 Min Gao1,2†
Min Gao1,2† Fengling Yu3,2†
Fengling Yu3,2† Rui Dong1,2
Rui Dong1,2 Kaihui Zhang1,2Yuqiang Lv1,2
Kaihui Zhang1,2Yuqiang Lv1,2 Jian Ma1,2
Jian Ma1,2 Dong Wang1,2Hongxia Zhang4,2*Zhongtao Gai1,2*
Dong Wang1,2Hongxia Zhang4,2*Zhongtao Gai1,2* Yi Liu1,2*
Yi Liu1,2*- 1Pediatric Research Institute, Children’s Hospital Affiliated to Shandong University, Jinan, China
- 2Shandong Provincial Clinical Research Center for Children’s Health and Disease, Jinan, China
- 3Clinical Laboratory, Children’s Hospital Affiliated to Shandong University, Ji’nan, China
- 4Department of Nephrology, Children’s Hospital Affiliated to Shandong University, Jinan, China
Background: Inherited kidney diseases (IKDs) are a group of kidney diseases characterized by abnormal kidney structure or function caused by genetic factors, but they are not easily diagnosed in childhood due to either nonspecific symptoms and signs or clinically silent symptoms in the early stages until the progressive stages, even end-stages. Early diagnosis of IKDs is very urgent for timely treatment and improving outcomes of patients. So far, the etiological diagnosis has been accelerated with the advance of clinical genetic technology, particularly the advent of next-generation sequencing (NGS) that is not only a powerful tool for prompt and accurate diagnosis of IKDs but also gives therapy guidance to decrease the risk of unnecessary and harmful interventions.
Methods: The patients presenting with urinalysis abnormalities or structural abnormalities from 149 Chinese families were enrolled in this study. The clinical features of the patients were collected, and the potentially causative gene variants were detected using exome sequencing. The clinical diagnostic utility of the genetic testing was assessed after more detailed clinical data were analyzed.
Result: In total, 55 patients identified having causative variants by exome sequencing were genetically diagnosed, encompassing 16 (29.1%) autosomal dominant IKDs, 16 (29.1%) autosomal recessive IKDs, and 23 (41.8%) X-linked IKDs, with 25 unreported and 45 reported variants. The diagnostic yield was 36.9%. The utility of the exome sequencing was accessed, 12 patients (21.8%) were confirmed to have suspected IKDs, 26 patients (47.3%) discerned the specific sub-types of clinical category, and 17 patients (30.9%) with unknown etiology or lack of typical manifestations were reclassified.
Conclusion: Our study supported that genetic testing plays a crucial role in the early diagnosis for children with IKDs, which affected follow-up treatment and prognostic assessment in clinical practice. Moreover, the variant spectrum associated with IKDs was expanded.
Introduction
Inherited kidney diseases (IKDs) are a group of kidney diseases characterized by abnormal kidney structure or function caused by genetic factors which present substantial clinical heterogeneity in clinical presentation, age of onset, severity, and progression of symptoms, accounting for at least 10%–15% of cases of kidney replacement therapy (Zhang et al., 2021; Torra et al., 2021). So far, there have been more than 150 known IKDs which can be divided into two categories of inherited renal structural abnormality and inherited renal dysfunction (Devuyst et al., 2014; Cunha et al., 2020).
IKDs are caused by different factors that can lead to permanent and irreversible deterioration of renal function, but they are not easily diagnosed in childhood due to either nonspecific symptoms and signs or clinically silent symptoms in the early stages until the progressive stages, even end-stages, when presenting characteristics of kidney impairment such as proteinuria and hematuria and extrarenal symptoms; thereby, early diagnosis of IKDs is very urgent for timely treatment and improving outcomes of patients. Renal biopsy has been the gold standard for diagnosis of kidney diseases since it was introduced in the 1950s by providing the injury pattern and prognosis information, while the limitation has become more and more obvious in the era of precision medicine as it is impossible to make an early, precise diagnosis of IKDs (Liapis and Gaut., 2013; Hogan et al., 2016; Murray et al., 2020). The etiological diagnosis has been accelerated with the advance of clinical genetic technology, particularly the advent of next-generation sequencing (NGS) that is not only a powerful tool for prompt and accurate diagnosis of IKDs but also gives therapy guidance to decrease the risk of unnecessary and harmful interventions (Groopman et al., 2019; Devarajan et al., 2022).
The genetic causes of more than 150 rare kidney diseases and variants in over 400 genes have been defined since the first case of a causal variant for a monogenic kidney disorder (Alport syndrome) was identified in 1990 (Barker et al., 1990; Knoers et al., 2022). NGS technology enabling the detection of all types of genetic variants and benefiting prompt and accurate diagnosis has been applied in detecting disease-causing genes of IKDs in children, but the data obtained from the clinical practice setting are rare, so recommendations have been made to use NGS techniques in the diagnosis of patients with kidney diseases (Groopman et al., 2019; Knoers et al., 2022).
Here, we present an investigation of causative gene variations in a well-characterized clinical cohort of 149 unrelated patients with urinalysis abnormalities or structural abnormalities from Shandong, a northern province in China, using exome sequencing. We identified 70 clinically diagnostic variants in 55 patients, and the diagnostic yield was 36.9%.
Materials and methods
Ethics statement
This work was approved by the Medical Ethics Committee of Children’s Hospital Affiliated to Shandong University. Informed consent was obtained from the patient’s parents before clinical and laboratory examinations were performed on the probands. The information of all the patients was anonymized prior to submission. All the procedures performed in the study were in accordance with the Declaration of Helsinki.
Subjects
A total of 149 probands with clinical suspected IKDs or unexplained proteinuria or/and hematuria who were referred for NGS in our institute of Children’s Hospital Affiliated to Shandong University (CHASU) from September 2017 to December 2021 were enrolled in this study. Their parents, except four unavailable fathers, were also enrolled in the study. All probands from Shandong, a northern province in China, were clinically examined and diagnosed by experienced pediatric nephrologists at the Department of Nephrology in CHASU following the proposal that all patients had urinalysis abnormalities or structural abnormalities and excluded the following causes or conditions: trauma, infectious diseases, inflammatory processes, malignant tumors, metabolic disorders, bleeding and blood cell disorders, and vascular disorders.
Exome sequencing
Genomic DNA was isolated from peripheral blood samples of the probands and their parents using the TIANamp Blood DNA Kit (TIANGEN, Beijing, China) following the manufacturer’s instructions and used for library preparation. The exon regions along with adjacent intron regions (±20 bp) of all genes contained in post-PCR libraries were then captured and enriched. Enriched sample libraries were quantified by Qubit dsDNA HS Assay (Thermal Fisher Scientific), and library size was checked by High Sensitivity D1000 Reagents (Agilent, United States). Final libraries were sequenced using the Human Exome Probes P039-Exome (MyGenostics, Beijing, China) on an Illumina NovaSeq 5000 sequencer (Illumina, United States).
Variant analysis and validation
The sequencing data were compared to the UCSC hg19 human reference genome sequence using NextGene V2.3.4 software, and the genetic variation was identified. The quality parameters such as coverage of the target area and average sequencing depth were collected. Annotations were made using NextGene V2.3.4 software. Potential effects of the variants on function were assessed using the following databases: UCSC Genome Bioinformatics (http://genome.ucsc.edu/) (comparing with the reference sequence of SMARCA2 gene); biological function prediction (Sorting Intolerant From Tolerant (http://sift.jcvi.org), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2), Mutation Taster (http://www.mutationtaster.org/)), frequency of normal population (1000 Genomes (http://browser.1000genomes.org), Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org), dbSNP database, Human Gene Mutation Database (http://www.hgmd.cf.ac.uk), ClinVar (http://www.ncbi.nlm.nih.gov/clinvar), and OMIM databases. Genetic variation was further screened using the following steps: 1) prioritize public attention genetic variants that may be associated with disease in the database, small insertions/deletions (INDEL), typical splice site changes, and missense variations; 2) in the normal population, the secondary allele frequency is <5% (except for the known pathogenicity of MAF ≥ 5%); and 3) the genetic variation included in the HGMD and ClinVar databases is prioritized. The pathogenicity assessment of genetic variations followed the Sequence Variation Interpretation Standards and Guidelines published by the American Society of Medical Genetics and Genomics (ACMG) and used the HGVS nomenclature.
Sanger sequencing was utilized to validate the potential clinically relevant gene variations in the probands and their parents. The primer sets for PCR were designed to target gene variants using Primer Premier v5.0 software. PCR amplification was carried out using AmpliTaq Gold* 360 DNA polymerase (Applied Biosystems, Shanghai, China). PCR products were then purified and sequenced on an ABI Prism 3700 automated sequencer (Applied Biosystems, CA, United States).
Statistical analysis
Diagnostic yield was calculated based on the counts of variants classified as “pathogenic” or “likely pathogenic.” Two-sided Fisher’s test was used to test the significance of the variants. The results were considered statistically significant when the p-value was <.05 and the confidence interval was 95%.
Result
Patients’ characteristics
In total, 149 patients enrolled in the study were Han Chinese from 149 unrelated families, including 142 (95.3%) probands with urinalysis abnormalities presenting with hematuria or/and proteinuria and 7 (4.7%) patients with structural abnormalities in kidneys detected by ultrasound. They consisted of 95 (63.8%) male and 54 (36.2%) female patients with a median age of 3 years ranging from 4 days to 13 years. In total, 44 (29.5%) probands had a family history related to kidney disease (Table 1).
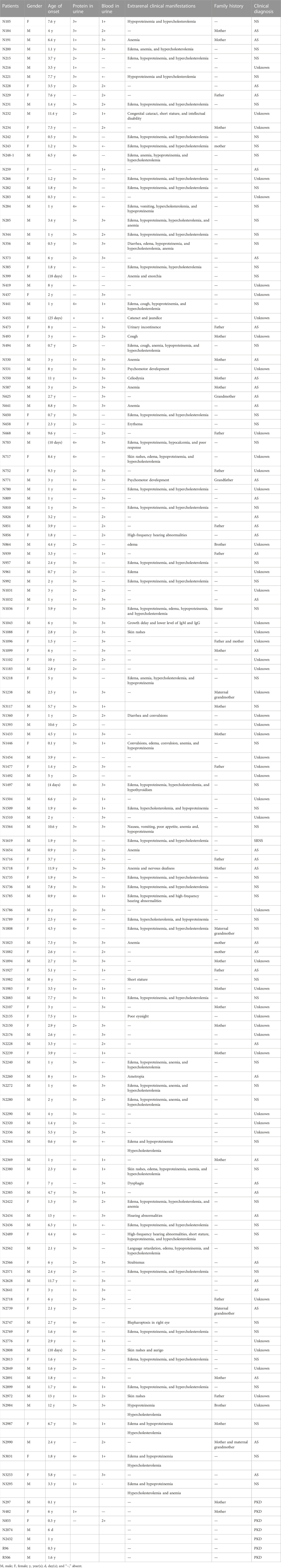
TABLE 1. Clinical details of 149 patients with suspected IKDs.
Genetic findings and diagnostic yield
Samples from the 149 families were tested, and 70 clinically diagnostic variants of 15 causal genes were discovered in 55 probands, encompassing 16 (29.1%) autosomal dominant IKDs, 16 (29.1%) autosomal recessive IKDs, and 23 (41.8%) X-linked IKDs. Of the 55 probands with clinically diagnostic variants, 7 (12.7%) belonged to inherited renal structural abnormality and 48 (87.3%) were inherited renal dysfunction. Diagnostic variants were found in 20 of the 32 patients (62.5%) who had a family history of kidney diseases, which was higher than those without family history: 35 diagnostic variants in the 117 patients (29.9%). The diagnostic yield was 36.9% (Table 2; Table 3).
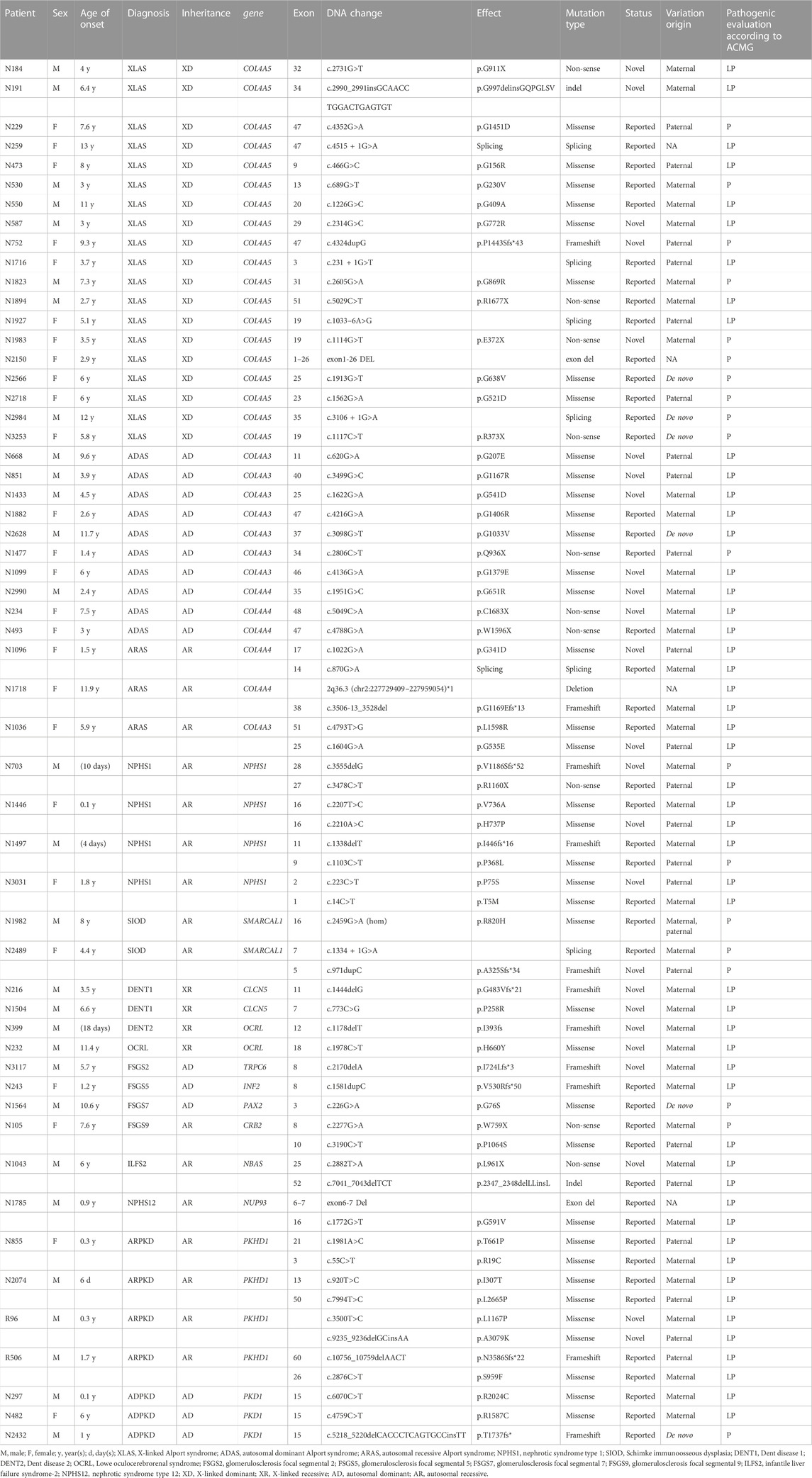
TABLE 2. Variants analyzed by exome sequencing in the patients from 55 Chinese families.
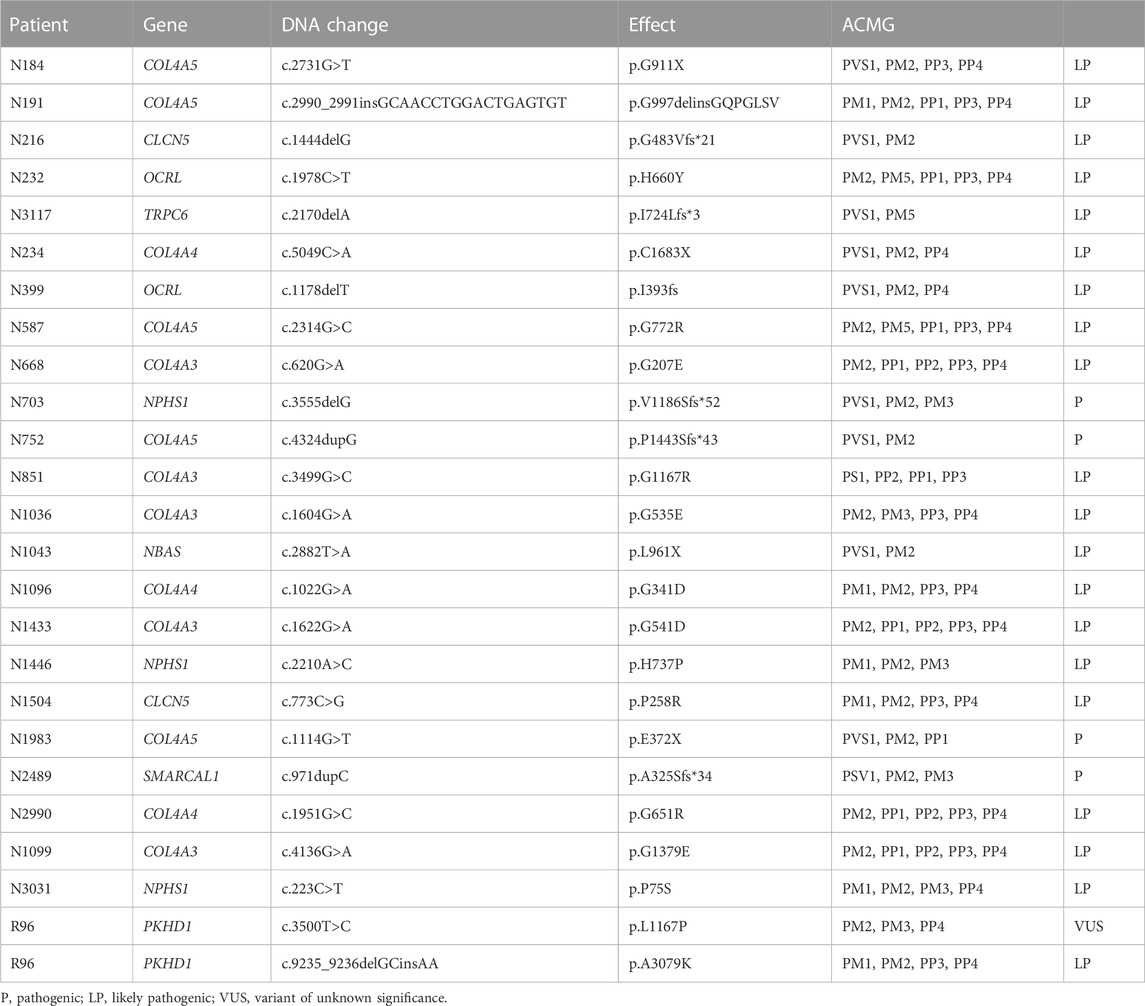
TABLE 3. Evaluation of novel variants according to the ACMG guidelines.
The seven probands with inherited renal structural abnormality included four autosomal recessive polycystic kidney disease (ARPKD) caused by the compound heterozygous variants in PKHD1 and three autosomal dominant polycystic kidney disease (ADPKD) caused by the heterozygous variants in PKD1.
Of the 48 probands with inherited renal dysfunction, 32 were Alport syndrome (AS) caused by the variants in the genes of COL4A5, COL4A3, or COL4A4 with different inheritance patterns; four inherited nephrotic syndrome type 1 (NPHS1) caused by the compound heterozygous variants in the gene of NPHS1, two Schimke immunoosseous dysplasia (SIOD) caused by the homozygous or compound heterozygous variants in the SMARCAL1 gene, two Dent disease type 1 (DENT1) caused by the heterozygous variants in the CLCN5 gene, one Dent disease type 2 (DENT2) caused by the heterozygous variant in the gene of OCRL, one Lowe oculocerebrorenal syndrome (OCRL) caused by the heterozygous variant in the OCRL gene, one glomerulosclerosis focal segmental 2 (FSGS2) caused by the heterozygous variant in the gene of TRPC6, one glomerulosclerosis focal segmental 5 (FSGS5) caused by the heterozygous variant in the gene of INF2, one glomerulosclerosis focal segmental 7 (FSGS7) caused by the heterozygous variant in the gene of PAX2, one focal segmental glomerulosclerosis 9 (FSGS9) caused by the compound heterozygous variant in the gene of CRB2, one infantile liver failure syndrome-2 (ILFS2) caused by the compound heterozygous variant in the gene of NBAS, and one nephrotic syndrome type 12 (NPSH12) caused by the compound heterozygous variant in the gene of NUP93.
The 70 clinically diagnostic variants of 15 causal genes comprised 11 variants in two genes leading to inherited renal structural abnormality and 59 resulting in inherited renal dysfunction in 13 genes with 25 unreported and 45 reported previously. Of the 70 variants, one was homozygous from both parents and six de novo, 22 parental, 37 maternal, and four unknown origin variants due to unavailable parents. The 11 variants causing seven inherited renal structural abnormalities included nine missense variants and two frameshift variants. The 59 variants resulting in inherited renal dysfunction consisted of 29 (49.2%) missense, 10 (16.9%) nonsense, 9 (15.2%) frameshift, 6 (10.2%) splicing, 2 (3.4%) indel, and 3 (5.1%) gross deletion variants (Table 2; Figure 1).
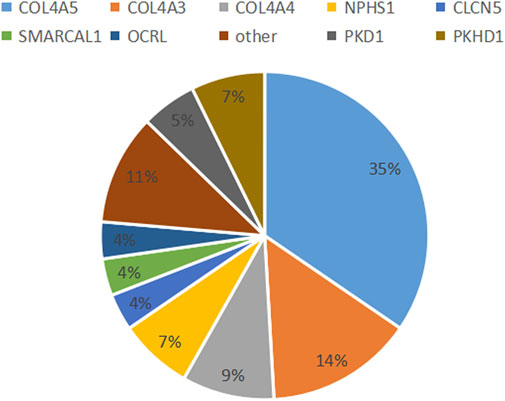
FIGURE 1. Most common diagnostic genetic findings. In total, 15 causative genes, representing 16 distinct monogenetic disorders, were detected in 55 patients.
Novel variants
Among the 55 patients with confirmed disease according to molecular diagnosis, 25 novel variants in 24 patients absent from population databases and our in-house database were identified (Table 3).
Of the 25 novel variants, 13 were found in the genes of COL4A5, COL4A4, and COL4A3 in 16 probands with Alport syndrome (AS). There were five novel variants in the COL4A5 gene in five probands, i.e., three male and two female patients, including one missense (p.G772R), two nonsense (p.G911X and p.E372X), one frameshift (p.P1443Sfs*43), and one indel (p.G997delinsGQPGLSV). Among the five probands, two female probands showed mild urinalysis abnormalities with no or little protein in urine, while three male probands indicated significant hematuria and proteinuria. They were genetically diagnosed with XLAS after exome sequencing. Five novel variants in the COL4A3 gene identified in five patients were all missense variants of p.G207E, p.G1167R, p.G541D, p.G535E, and p.G1379E. Furthermore, the variant of p.G535E combined with a reported one of p.L1598R, was detected in a 5.9-year-old girl (N1036) who presented with distinct hematuria and proteinuria with level 3 originally clinically diagnosed as nephrotic syndrome (NS). Both the variants of p.G535E and p.L1598R were obtained from her father and mother, separately, which constituted a compound heterozygous mutation effect; thus, the patient was finally diagnosed with ARAS. The remaining four variants were in the COL4A3 gene with two paternal and two maternal variation origins in other four patients who mainly presented with hematuria with strong family history, and they were diagnosed with ADAS. There were three novel variants in COL4A4 in three probands who manifested remarkable hematuria and were genetically diagnosed with ADAS.
Three novel variants in the NPHS1 gene were identified as one frameshift (p.V1186Sfs*52) in Patient N703 and two missense (p.H737P and p.P75S) in patients N1446 and N3031. Three patients were very young, presenting with severe proteinuria with primary diagnosis of NS, and were finally confirmed as having nephrotic syndrome type 1 (NPHS1).
We detected two novel variants in the OCRL gene in two probands. Patient N399, an 18-day-old boy, showed recurrent proteinuria, and his genetic investigation revealed a maternally inherited variant (p.I393fs) in the OCRL gene. He was diagnosed with DENT2 after genetic testing, whereas the other was a maternally inherited missense variant (p.H660Y) in patient N232, who presented with hematuria, proteinuria, and extrarenal symptoms of congenital cataract, short stature, and intellectual disability that were consistent with the Lowe oculocerebrorenal syndrome (OCRL).
Other seven novel variants were identified in five genes, namely, SMARCAL1, CLCN5, NBAS, TRPC6, and PKHD1 in six probands, who were diagnosed as SIOD, DENT1, ILFS2, FSGS2, and ARPKD (Table 3).
Clinical implications of genetic diagnosis
In this study, the 55 probands detected with causative variants by NGS were genetically diagnosed, and the clinical diagnostic utility of the exome sequencing was assessed after more detailed clinical data were analyzed. Among them, 12 patients (21.8%) were confirmed suspected IKD cases, 26 patients (47.3%) discerned the specific subtypes of clinical category, and 17 patients (30.9%) with unknown etiology or lack of typical manifestations were reclassified so that the specific intervention was managed in a timely manner (Table 4; Figure 2). Taking the N229 family, for instance, the proband, a 7.6-year-old girl and the only child of her parents, visited our hospital for the first time in December 2019 due to her positive urine occult blood test (level 2) and strong family history, while there were no obvious symptoms. Her 25-year-old father had suffered from uremia and was on kidney dialysis regularly. Both the patients and her father were detected the variant of c.4352G>A (p.G1451D) in the COL4A5 gene and finally diagnosed with XLAS (Figure 3). From then on, the patient has received enalapril maleate tablet (angiotensin-converting enzyme inhibitor) therapy, plus regular urine test for three years, demonstrating a good condition without proteinuria since then.
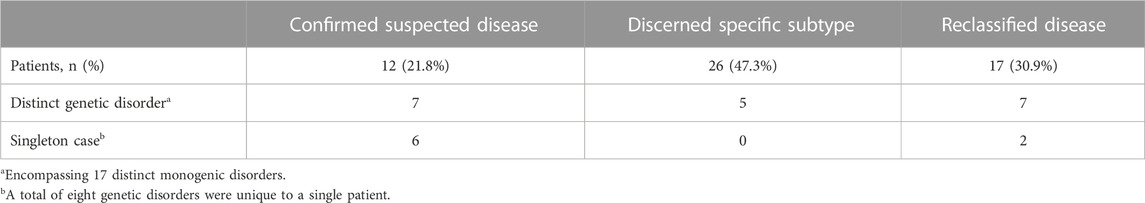
TABLE 4. Diagnostic utility in 55 patients with genetic diagnosis of IKDs.
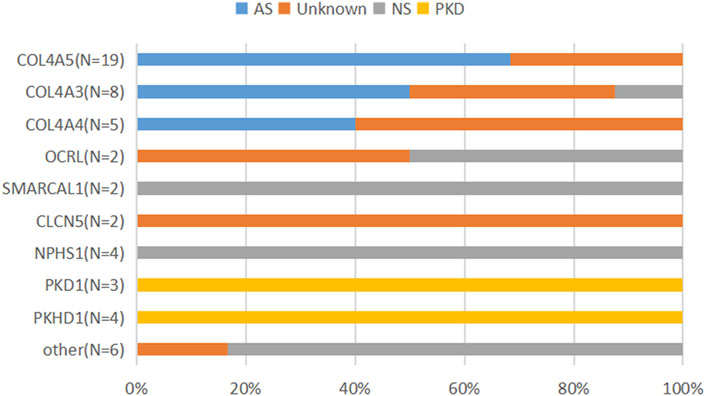
FIGURE 2. Clinical diagnostic spectrum of patients with genetic diagnosis of IKDs. The patients who were clinically diagnosed with NS were associated with CLO4A3, OCRL, SMARCAL1, and NPHS1 or other genes; the patients who were clinically diagnosed with AS were associated with COL4A3, COL4A4, and COL4A5; the patients clinically undiagnosed (unknown) were associated with the genes of COL4A5, COL4A3, COL4A4, OCRL, and CLCN5 and others.
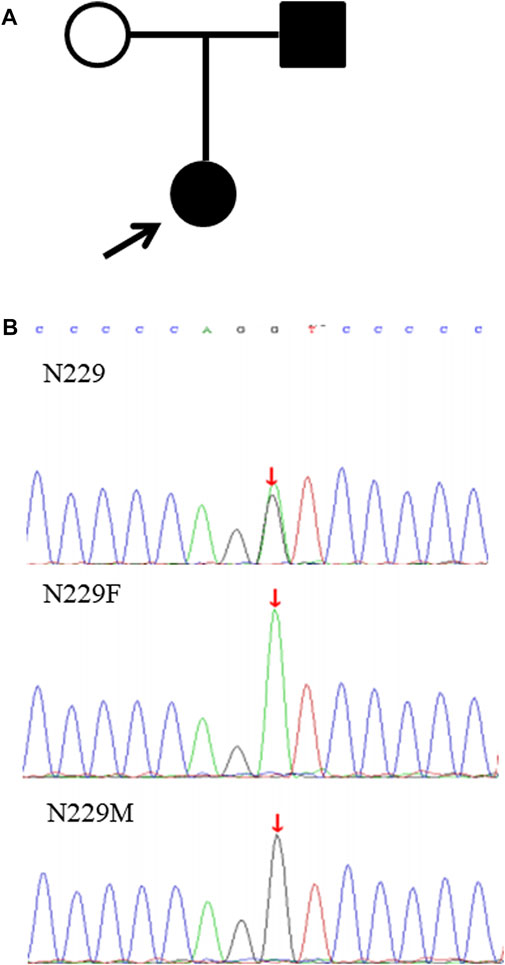
FIGURE 3. Pedigree chart and verification of the variant in COL4A5 in the N229 family. (A) Pedigree chart of the N229 family; (B) the variant c.4352G>A in COL4A5 validated by Sanger sequencing in the proband (N229), father (N229F), and mother (N229M).
Discussion
Several studies indicated that NGS has particular benefits for children with IKDs by Providing early diagnosis, which would affect follow-up treatment and improve outcomes in clinical practice (Groopman et al., 2019; Zamani et al., 2021; Knoers et al., 2022; Kashtan, 2021). In the present study, we investigated the genetic cause of 149 child probands with clinically suspected IKDs or unexplained proteinuria or/and hematuria using exome sequencing. In total, 55 probands were found with clinically significant variants involving 15 causative genes and were then diagnosed with 16 monogenic disorders. Of them, 14 patients (25.5%) presenting with mild urinalysis abnormalities of trace blood or protein in urine were precisely identified so that the intervention was performed and potential extrarenal symptoms or complications could be monitored. The diagnostic yield was 36.9% (55/149), which is higher than that in Groopman’s report (Groopman et al., 2019) while is similar as two previous studies in children in two Asian populations of Seoul, South Korea, and Shanghai, South China, with the diagnostic yields of 39.2% and 39.9%, respectively (Chen et al., 2021; Oh et al., 2021), further demonstrating that exome sequencing is a powerful tool for the early genetic profiling of IKDs and genetic variability among ethnic and geographical regions.
It is reported that genetic diagnosis contributed to the reclassification of original diagnosis in 10%–22% of cases (Groopman et al., 2019; Knoers et al., 2022). In this study, a higher number of 32.7% (18/55) patients with unknown etiology or lack of typical manifestations were reclassified for which we considered two reasons: first, the patients with IKDs have heterogeneity of genotype and phenotype in different populations, particularly in children at early stages of IKDs; second, most IKDs are rare diseases, some of which are unknown even to pediatric nephrologists (Groopman et al., 2019; Murray et al., 2020; Torra et al., 2021). For example, patient N216 was a 3.5-year-old boy manifesting only little protein in urine without other specific symptoms, who presented with a potential kidney disease of an unknown cause until the genetic result was received.
Genetic testing plays a crucial role in precise genetic counseling by providing information about hereditary modes, accessing recurrence risks, and avoiding giving birth to an affected child with the same disease (de Haan et al., 2019; Wang and Weng., 2017). For instance, Alport syndrome (AS) is a progressive hereditary renal disease accompanied by sensorineural hearing loss and ocular abnormalities, caused by the pathogenic variants in the genes of COL4A3, COL4A4, and COL4A5 encoding type IV collagen α3, α4, and α5 chains, respectively (Nozu et al., 2020; Kashtan, 2021). In this study, 32 probands with AS were reclassified into specific subtypes of 19 X-linked Alport syndrome (XLAS), 10 autosomal dominant Alport syndrome (ADAS), and three autosomal recessive Alport syndrome (ARAS) based on the exome sequencing. In total, of 35 variants found in 32 probands, only four variants were de novo. Of the 32 probands, 22 had a family history, with four parents presenting with uremia or renal failure. Hence, genetic counseling is particularly essential for families to perform prenatal diagnosis in subsequent pregnancies.
Alport syndrome (AS) has been reported as the second most common IKD following ADPKD in Europe and the US during adulthood (Groopman et al., 2019; Oberdhan et al., 2022), with most of them (about 80%) being XLAS caused by the pathogenic variants in the COL4A5 gene (Nozu et al., 2020), which leads to heterogeneous renal manifestations from hematuria alone to end-stage kidney disease. In male patients with XLAS, there is an evident genotype–phenotype correlation: patients with nonsense variants having the most severe phenotypes, patients with splicing variants having moderate phenotypes, and patients with missense variants presenting with mild phenotypes (Aoto et al., 2022). Our study showed that AS was the most common IKD in children in Shandong, China, and the percentage of patients with XLAS was 59.4% (19/32), which was lower than that found in the previous report. As the most common variants were missense, the patients with AS often presented with mild manifestations without extrarenal phenotypes, which could lead to the parents or even the clinicians failing to recognize it in a timely manner. This might be corresponding to the lower percentage of AS patients with the COL4A5 variant.
Exome sequencing is demonstrated to be a powerful genetic tool in clinical practice settings, but there are still some limitations. The introns and promoter regions of genes were not included in the exome sequencing carried out in our study, and the variants in low coverage areas of exons might be filtered out. In addition, new genes of unknown function might be found in these patients. Finally, HLA-DR/DQ alleles and haplotypes might be associated with some patients. All of the aforementioned factors need to be explored in the future.
In conclusion, nearly 36.9% (55/149) of children with abnormal kidney structure or function received a precise diagnosis by genetic testing. AS is the most common disease in Chinese children, mostly presenting with mild symptoms, and 25 novel variants in 24 patients were identified, which expands the variant spectrum of causative genes in IKDs. For clinical diagnostic applications in children with suspected IKDs, genetic testing could be performed to provide an accurate molecular diagnosis and more guidance in follow-up treatment and prognostic assessment.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/genbank/,ON381715-ON381719.
Ethics statement
Written informed consent was obtained from the individual(s) and minor(s)’legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
ZG and YL conceived and designed this study. Experiments were conducted by MG, DW, KZ, YL, DW, and JM. Data were analyzed by DW, HZ, and YL. Clinical diagnosis of the patients was undertaken by HZ, FY, and ZG. The manuscript was written by MG and YL.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aoto, Y., Horinouchi, T., Yamamura, T., Kondo, A., Nagai, S., Ishiko, S., et al. (2022). Last nucleotide substitutions of COL4A5 exons cause aberrant splicing. Kidney Int. Rep. 7, 108–116. doi:10.1016/j.ekir.2021.10.012
Barker, D. F., Hostikka, S. L., Zhou, J., Chow, L. T., Oliphant, A. R., Gerken, S. C., et al. (1990). Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science 248, 1224–1227. doi:10.1126/science.2349482
Chen, J., Lin, F., Zhai, Y., Wang, C., Wu, B., Ma, D., et al. (2021). Diagnostic and clinical utility of genetic testing in children with kidney failure. Pediatr. Nephrol. 36, 3653–3662. doi:10.1007/s00467-021-05141-5
Cunha, M., Sevignani, G., Pavanelli, G. M., Carvalho, M., and Barreto, F. C. (2020). Rare inherited kidney diseases: An evolving field in Nephrology. J. Bras. Nefrol. 42, 219–230. doi:10.1590/2175-8239-JBN-2018-0217
De Haan, A., Eijgelsheim, M., Vogt, L., Knoers, N., and De Borst, M. H. (2019). Diagnostic yield of next-generation sequencing in patients with chronic kidney disease of unknown etiology. Front. Genet. 10, 1264. doi:10.3389/fgene.2019.01264
Devarajan, P., Chertow, G. M., Susztak, K., Levin, A., Agarwal, R., Stenvinkel, P., et al. (2022). Emerging role of clinical genetics in CKD. Kidney Med. 4, 100435. doi:10.1016/j.xkme.2022.100435
Devuyst, O., Knoers, N. V., Remuzzi, G., Schaefer, F., European, D., and Transplant, A. (2014). Board of the working group for inherited kidney diseases of the European RenalRare inherited kidney diseases: Challenges, opportunities, and perspectives. Lancet 383, 1844–1859. doi:10.1016/S0140-6736(14)60659-0
Groopman, E. E., Marasa, M., Cameron-Christie, S., Petrovski, S., Aggarwal, V. S., Milo-Rasouly, H., et al. (2019). Diagnostic utility of exome sequencing for kidney disease. N. Engl. J. Med. 380, 142–151. doi:10.1056/NEJMoa1806891
Hogan, J. J., Mocanu, M., and Berns, J. S. (2016). The native kidney biopsy: Update and evidence for best practice. Clin. J. Am. Soc. Nephrol. 11, 354–362. doi:10.2215/CJN.05750515
Kashtan, C. E. (2021). Alport syndrome: Achieving early diagnosis and treatment. Am. J. Kidney Dis. 77, 272–279. doi:10.1053/j.ajkd.2020.03.026
Knoers, N., Antignac, C., Bergmann, C., Dahan, K., Giglio, S., Heidet, L., et al. (2022). Genetic testing in the diagnosis of chronic kidney disease: Recommendations for clinical practice. Nephrol. Dial. Transpl. 37, 239–254. doi:10.1093/ndt/gfab218
Liapis, H., and Gaut, J. P. (2013). The renal biopsy in the genomic era. Pediatr. Nephrol. 28, 1207–1219. doi:10.1007/s00467-012-2356-9
Murray, S. L., Fennelly, N. K., Doyle, B., Lynch, S. A., and Conlon, P. J. (2020). Integration of genetic and histopathology data in interpretation of kidney disease. Nephrol. Dial. Transpl. 35, 1113–1132. doi:10.1093/ndt/gfaa176
Nozu, K., Takaoka, Y., Kai, H., Takasato, M., Yabuuchi, K., Yamamura, T., et al. (2020). Genetic background, recent advances in molecular biology, and development of novel therapy in Alport syndrome. Kidney Res. Clin. Pract. 39, 402–413. doi:10.23876/j.krcp.20.111
Oberdhan, D., Schaefer, F., Cole, J. C., Palsgrove, A. C., Dandurand, A., and Guay-Woodford, L. (2022). Polycystic kidney disease-related disease burden in adolescents with autosomal dominant polycystic kidney disease: An international qualitative study. Kidney Med. 4, 100415. doi:10.1016/j.xkme.2022.100415
Oh, J., Shin, J. I., Lee, K., Lee, C., Ko, Y., and Lee, J. S. (2021). Clinical application of a phenotype-based NGS panel for differential diagnosis of inherited kidney disease and beyond. Clin. Genet. 99, 236–249. doi:10.1111/cge.13869
Torra, R., Furlano, M., Ortiz, A., and Ars, E. (2021). Genetic kidney diseases as an underrecognized cause of chronic kidney disease: The key role of international registry reports. Clin. Kidney J. 14, 1879–1885. doi:10.1093/ckj/sfab056
Wang, M. H., and Weng, H. (2017). Genetic test, risk prediction, and counseling. Adv. Exp. Med. Biol. 1005, 21–46. doi:10.1007/978-981-10-5717-5_2
Zamani, M., Seifi, T., Sedighzadeh, S., Negahdari, S., Zeighami, J., Sedaghat, A., et al. (2021). Whole-exome sequencing application for genetic diagnosis of kidney diseases: A study from southwest of Iran. Kidney360 2, 873–877. doi:10.34067/KID.0006902020
Keywords: exome sequencing, inherited kidney diseases, clinical diagnosis, diagnostic utility, early diagnosis
Citation: Gao M, Yu F, Dong R, Zhang K, Lv Y, Ma J, Wang D, Zhang H, Gai Z and Liu Y (2023) Diagnostic application of exome sequencing in Chinese children with suspected inherited kidney diseases. Front. Genet. 13:933636. doi: 10.3389/fgene.2022.933636
Received: 09 August 2022; Accepted: 15 December 2022;
Published: 06 January 2023.
Edited by:
Corrado Romano, University of Catania, ItalyReviewed by:
Tiziana Vaisitti, University of Turin, ItalyShahid Mahmood Baig, National Institute for Biotechnology and Genetic Engineering, Pakistan
Copyright © 2023 Gao, Yu, Dong, Zhang, Lv, Ma, Wang, Zhang, Gai and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongxia Zhang, eHVhbnh1YW5fMDMwMjE1QDE2My5jb20=; Zhongtao Gai, Z2Fpemhvbmd0YW9Ac2luYS5jb20=; Yi Liu, eV9saXU5OUBzaW5hLmNvbQ==
†These authors have contributed equally to this work