribute to severe cataract, nephropathy, and possibly uveitis. Although symptoms of leiomyomatosis were not found at this young age, close follow-ups are necessary. Female carriers of these combined deletions reported only a mild clinical presentation, even free of nephropathy (Zhou et al., 2021). This is concordant with our study that although the little boy was affected with ocular and nephropathy, his mother is without any ocular or renal involvement.
Anterior ocular findings associated with Alport syndrome include cataracts, anterior lenticonus, and posterior polymorphous corneal dystrophy (Cheong et al., 1994). Retinal findings such as perimacular dot-and-fleck retinopathy, peripheral confluent retinopathy, retinal thinning, bull’s eye maculopathy, and vitelliform macular detachment have been reported (Fawzi et al., 2009; Savige and ColvilleOpinion, 2009). Uveitis had never been previously reported in Alport syndrome; as a result, we could not differentiate it from infection until the negative results from the metagenomics and microbiological culture were obtained. Tubulointerstitial nephritis and uveitis (TINU) is usually manifested as bilateral sudden onset non-granulomatous anterior uveitis and acute nephritis and can be differentiated from Alport syndrome by typical pathologic findings.
The relationship between Alport syndrome and inflammation has raised interest in recent years. Evidence shows that in chronic kidney disease, the resident kidney cells induce sterile inflammation by activating and producing proinflammatory cytokines and chemokines (Kurts et al., 2013). In our case, the sample from the aqueous humor showed an elevated level of VCAM, IL-6, and IL-8, which indicate a strong inflammatory reaction in the affected eye. We hypothesize that ocular inflammation may result from the breakdown of the blood–retina/aqueous humor barrier, with an autoimmune reaction to abnormal collagen products in the eye.
In an animal study, treatment with the epithelial growth factor receptor (EGFR) inhibitor erlotinib suppressed the expression of inflammatory cytokines in the kidney tissue of AS mice but did not improve the renal pathology (Omachi et al., 2017). In our case, the use of steroids resolved the inflammation in the eye but did not improve the patient’s renal function.
Early diagnosis of Alport syndrome in patients without a related family history is challenging. Extrarenal manifestations such as perceptive hearing loss, lenticonus, and temporal retinal thinning are of limited diagnostic value for these patients as they are generally asymptomatic until a certain age. In our case, the presence of cataract was the original finding. Further diagnosis revealed uveitis. Microscopic hematuria was the only extraocular manifestation and was discovered coincidentally in routine examinations. The confirmative diagnosis was made based on the electron microscopic findings and genetic analysis. The atypical findings (cataracts and uveitis) and asymptomatic findings (hematuria) in patients with Alport syndrome may be underestimated by physicians. We suggest that for patients with ocular manifestations and microscopic hematuria, Alport syndrome should be included in the list of differential diagnoses.
In conclusion, this is the first reported case of non-infectious uveitis associated with Alport syndrome. Anti-inflammatory treatment with steroids is useful for uveitis but not for nephropathy.
The sequence data reported in this paper have been deposited in the Genome Sequence Archive (GSA) database with the accession no. GVM000399.
Written informed consent was obtained from the patient's legal guardian, for the publication of any potentially identifiable images or data included in this article.
YT and LZ: conceived and designed the manuscript; YT, XW, and ZL: clinical data acquisition; YL performed the genetic analysis; WH interpreted the genetic data; FM revised the manuscript; LZ drafted and revised the manuscript. All authors contributed to the manuscript and approved the final version.
This work was supported by the Natural Science Foundation of Hunan Province, China (grant no. 2021JJ30982).
The authors are grateful to the patient and his family for their cooperation and consent to this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alport, A. C. (1927). Hereditary familial congenital haemorrhagic nephritis. Br. Med. J. 1, 504–506. doi:10.1136/bmj.1.3454.504
Cheong, H. I., Kashtan, C. E., Kim, Y., Kleppel, M. M., and Michael, A. F. (1994). Immunohistologic studies of type IV collagen in anterior lens capsules of patients with Alport syndrome. Lab. Invest. 70, 553–557.
Fawzi, A. A., Lee, N. G., Eliott, D., Song, J., and Stewart, J. M. (2009). Retinal findings in patients with Alport syndrome: Expanding the clinical spectrum. Br. J. Ophthalmol. 93, 1606–1611. doi:10.1136/bjo.2009.158089
Kashtan, C. E. (2021). Alport syndrome: Achieving early diagnosis and treatment. Am. J. Kidney Dis. 77, 272–279. doi:10.1053/j.ajkd.2020.03.026
Kurts, C., Panzer, U., Anders, H. J., and Rees, A. J. (2013). The immune system and kidney disease: Basic concepts and clinical implications. Nat. Rev. Immunol. 13, 738–753. doi:10.1038/nri3523
Martin, P., Heiskari, N., Zhou, J., Leinonen, A., Tumelius, T., Hertz, J. M., et al. (1998). High mutation detection rate in the COL4A5 collagen gene in suspected Alport syndrome using PCR and direct DNA sequencing. J. Am. Soc. Nephrol. 9, 2291–2301. doi:10.1681/ASN.V9122291
Mehine, M., Kaasinen, E., Makinen, N., Katainen, R., Kampjarvi, K., Pitkanen, E., et al. (2013). Characterization of uterine leiomyomas by whole-genome sequencing. N. Engl. J. Med. 369, 43–53. doi:10.1056/NEJMoa1302736
Mothes, H., Heidet, L., Arrondel, C., Richter, K. K., Thiele, M., Patzer, L., et al. (2002). Alport syndrome associated with diffuse leiomyomatosis: COL4A5-COL4A6 deletion associated with a mild form of Alport nephropathy. Nephrol. Dial. Transpl. 17, 70–74. doi:10.1093/ndt/17.1.70
Murata, T., Katayama, K., Oohashi, T., Jahnukainen, T., Yonezawa, T., Sado, Y., et al. (2016). COL4A6 is dispensable for autosomal recessive Alport syndrome. Sci. Rep. 6, 29450. doi:10.1038/srep29450
Omachi, K., Miyakita, R., Fukuda, R., Kai, Y., Suico, M. A., Yokota, T., et al. (2017). Long-term treatment with EGFR inhibitor erlotinib attenuates renal inflammatory cytokines but not nephropathy in Alport syndrome mouse model. Clin. Exp. Nephrol. 21, 952–960. doi:10.1007/s10157-017-1386-9
Qian, Z., Zhang, Y., Wang, L., Li, Z., Wang, H., Kang, H., et al. (2022). Application of metagenomic next-generation sequencing in suspected intraocular infections. Eur. J. Ophthalmol. doi:10.1177/11206721221107311
Savige, J., and ColvilleOpinion, D. (2009). Opinion: Ocular features aid the diagnosis of Alport syndrome. Nat. Rev. Nephrol. 5, 356–360. doi:10.1038/nrneph.2009.65
Savige, J., Gregory, M., Gross, O., Kashtan, C., Ding, J., and Flinter, F. (2013). Expert guidelines for the management of Alport syndrome and thin basement membrane nephropathy. J. Am. Soc. Nephrol. 24, 364–375. doi:10.1681/ASN.2012020148
Savige, J., and Harraka, P. (2021). Pathogenic variants in the genes affected in Alport syndrome (COL4A3-COL4A5) and their association with other kidney conditions: A review. Am. J. Kidney Dis. 78, 857–864. doi:10.1053/j.ajkd.2021.04.017
Thielen, B. K., Barker, D. F., Nelson, R. D., Zhou, J., Kren, S. M., and Segal, Y. (2003). Deletion mapping in Alport syndrome and Alport syndrome-diffuse leiomyomatosis reveals potential mechanisms of visceral smooth muscle overgrowth. Hum. Mutat. 22, 419. doi:10.1002/humu.9191
Yang, S., Yao, G., Chen, X., Shi, H., Lou, C., Ren, S., et al. (2022). A novel mutation of KCNJ1 identified in an affected child with nephrolithiasis. BMC Nephrol. 23, 227. doi:10.1186/s12882-022-02783-x
Keywords: Alport syndrome, uveitis, COL4A5, pediatric, collagen
Citation: Tian Y, Wu X, Li Y, He W, Liu Z, Myers FL and Zhou L (2022) Case report: Unilateral panuveitis as a manifestation of Alport syndrome in a Chinese pediatric patient. Front. Genet. 13:934829. doi: 10.3389/fgene.2022.934829
Received: 03 May 2022; Accepted: 24 October 2022;
Published: 14 November 2022.
Edited by:
Babak Behnam, National Sanitation Foundation International, United StatesReviewed by:
Michelle Grunin, Case Western Reserve University, United StatesCopyright © 2022 Tian, Wu, Li, He, Liu, Myers and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liang Zhou, WmhvdWxpYW5nMTJAY3N1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.