
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 18 July 2022
Sec. Evolutionary and Population Genetics
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.932173
This article is part of the Research Topic Fisheries and Aquaculture Genetics View all 18 articles
 Baohua Duan1
Baohua Duan1 Shumei Mu1Yueqiang Guan1Weibiao Liu1Tongxu Kang1Yana Cheng1Zejian Li2Yang Tian3
Shumei Mu1Yueqiang Guan1Weibiao Liu1Tongxu Kang1Yana Cheng1Zejian Li2Yang Tian3 Xianjiang Kang1,4,5*
Xianjiang Kang1,4,5*P. trituberculatus is an economically important mariculture species in China. Evaluating its genetic diversity and population structure can contribute to the exploration of germplasm resources and promote sustainable aquaculture production. In this study, a total of 246,243 SSRs were generated by transcriptome sequencing of P. trituberculatus. Among the examined 254,746 unigenes, 66,331 had more than one SSR. Among the different SSR motif types, dinucleotide repeats (110,758, 44.98%) were the most abundant. In 173 different base repeats, A/T (96.86%), AC/GT (51.46%), and ACC/GGT (26.20%) were dominant in mono-, di-, and trinucleotide, respectively. GO annotations showed 87,079 unigenes in 57 GO terms. Cellular process, cell, and binding were the most abundant terms in biological process, cellular component, and molecular function categories separately. A total of 34,406 annotated unigenes were classified into 26 functional categories according to the functional annotation analysis of KOG, of which “general function prediction only” was the biggest category (6,028 unigenes, 17.52%). KEGG pathway annotations revealed the clustering of 34,715 unigenes into 32 different pathways. Nineteen SSRs were identified as polymorphic and, thus, used to assess the genetic diversity and structure of 240 P. trituberculatus individuals from four populations in the Bohai Sea. Genetic parameter analysis showed a similar level of genetic diversity within wild populations, and the cultured population indicated a reduction in genetic diversity compared with wild populations. The pairwise FST values were between 0.001 and 0.04 with an average of 0.0205 (p < 0.05), suggesting a low but significant level of genetic differentiation among the four populations. Structure analysis demonstrated that the four populations were classified into two groups including the cultured group and other populations. The phylogenetic tree and PCA revealed that a vast number of samples were clustered together and that cultivated individuals were distributed more centrally than wild individuals. The findings contribute to the further assessment of germplasm resources and assist to provide valuable SSRs for marker-assisted breeding of P. trituberculatus in the future.
Swimming crab, P. trituberculatus, is an edible portunid of great commercial significance, which has been widely farmed in China. Due to its high nutritional value and rapid growth, P. trituberculatus has become one of the most important economic crab species in marine aquaculture (Lv et al., 2017). Indeed, P. trituberculatus is one of the most heavily fished brachyurans in the world with approximately 95% of the total catch occurring in China (Liu et al., 2013; Hui et al., 2018). The total catch was 424,630 tons in 2020 (Bureau of Fisheries of Ministry of Agriculture, PRC, 2021). With increasing serious problems such as mass outbreaks of disease, overfishing, and water pollution in recent years, however, the P. trituberculatus aquaculture and fishing industry face great pressure. Despite the improved artificial propagation and rearing techniques, the crab industry still relies on the collection of wild specimens to provide parental stock (Wang et al., 2012). It is urgent to determine the genetic diversity and population structure of P. trituberculatus for the exploration of germplasm resources and conservation management of this species.
Genetic diversity is the material basis necessary for populations to deal with changing environment, and it can trace the history of biological evolution and explore the evolutionary potential of existing organisms (Liu et al., 2021a). However, for the breeding of shrimp and crab, long-term artificial directional selection eventually leads to a decline in genetic diversity in the population (Wang et al., 2018a). Moreover, it is difficult for natural stocks to recover from declining genetic diversity caused by overfishing (Liu et al., 2009). Benefitting from the rapid advance of high-throughput sequencing and genotyping technologies, an increasing number of molecular markers are developed and applied to genetic analyses in aquatic species. To date, molecular markers including isozyme (Fan et al., 2009), random amplified polymorphic DNA (RAPD) (Chi et al., 2010), amplified fragments length polymorphism (AFLP) (Liu et al., 2013; Liu et al., 2014), mitochondrial DNA (Guo et al., 2012; Shan et al., 2017; Hui et al., 2018), microsatellites DNA (Lee et al., 2013; Yue et al., 2022), and single nucleotide polymorphism (SNP) (Duan et al., 2022a) were developed and used in population genetic analysis of P. trituberculatus. Among these markers, microsatellite DNA markers (simple sequence repeats, SSRs) have become an ideal molecular marker in population genetics research because of their co-dominant inheritance, high polymorphism, reproducibility, hyper-variable, transferability, random distribution in the genome, and ease of analysis via PCR (Gou et al., 2020; Pavan Kumara et al., 2020; Zhu et al., 2021; Lu et al., 2022). Such markers are often used to obtain genetic diversity coefficients, which can provide a basis for genetic protection strategies. However, the traditional methods of developing SSRs are usually time-consuming and labor-intensive because of establishing of the genomic library to get the fragmented sequence and hybridization in situ with probes. In recent years, the increased access and affordability of high-throughput sequencing technologies have enabled genomic and transcriptomic research on many marine species, thus leading to more rapid and accurate identification of SSR markers (Yang et al., 2018; Liu et al., 2022).
In recent years, transcriptome sequencing (RNA-seq) is widely used in the study of species genetics because of its wide dynamic range, precise, sensitivity, unbiased quantification of transcripts, and comprehensive coverage of all expressed sequences in a given tissue sample (Chakraborty et al., 2022). Now, RNA-seq is a very updated and efficient method for discovering new genes, expression pattern identification, and development of SSR markers with higher throughput and much lower cost (Li et al., 2018; Tulsania et al., 2020). SSR markers acquired from RNA-seq have intrinsic advantages over genomic SSRs because of high efficiency, strong transferability, and correlation with potential genes, as well as their applicability as anchor markers for comparative mapping and evolutionary studies (Zheng et al., 2014; Zhao et al., 2019). Transcriptomic SSRs have been extensively explored and applicated in various aquatic species, such as giant freshwater prawn (Macrobrachium rosenbergii) (Jung et al., 2011; Yu et al., 2019), oriental river prawn (Macrobrachium nipponense) (Ma et al., 2012), mud crab (Scylla paramamosain) (Ma et al., 2014), Paphia textile (Chen et al., 2016), Penaeus monodon (Nguyen et al., 2016), and pearl oyster (Pinctada maxima) (Wang et al., 2019a). Transcriptomics have also played an important role in advancing portunid aquaculture (Waiho et al., 2022). Nevertheless, the current transcriptome studies involving P. trituberculatus mainly focus on its nutrition (Zhou et al., 2019a; Fang et al., 2021a), development (Liu et al., 2018a; Liu et al., 2019), reproduction (Wang et al., 2018b), and sex determination (Zhang et al., 2022), while molecular research is scarce.
The present study aimed to 1) develop SSR markers with RNA-seq technology; 2) characterize the transcriptome of P. trituberculatus; and 3) evaluate the genetic diversity and structure among different populations using the polymorphic SSR markers from transcriptome sequencing. Our findings not only contribute to molecular genetic analyses but also provide valuable information for effective breeding and conservation strategies of the P. trituberculatus aquaculture.
A total of 240 swimming crab samples from four populations were collected (Supplementary Table S1; Figure 1), including three wild populations [Qinhuangdao (QHD), Huanghua (HW), and Penglai (PL)] from the Bohai Sea and one cultured stock (HC) from the national breeding farm of swimming crabs in Huanghua (Hebei, China) which is adjacent to the Bohai Sea. Claws of each sample were obtained and preserved in absolute alcohol and stored at −20°C until DNA extraction. Genomic DNA was isolated from the claw muscle using the TIANamp Marine animal DNA extraction kit (TIANGEN, Beijing, China) following the manufacturer’s protocols. The quality and concentration of the extracted DNA were determined using the NanoDrop ND-1000 spectrophotometer (Thermo Scientific, Wilmington, DE, United States) and 1% agarose electrophoresis gel, and then diluted to 100 ng/μl and stored at −20°C for polymerase chain reaction (PCR) amplification.
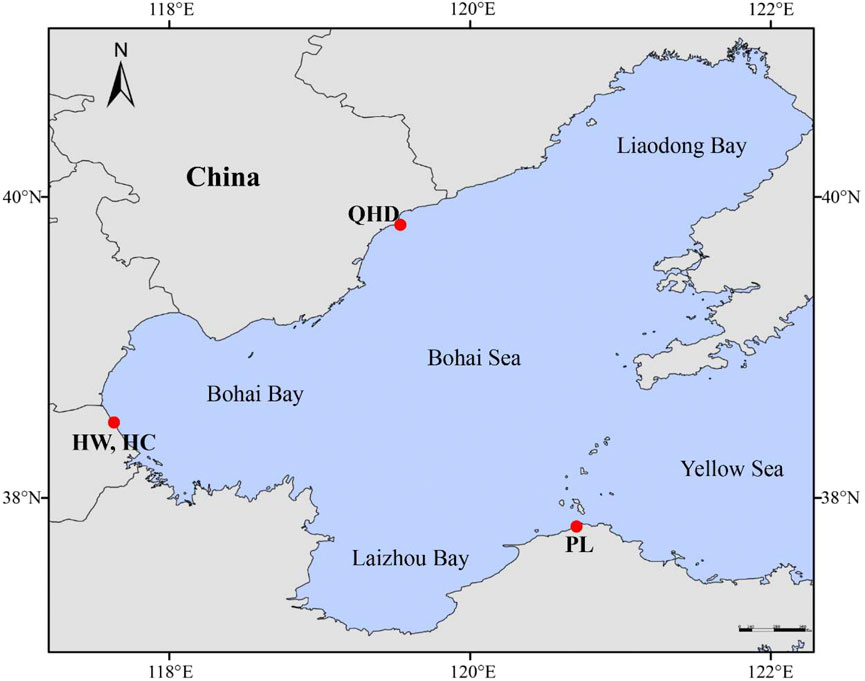
FIGURE 1. Collection map of P. trituberculatus samples.
A total of 18 individuals P. trituberculatus (nine wild females and nine cultured females) were collected from the national breeding farm of swimming crabs in Huanghua, China. The crabs were all anesthetized on ice and dissected to collect muscle and ovaries samples. All of the samples were rapidly flash-frozen in liquid nitrogen and stored at −80°C for RNA extraction. An equal amount of either muscle tissue or ovaries tissue was dissected from each of three individuals and pooled into one mixed sample. A total of 12 mixed samples were produced, including 6 mixed muscle samples (3 wild and 3 farmed) and 6 mixed ovarian samples (3 wild and 3 farmed), each containing three biological replicates. Total RNA was extracted from each mixed sample using the TRIzol reagent (Invitrogen, Carlsbad, CA, United States) following the manufacturer’s protocol. RNA degradation and contamination were monitored on 1% agarose gels, and RNA purity was checked using the NanoPhotometer® spectrophotometer (IMPLEN, CA, United States). RNA concentration and integrity were measured using the Qubit® RNA Assay Kit in Qubit® 2.0 Flurometer (Life Technologies, CA, United States) and the RNA Nano 6000 Assay Kit of the Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, United States), respectively.
A total amount of 1.5 μg RNA from each sample was used as input material for the RNA sample preparations. Sequencing libraries were generated using the NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (NEB, United States) following the manufacturer’s recommendations and index codes were added to attribute sequences to each sample. Briefly, mRNA was purified from total RNA using poly-T oligo-attached magnetic beads. Fragmentation was carried out using divalent cations under elevated temperature in NEBNext First Strand Synthesis Reaction Buffer (5X). First-strand cDNA was synthesized using random hexamer primer and M-MuLV Reverse Transcriptase (RNase H−). Second strand cDNA synthesis was subsequently performed using DNA Polymerase I and RNase H. Remaining overhangs were converted into blunt ends via exonuclease/polymerase activities. After adenylation of 3′ ends of DNA fragments, NEBNext Adaptor with hairpin loop structure was ligated to prepare for hybridization. In order to select cDNA fragments of preferentially 250–300 bp in length, the library fragments were purified with AMPure XP system (Beckman Coulter, Beverly, United States). Then 3 μl USER Enzyme (NEB, United States) was used with size-selected, adaptor-ligated cDNA at 37°C for 15 min followed by 5 min at 95°C before PCR. Subsequently, PCR was performed with Phusion High-Fidelity DNA polymerase, Universal PCR primers, and Index (X) Primer. Then PCR products were purified (AMPure XP system) and library quality was assessed on the Agilent Bioanalyzer 2100 system. At last, the library preparations were sequenced on an Illumina HiSeq 4000 platform and paired-end reads were generated at Novogene Corporation (Tianjin, China).
Raw data (raw reads) of fastq format were firstly processed through in-house perl scripts. In this step, the clean data (clean reads) were obtained by removing reads containing adapter, reads containing ploy-N, and low-quality reads (quality score < 20) from raw data. At the same time, Q20, Q30, GC-content, and sequence duplication levels of the clean data were calculated. All the downstream analyses were based on clean data with high quality. Transcriptome assembly of the high-quality clean reads was accomplished using Trinity software with default settings (Grabherr et al., 2011). In order to annotate the assembled unigenes, a BLASTX search with an E-value < 10−5 (Camacho et al., 2009) was performed against several public databases, including NCBI non-redundant protein sequences (Nr), NCBI non-redundant nucleotide sequences (Nt), Protein family (PFAM), euKaryotic Ortholog Groups (KOG), Swiss-Prot protein, KEGG Ortholog database (KO) and Gene Ontology (GO). Assigning the GO terms to the unigenes was implemented on Blast2GO software (Götz et al., 2008).
SSR loci were identified throughout all unigenes generated by the P. trituberculatus transcriptome sequencing using MISA software version 1.0 (http://pgrc.ipk-gatersleben.de/misa/misa.html). The minimum number of repeats was defined as ten for mononucleotide repeats, six for dinucleotide repeats, five for tri-, tetra-, penta-, and hexanucleotide repeats. Primer pairs for each SSR locus were designed using Primer3 (http://primer3.sourceforge.net/releases.php) according to the following criteria: 1) primer length of 18–25 bp; 2) annealing temperature (Tm) between 55°C and 62°C; 3) GC content from 40% to 60%; 4) PCR product length of 90–250 bp; 5) avoidance of primer dimers and hairpin structures. SSR primers were synthesized by General Biosystems Co., Ltd. (Anhui, China). Twelve samples were used to identify the polymorphism of the selected SSR primers through PCR amplification and 8% non-denaturing polyacrylamide gel electrophoresis with pBR322 DNA/MspI (MBI) as a standard DNA marker.
A total of 19 pairs of polymorphic SSR primers were identified and used for subsequent analysis (Table 1). All forward primers were labeled with the fluorescent dye, 6-carboxy-fluorescein (FAM). PCR amplification was performed in 20 μl reaction volumes containing 2 μl of template DNA, 2 μl of each primer (2.5 μmol/L each), 10 μl of 2 × Es Taq Master Mix (CWBIO, Beijing, China), and 4 μl of ddH2O. Amplification cycles consisted of initial denaturation (5 min at 95°C), followed by 34 cycles of denaturation (30 s at 95°C), annealing (30 s at 60°C), extension (30 s at 72°C), and further extension (10 min at 72°C). After amplification, PCR products were diluted 10 times in sterile water. The pooled sample was composed of 20 μl Hi-Di formamide and 0.2 μl GeneScan 500 ROX Size Standard. An ABI 3730XL Genetic Analyzer (Applied Biosystems, Foster City, CA, United States) was used to conduct capillary electrophoresis (CE) following the manufacturer’s instructions. Each CE sample contained 1 μl diluted PCR product and 15 μl pooled sample. Allele sizes (in base pairs) were determined with GeneMarker® Fragment Analysis Software (Softgenetics LLC®, State College, PA, United States) on the comparison of the position of the internal size standard in each lane with the position of the peak value of each sample.
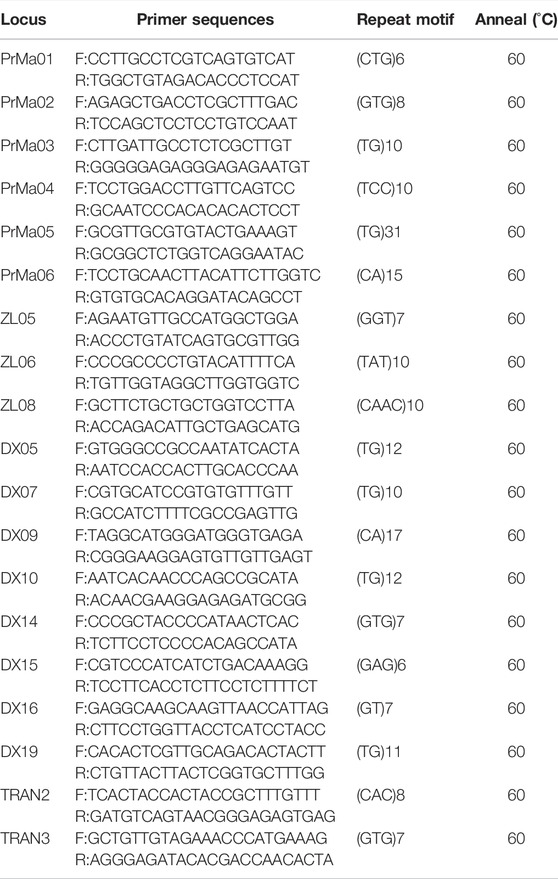
TABLE 1. Characteristics of 19 SSR loci for P. trituberculatus.
Genetic diversity was estimated by determining the genetic parameters, such as the number of alleles (Na), the effective number of alleles (Ne), the observed heterozygosity (HO) and expected heterozygosity (HE) using POPGENE version 1.3 (Yeh et al., 1999). Based on allele frequency, the polymorphism information content (PIC) per locus was estimated by PIC-CALC software (Nagy et al., 2012). The null allele frequency (Fna) for loci was calculated using GenePOP (Rousset, 2008). p values were calculated for determining the Hardy–Weinberg equilibrium (HWE) at each locus with POPGENE version 1.3. Genetic differentiation index (FST) between populations and analysis of molecular variance (AMOVA) were calculated using GenAlEx 6.5 (Peakall and Smouse, 2012).
The phylogenetic tree was constructed using the neighbor-joining (NJ) method as implemented in MEGA7 (Kumar et al., 2016). Principal component analysis (PCA) was carried out using Canoco 4.5 to elucidate genetic relationships within and among populations. Bayesian model-based population genetic structure was inferred by STRUCTURE version 2.3.4 (Pritchard et al., 2000). The putative number of populations (K) was set from 1 to 10 with 3 replicate simulations for each K value using 100,000 MCMC (Markov Chain Monte Carlo) iterations after an initial 100,000 burn-in period. With the log probability of data (LnP(D)) and an ad hoc statistic ΔK based on the rate of change in LnP(D) between successive K-values, the structure output was entered into Structure Harvester (Evanno et al., 2005; Earl and Vonholdt, 2012) to determine the optimum K value.
The transcriptome sequencing of 12 mixed samples from muscle and ovary of P. trituberculatus was conducted to generate RNA sequences, and the statistical data has been shown in Table 2. Illumina sequencing generated 661,922,456 raw reads. The raw reads produced in this study have been deposited in the Short Read Archive of the National Center for Biotechnology Information (NCBI) with accession numbers SUB11453401 and PRJNA836158. After stringent quality filtering, a total of 637,983,466 clean reads were obtained, accounting for 99.38% of the total raw reads. GC content ranged from 48.5% to 53.47% with an average of 50.88%, and the mean Q20 and Q30 were 96.3% and 91.07%, respectively. A total of 338,285 transcripts were identified with an average length of 879 bp (N50 length of transcript = 1,730 bp, which is defined as the shortest sequence length of 50% of total contigs and is used to evaluate the quality of assembled sequences), of which 11,0596 (32.69%) were less than 301 bp in length; 84,886 (25.09%) were 301–500 bp; 61,915 (18.3%) were 501–1,000 bp; 43,085 (12.74%) were 1,001–2,000 bp; 37,803 (11.18%) were over 2,000 bp (Supplementary Figure S1). Totaling 254,746 unigenes were assembled with an average length of 1,077 bp (N50 length of unigenes is 1,936 bp), among which 47,174 (18.52%) were less than 301 bp in length; 67,149 (26.36%) were 301–500 bp; 59,692 (23.43%) were 501–1,000 bp; 42,928 (16.85%) were 1,001–2,000 bp; 37,803 (14.84%) were over 2,000 bp (Supplementary Figure S1).

TABLE 2. Summary statistics for transcriptome sequencing of P. trituberculatus.
GO database was the largest matched database with 87,079 unigenes (34.18% of all unigenes) annotated, followed by PFAM (86,669, 34.02%), Nr (77,856, 30.56%), SwissProt (58,305, 22.88%), KO (34,715, 13.62%), KOG (34,406, 13.5%), and Nt (29,269, 11.48%) database (Supplementary Figure S2). In all, 118,572 (46.54%) unigenes were annotated in at least one database and 9,901 (3.88%) were annotated in all databases. In the Nr databases, 77,856 unigenes were annotated from 835 species. The top-hit species in similarity search against the Nr database included Zootermopsis nevadensis (9,854, 12.7%), Daphnia pulex (4,798, 6.2%), Tribolium castaneum (2,730, 3.5%), Stegodyphus mimosarum (2,399, 3.1%), Crassostrea gigas (2,389, 3.1%), and other (55,686, 71.5%) (Supplementary Figure S3).
For the functional annotation and classification of the assembled unigenes, 87,079 unigenes were assigned to 57 GO terms which included three ontology categories: biological process (258,416 unigenes), cellular component (178,933), and molecular function (102,370) (Figure 2A). The main components within biological process category contained cellular process (52,412, 60.19%), metabolic process (43,020, 49.40%), and single-organism process (39,395, 45.24%). Cell (32,536, 37.36%) and cell part (32,535, 37.36%) were the most frequent proportion in cellular component category. In the molecular function category, the largest potion was binding (45,970, 52.79%), followed by catalytic activity (32,373, 37.18%). According to KOG annotations, 34,406 annotated unigenes were classified into 26 functional categories (Figure 2B). Among these categories, “general function prediction only” was the biggest category (6,028 unigenes, 17.52%), followed by “signal transduction mechanisms” (5,223, 15.18%), and “posttranslational modification, protein turnover, chaperones” (3,466, 10.02%) category.
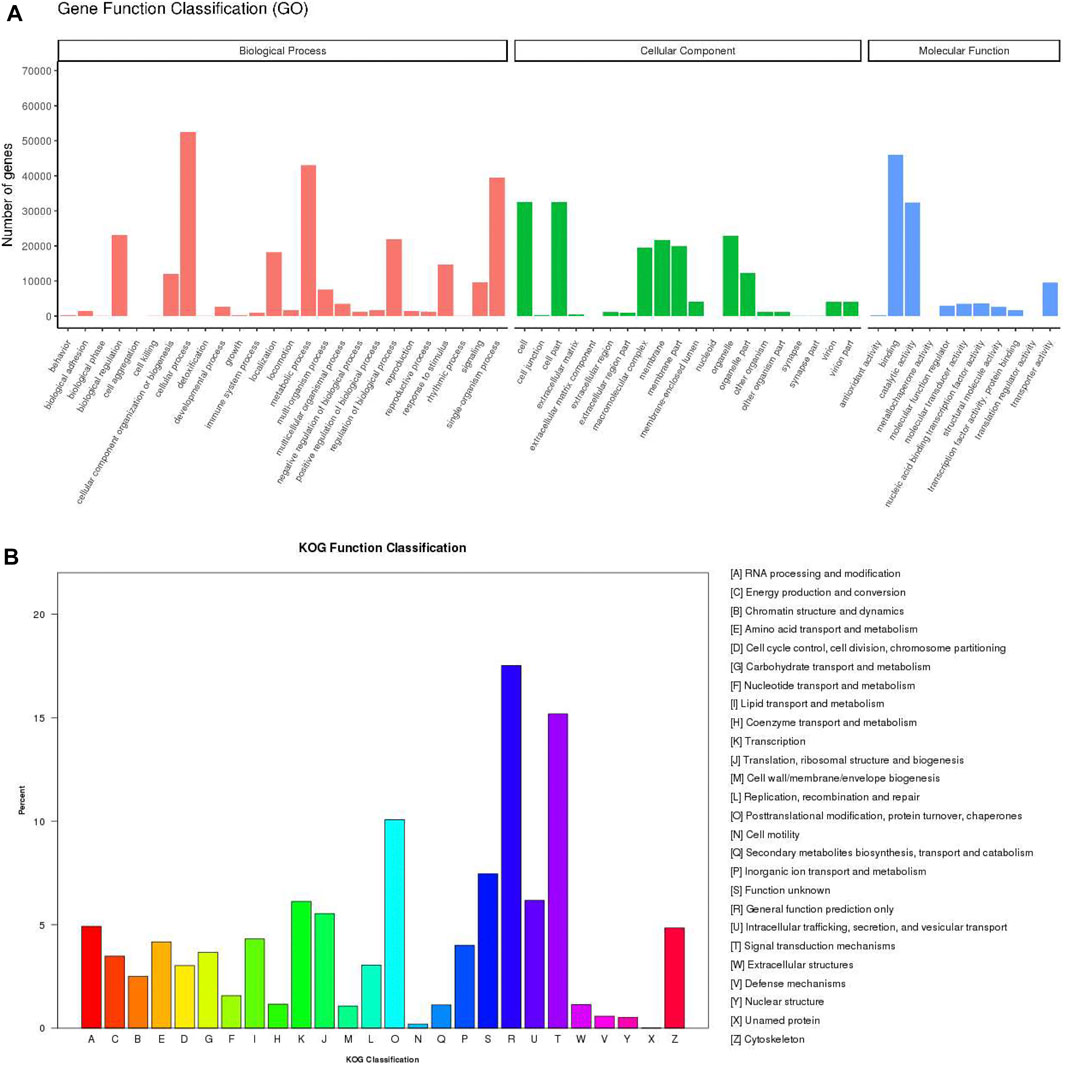
FIGURE 2. Annotation of the P. trituberculatus transcriptome. (A) GO annotation and classification of unigenes. (B) KOG annotation and classification of unigenes. The x-axis indicates the categories, and the y-axis indicates the number of the unigenes.
Identification of the biological pathways was performed according to the KEGG annotations, which showed the clustering of 34,715 unigenes into 32 pathways (Figure 3). Detailedly, these unigenes were categorized into five KEGG biochemical pathways: Cellular Processes (A), Environmental Information Processing (B), Genetic Information Processing (C), Metabolism (D), and Organismal Systems (E). This analysis revealed that the top five pathways included signal transduction (4,665 unigenes, 13.44%), endocrine system (2,629, 7.57%), transport and catabolism (2,491, 7.18%), translation (2,100, 6.05%), and cellular community (1,852, 5.33%).
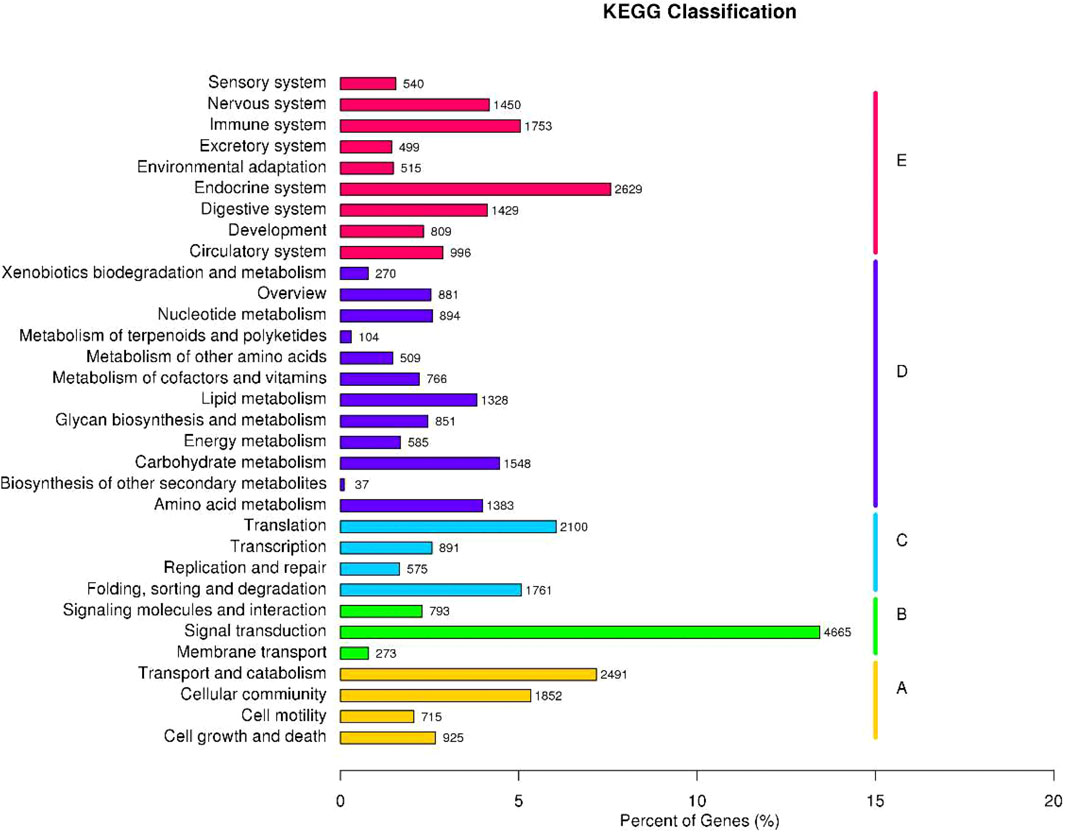
FIGURE 3. Function annotation and classification of P. trituberculatus unigenes in the KEGG category.
A total of 246,243 SSRs were identified from 132,908 SSR-containing unigenes of P. trituberculatus. The distribution density was one SSR per 1.11 kb, and 66,331 sequences contained more than one SSR (Table 2). Dinucleotide repeats were the most abundant type (110,758, 44.98%), followed by mono- (64,679, 26.27%), tri- (61,923, 25.15%), tetra- (7,154, 2.91%), penta- (1,343, 0.55%), and hexanucleotide (386, 0.16%) (Supplementary Table S1). The number of tandem repeats of microsatellite motifs ranged from 5 to 103. Microsatellites with six tandem repeats (28371, 11.52%) were the most common, followed by five tandem repeats (25,920, 10.53%), ten tandem repeats (24,777, 10.06%), and eleven tandem repeats (24,136, 9.88%). Microsatellite motifs with > 16 tandem repeats accounted for 22.39% (55,129) (Supplementary Table S2).
Of the 246,243 SSR loci, 173 different repeat motifs were detected (Supplementary Table S3). Among the two types of mononucleotide repeats, A/T was the most abundant (62,646, 96.85%) when compared to C/G. AC/GT (56,994, 51.46%) was the dominant motif type in the dinucleotide repeat, followed by AG/CT (47,487, 42.87%), and AT/AT (6,046, 5.46%). The most abundant types in the trinucleotide were ACC/GGT (16,224, 26.20%). Moreover, a low percentage (3.62%) of tetra-, penta- and hexanucleotide repeat motifs were observed in all identified microsatellite motifs. The physical positions of these SSR markers in the unigenes were also identified that 1708, 8,644, and 5,067 SSRs were located in the coding sequence (CDS), 3′untranslated region (UTR) and 5′UTR, respectively. In CDS, trinucleotide repeats (1,279, 74.88%) were the dominant type. Most of the mono- and dinucleotide repeats (6,290, 72.77%) were located in 3′UTR, and 5′UTR contained the majority of di- and trinucleotide repeats (3,283, 64.79%) (Supplementary Figure S4).
A total of 104,424 pairs of SSR primers were designed, 150 of which were randomly selected to identify polymorphism (Supplementary Table S4). Finally, 19 pairs of SSR primers showed high polymorphism in 8% non-denaturing polyacrylamide gel electrophoresis (Table 1), and they were used for subsequent analysis in P. trituberculatus.
All parameters of the 19 SSR loci were calculated (Table 3). The PIC values ranged from 0.598 (TRNA3) to 0.948 (PrMa05) with a mean of 0.802, showing that these SSRs has high polymorphism (PIC > 0.5) and are suitable for the evaluation of genetic diversity in P. trituberculatus populations. A total of 243 alleles were found with an average of 12.79 per locus. The Ne values ranged from 2.751 to 20.156 with a mean of 7.095. The HO and HE ranged from 0.467 to 0.938 (mean: 0.692) and from 0.636 to 0.95 (mean: 0.824), respectively. The three wild populations (HW, PL, and QHD) showed a similar level of genetic diversity, while the cultivated population indicated a reduction in genetic diversity compared with them due to the relatively smaller genetic parameters (Table 4). Notably, the majority of SSR loci had null alleles and presented significant deviations from HWE, and heterozygote deficiency (HO < HE) was observed, with the exception of PrMa01, DX05, and DX19.
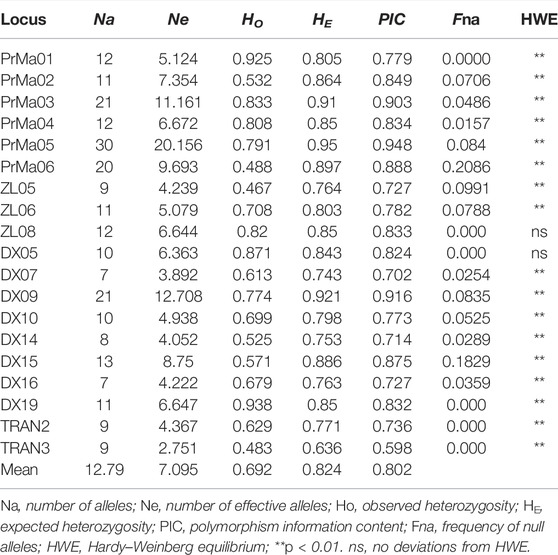
TABLE 3. Genetic diversity parameters for 19 SSR loci.
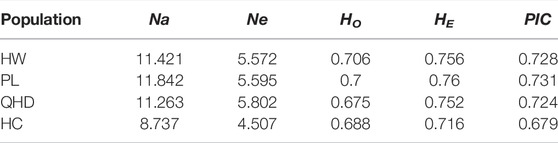
TABLE 4. Mean genetic parameters of four P. trituberculatus populations.
The populations HW and HC showed highest FST (0.040) (p < 0.05) whereas the lowest FST of 0.001 (p < 0.05) was observed between populations PL and QHD (Table 5). The mean FST was observed to be 0.0205, indicating low but significant levels of genetic differentiation among the four populations. The results of AMOVA revealed that only 2% of genetic variation was partitioned among populations while 98% of the variation was concentrated within populations (Table 6).
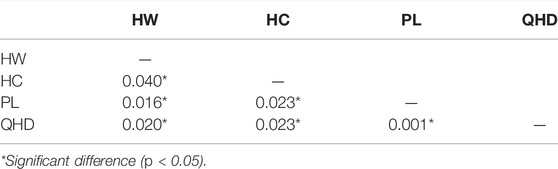
TABLE 5. Estimates of pairwise FST values among the four P. trituberculatus populations.

TABLE 6. Analysis of molecular variance (AMOVA) from four P. trituberculatus populations.
The genetic structural analysis of 240 P. trituberculatus samples was performed to infer the optimal K value with the ΔK method. When the highest ΔK value was observed, the optimal K value was 2 (Figure 4A), indicating that all individuals were clustered into two groups, including wild group (green) and the cultivated group (red) (Figure 4B). This was consistent with the population-level phylogenetic tree that the four populations were divided into 2 main clusters, in which cluster 1 contained only HC and cluster 2 contained all wild populations (Supplementary Figure S5). A certain degree of biological mixing, however, was also observed between wild and cultivated samples. The PCA revealed that the first two principal components (PCs) explained 7.9% (PC1) and 6.62% (PC2) of total variation respectively (Figure 5). The majority of samples were clustered together, and no obvious geographical patterns were observed. The cultivated individuals were mainly clustered towards the right side (positive values) of PC1. The individual-level phylogenetic tree was constructed based on NJ method, in which all individuals were clustered into two clades, and no significant clustering patterns related to geographical locations were found, but cultivated individuals were distributed more centrally than wild individuals (Figure 6).
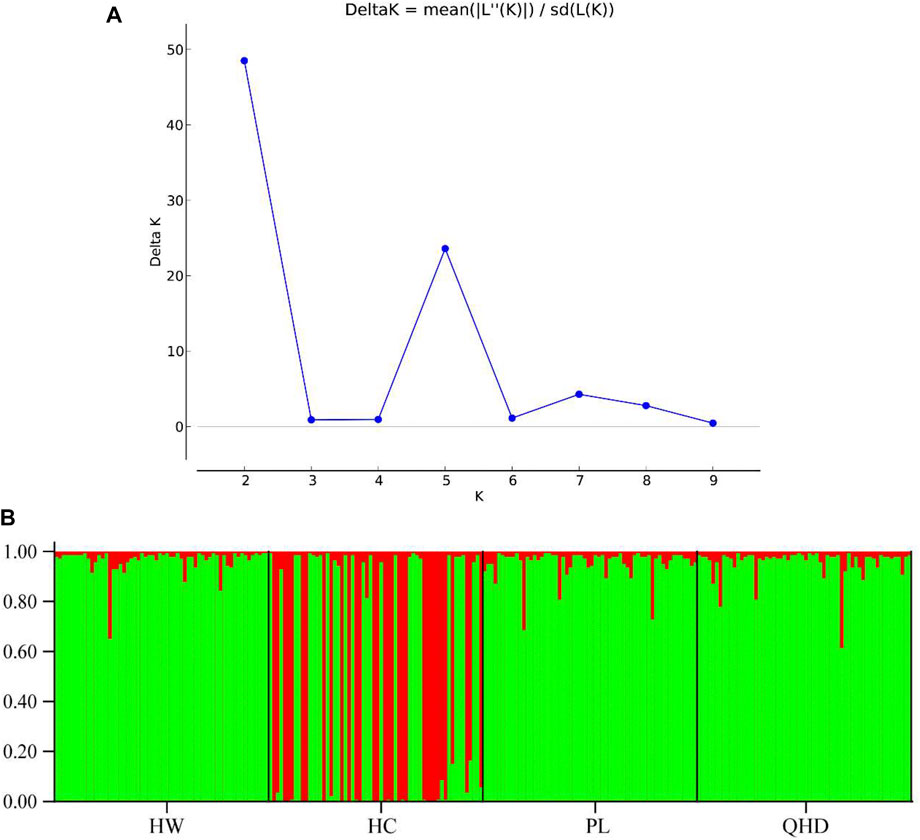
FIGURE 4. (A) Relationships between the number of clusters (K) and the corresponding. ΔK statistics calculated based on STRUCTURE analysis. (B) Population genetic structure based on the Bayesian clustering model among 240 P. trituberculatus samples at K = 2.
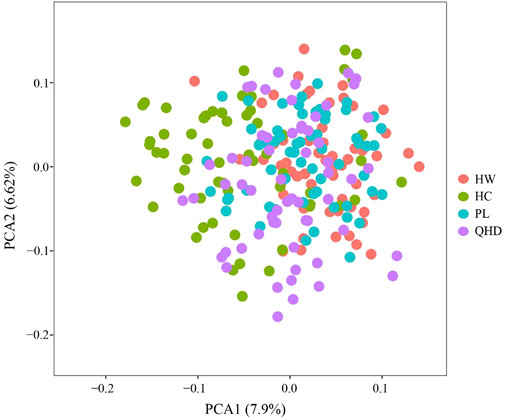
FIGURE 5. PCA analyses based on SSR data for 240 individuals from 4 P. trituberculatus populations.
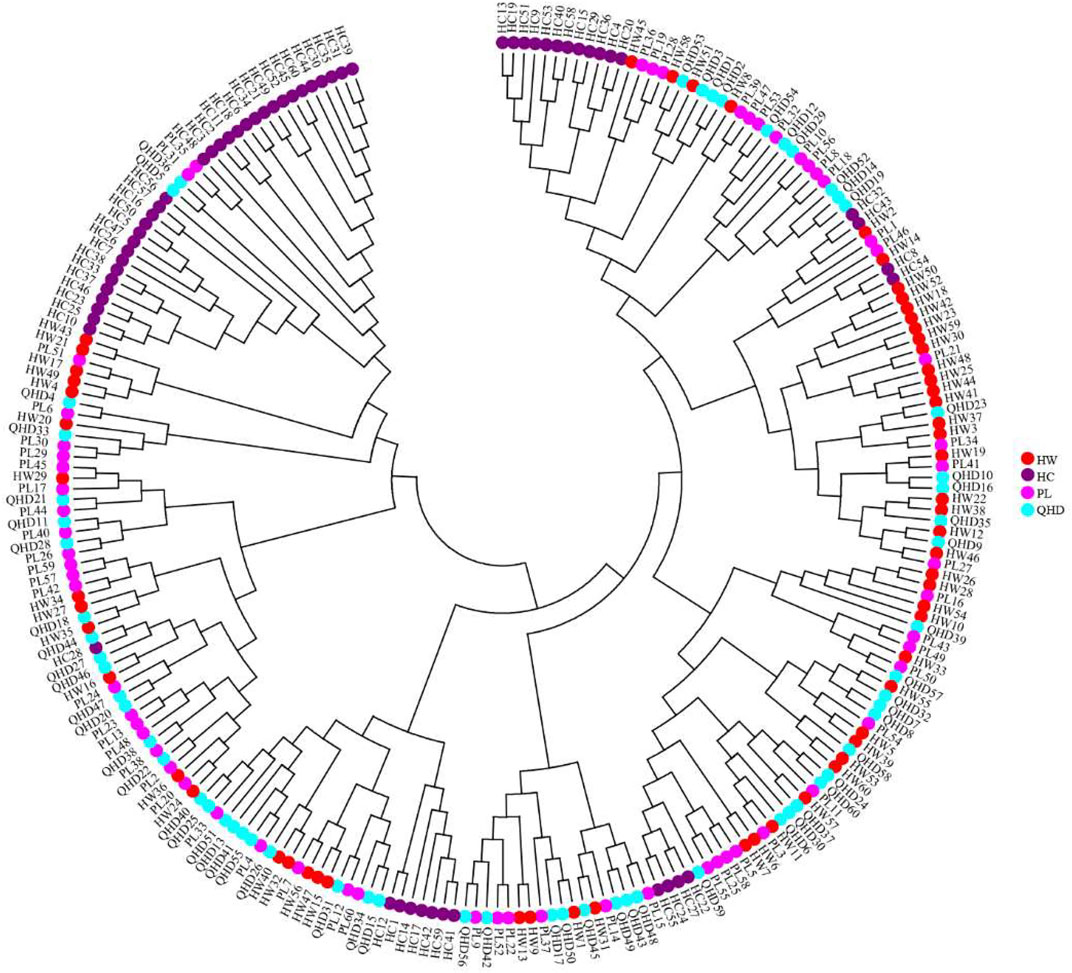
FIGURE 6. Neighbor-joining phylogenetic tree of 240 individuals of P. trituberculatus based on SSR data.
P. trituberculatus is a typical mariculture species with significant economic value. The Bohai Sea is an important habitat and fishing area for this species. The germplasm resources from the Bohai Sea form a vital foundation for the protection and breeding of P. trituberculatus in the national breeding farms of swimming crabs. Large-scale development of molecular markers and advancement of high-throughput sequencing technologies provide a solid support for germplasm resources assessment of P. trituberculatus in recent years (Shan et al., 2017; Liu et al., 2021b). Lv et al. (2014) identified 22,673 SSR markers of P. trituberculatus with transcriptome sequencing, which provided a material basis for future genetic linkage and quantitative trait loci analyses in this species. In this study, Illumina sequencing of P. trituberculatus was performed for the development of SSR markers, which generated new high-throughput data for transcriptomics, providing valuable information for germplasm conservation and selective breeding of P. trituberculatus.
A total of 246,243 SSRs were identified with the Illumina HiSeq 4000 platform, which was far higher than that of yesso scallop (Patinopecten yessoensis) (2,748 SSRs, Hou et al., 2011), yellow drum (Nibea albiflora) (12,254, Gong et al., 2016), Xingguo red carp (Cyprinus carpio var.singuonensis) (13,652, Yue et al., 2016), freshwater ornamental shrimps (Neocaridina denticulate) (25,355, Huang et al., 2020), and clam (Cyclina sinensis) (12,418, Fang et al., 2020). The possible reason is that P. trituberculatus has more chromosomes (2n = 106) than shellfish, fish, and other crustaceans (Liu et al., 2012), thus containing more SSR sequences. The average density of SSRs was 1/1.11 kb, which was higher than that in P. maxima (1/29.73 kb) (Wang et al., 2019a), sea cucumber (Apostichopus japonicus) (1/29.2 kb) (Du et al., 2012), mandarin fish (Siniperca chuatsi) (1/26.28 kb) (Sun et al., 2019), large-scale loach (Paramisgurnus dabryanus) (1/6.99 kb) (Li et al., 2015a), giant wrasse (Cheilinus undulatus) (1/5.35 kb) (Liu et al., 2020), burbot (Lota lota) (1/4.25 kb) (Meng et al., 2019), and the Bombay duck (Harpadon nehereus) (1/3 kb) (Huang et al., 2021), while lower than that of the South African abalone (Haliotis midae) (1/0.756 kb) (Franchini et al., 2011), tu-chung (Eucommia ulmoides) (1/0.73 kb) (Huang et al., 2013). The differences between SSR frequency and density may be attributed to genome structure, SSR mining tools, dataset size, and search criteria (Liu et al., 2021c). In addition, the most abundant types of SSR are dinucleotide repeats, which was consistent with the conclusions obtained by other high-throughput sequencing technologies in aquatic animals (Kong et al., 2019; Zhai et al., 2020).
In 173 different repeat motifs from the identified microsatellites in this study, A/T was the most abundant motif type higher than C/G in mononucleotide, which was congruent with the previous studies (Song et al., 2008; Fang et al., 2020; Tian et al., 2021). Ni et al. (2018) indicated that DNA recombination and replication sliding mechanisms in PCR amplification might result in high A/T content. In addition, methylated cytosine C is easily mutated into thymine T through deamination, which makes G/C mutate A/T in the process of DNA replication and transcription (Schlötterer and Tautz, 1992). In dinucleotide repeats, AC/GT exhibited the highest frequency, which was coincident with the result obtained by Lv et al. (2013). In the development of SSR markers based on transcriptome sequencing in blood clam (Scapharca kagoshimensis), AC/GT was also predominant among dinucleotide repeats (Chen et al., 2022). However, the opposite results were found in the Zhikong scallop (Chlamys farreri) (Zhan et al., 2008) and bay scallop (Argopecten irradians) (Li et al., 2009) from the SSR-enriched library, which showed that the proportion of AG/CT was higher than that of AC/GT in the genome. This difference may be related to the SSR screening method, base composition preference of different coding genes, and methylase activity in vivo (Chen et al., 2022).
The location of SSR loci determines their functional roles. SSRs in CDS affect the inactivated or activated genes or protein synthesis process, and SSRs in 3′UTR are involved in transcription slippage or gene silencing, and SSRs in 5′UTR impact gene transcription and translation (Xia et al., 2014; Xu et al., 2020; Liu et al., 2021c). In the present study, 88.92% of microsatellites were located in UTRs, which was much higher than that of CDS regions. One possible reason is that microsatellites located in UTRs are subject to fewer evolutionary constraints and natural selection pressure, thus easily leading to phenotype changes (Xu et al., 2020; Vidya et al., 2021). Moreover, 74.88% of trinucleotide repeats were found to be accumulated in CDSs regions. This might explain that non-trinucleotides negatively selected frameshift mutations, while trinucleotide did not cause frame shift mutation and failed to affect gene expression (Xia et al., 2014; Liu et al., 2021c).
To explore the potential functions of the obtained unigenes, the functional annotation and classification of these unigenes were conducted through BLASTX search in the public databases. GO annotations showed a lot of unigenes distributed to cellular process, metabolic process, cell, cell part, binding, and catalytic activity terms. This suggests that genes encoding these functions may be more conserved across different species and are thus easier to annotate in the database. In addition, KEGG and KOG annotations revealed that many unigenes might participate in the life activities and basal metabolism of P. trituberculatus with various biological functions. In summary, these annotation analyses contribute to finding potential genes associated with the growth and development of P. trituberculatus for breeding programs. Further studies also should be carried out to identify the molecular functions of these putative genes.
Genetic diversity is the foundational core of ecosystems and species diversity and can reveal population connectivity and adaptive potential of a species as well as provide insight into past events (Fang et al., 2021b; Ma et al., 2021). It is affected by many factors, including artificial selection, genetic drift, migration, and breeding systems and is usually evaluated by the polymorphism information content (PIC) and heterozygosity (Zhou, et al., 2019b). Values of PIC above 0.5 indicate high polymorphism (Singh et al., 2020). Heterozygosity is an important index to evaluate population variation at the genetic level, and the greater its value, the higher the population genetic diversity (Qin et al., 2013). Li et al. (2011) investigated the genetic diversity of five P. trituberculatus populations with eight SSR markers and observed that the mean HE values ranged from 0.7283 to 0.7704, which revealed a high level of genetic diversity in the wild resources. The current study reports PIC values of all SSR loci greater than 0.5, indicating the high polymorphic nature of the loci and their suitability for assessing genetic diversity in the four P. trituberculatus populations. The observed and expected heterozygosity values indicated a similar level of genetic diversity among the wild populations, and compared with the wild populations, the genetic diversity of the cultivated population showed a reduction because of lower genetic coefficients. In the estimation of genetic diversity of P. trituberculatus populations from Shandong peninsula, Liu et al. (2012) found a similar result. It is possible that genetic decline, genetic drift, and inbreeding result in low genetic variability in farmed stocks (Jorge et al., 2018). Additionally, the domesticated stocks are subjected to artificial selection in a selective breeding program, which may show reduced effective population size, thus leading to a decline in genetic diversity (Wong et al., 2022).
In general, the expected heterozygosity (HE) is more accurate than the observed heterozygosity (HO) for evaluating the level of population genetic diversity because Ho is easily influenced by sample sizes (Qin et al., 2013). Based on polymorphic SSR markers, middle (HE = 0.73–0.84) to high (HE = 0.916–0.918) genetic diversity of P. trituberculatus was revealed by Guo et al. (2013) and Xu and Liu (2011). In this study, the mean HE ranged from 0.675 to 0.706 which is lower than those observed in the above studies, thus showing a lower level of genetic diversity among the four populations. This may be attributable to the special geographical location of the Bohai Sea. The Bohai Sea is a semi-enclosed shallow water body and has limited connectivity to the Yellow Sea by the Bohai Strait, which restricts the dispersal of crabs and thus results in low genetic diversity (Liu et al., 2012). Moreover, the phenomena of eutrophication and hypoxia, as well as the serious interference from anthropogenic activities such as land-source pollution, aquaculture pollution, and reclamation in the Bohai Sea also reduced the genetic diversity of species (Wang et al., 2021).
FST is an important gauge of genetic differentiation between populations and is crucial for a better understanding of the genetic relationships. A value of FST which falls between 0 and 0.05 shows a low level of genetic differentiation (Wang et al., 2019b). The current study reports the mean FST values of 0.021 (p < 0.05) which is less than 0.05 (Table 5), indicating low levels of genetic differentiation among the four P. trituberculatus populations, which is conformed to the result described by Xu and Liu (2011). Based on the pairwise FST ranging from 0.0142 to 0.0498, six SSR loci showed no genetic difference between wild and cultivated populations of P. trituberculatus from the Zhejiang coastal region (Li et al., 2015b). This genetic similarity may be accounted for release of hatchery-produced offspring, which results in hybrid germplasm. Liu et al. (2018b) and Liu et al. (2021b) used SSR markers to evaluate the population structure of P. trituberculatus in Panjin and Yingkou (Liaoning, China) adjacent to Liaodong Bay, respectively, and found low but significant levels of genetic differentiation (FST < 0.05, p < 0.05), suggesting that large-scale stock enhancement of P. trituberculatus presents potential genetic risks on wild populations, and that the relevant management measures should be formulated to achieve successful stock enhancement and resource restoration for the swimming crab.
During the current study, AMOVA results revealed that total variance within populations (98%) was significantly greater than that among populations (2%). The result corresponded to the genetic variation found in blue swimmer crab (Portunus pelagicus) (Chai et al., 2017). Most loci showed a deficit of heterozygotes, which might result from the presence of null alleles, artificial selection, migration, and inbreeding in the population (Guo et al., 2013). Additionally, a majority of SSR loci deviated from the Hardy-Weinberg equilibrium, and this finding might be ascribed to null alleles or a small number of samples. Hence, designing more effective SSR primers to eliminate null alleles, and combining more molecular markers with a larger sample size are essential to elaborate the genetic diversity of P. trituberculatus populations in the Bohai Sea. In addition, genetic monitoring is required to preserve the genetic variations for preventing germplasm degradation and making full use of the genetic resources of P. trituberculatus.
A stable genetic structure is central to species survival. Its disintegration will lead to decreased populations or even extinction. Given the economic significance of P. trituberculatus, the understanding genetic structure is crucial for the development of effective management strategies and can provide a genetic tool for breeding and offer a scientific support for resource conservation of this species (Liu et al., 2009). The results of the current study establish that K = 2 is the most likely number of clusters when ΔK is at its highest. This finding confirms that the P. trituberculatus specimens from the four populations cluster into two groups including the cultured group and the wild group (Figure 4). Some of the genetic information gained from the cultured samples has been assigned to wild populations, indicating that the ancestral generation of these wild individuals may derive from cultivated populations because of the hatchery-reared seed release activities. In a cultivated group, some genetic information that derives from wild samples can be observed. This observation indicates some degree of introgression of wild populations into the cultivated population, which may be accounted for the fact that fertilized female crabs are caught as broodstocks from the wild to use to artificially culture and produce seeds (Duan et al., 2022b).
The individual-level phylogenetic tree and PCA illuminated that all individuals showed some degree of genetic connectivity, and that the cultured individuals were relatively concentrated in comparison with wild individuals. Despite the annual release activities, the gene exchange between cultivated and wild populations is limited when compared to that between wild populations in the open sea, thus leading to the separation of cultured individuals from all individuals, showing more obvious particularity. Therefore, it is vital to further investigate the genetic structure of wild and cultured populations of P. trituberculatus in the Bohai Sea for formulating scientific management measures to prevent mutual interference between them.
Overall, this study performed assembly of transcriptome sequences, functional annotation, and SSR markers discovery of P. trituberculatus. Nineteen polymorphic SSRs were identified and used to investigate the genetic variation and structure of the four P. trituberculatus populations from the Bohai Sea. The findings revealed a lower level of genetic diversity in P. trituberculatus populations from the Bohai Sea when compared to the other populations from the Yellow Sea and the East China Sea. The pairwise FST values showed low but significant genetic differentiation between populations. The population structure analysis, phylogenetic tree, and PCA showed a mixing of wild and cultivated individuals, which corroborated the genetic connectivity between them, but cultivated individuals were distributed more centrally than wild individuals. In addition, heterozygote deficiencies, null alleles, and significant deviation from HWE at many SSR loci were observed. Therefore, practical and effective measures are expected to be taken to reinforce the identification and protection of genetic diversity and prevent degeneration of P. trituberculatus germplasm. For example, developing high-quality markers such as SNPs using a chromosome-level genome of P. trituberculatus (Tang et al., 2020; Lv et al., 2021), and carrying out a large-scale investigation to fully elucidate the genetic diversity and population structure of P. trituberculatus in the Bohai Sea. Additionally, increasing the scale of swimming crab aquaculture, extending the fishing moratorium, and performing long-term genetic monitoring is also helpful for the conservation and utilization of germplasm resources in P. trituberculatus. In conclusion, the results improve our understanding of the population genetic structure of P. trituberculatus in the Bohai Sea and provide valuable information for the selection breeding of this species.
The original contributions presented in the study are publicly available. This data can be found in NCBI under accession numbers SUB11453401 and PRJNA836158.
BD: writing—original draft, formal analysis, investigation, and methodology. SM and YG: data curation and formal analysis. WL, TK, and YC: investigation, methodology, and validation. ZL and YT: resources. XK: supervision, project administration, conceptualization, and writing—review and editing. All authors contributed to the article and approved the submitted version.
This research was supported by the Natural Science Foundation of Hebei Province (C2016201249); Science and Technology Innovation Project of Modern Seed Industry (21326307D); Institute of Life Science and Green Development (Hebei University), and Innovation Center for Bioengineering and Biotechnology of Hebei Province.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.932173/full#supplementary-material
Bureau of Fisheries of Ministry of Agriculture, PRC (2021). China Fishery Statistical Yearbook of 2021. Beijing, China: Agricultural press.
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: Architecture and Applications. BMC Bioinforma. 10, 421. doi:10.1186/1471-2105-10-421
Chai, C. J., Bin Esa, Y., Ismail, S., and Kamarudin, M. S. (2017). Population Structure of the Blue Swimmer Crab Portunus Pelagicus in Coastal Areas of Malaysia Inferred from Microsatellites. Zool. Stud. 56, e26. doi:10.6620/ZS.2017.56-26
Chakraborty, D., Sharma, N., Kour, S., Sodhi, S. S., Gupta, M. K., Lee, S. J., et al. (2022). Applications of Omics Technology for Livestock Selection and Improvement. Front. Genet. 13, 774113. doi:10.3389/fgene.2022.774113
Chen, L., Li, L., Shi, X., Qin, Y., Liu, L., and Guo, Y. (2022). Development and Evaluation of SSR Markers Based on Transcriptome Sequencing in Scapharca Kagoshimensis. Prog. Fish. Sci. 43, 129–137. doi:10.19663/j.issn2095-9869.20210206003
Chen, X., Li, J., Xiao, S., and Liu, X. (2016). De Novo assembly and Characterization of Foot Transcriptome and Microsatellite Marker Development for Paphia Textile. Gene 576, 537–543. doi:10.1016/j.gene.2015.11.001
Chi, D., Yan, B., Shen, S., and Gao, H. (2010). Rapd Analysis between Color-Different Crab Individuals of Portunus Trituberculatus. Mar. Sci. Bull. 12, 47–54. doi:10.3969/j.issn.1000-9620.2010.02.006
Du, H., Bao, Z., Hou, R., Wang, S., Su, H., Yan, J., et al. (2012). Transcriptome Sequencing and Characterization for the Sea Cucumber Apostichopus Japonicus (Selenka, 1867). PLoS One 7, e33311. doi:10.1371/journal.pone.0033311
Duan, B., Mu, S., Guan, Y., Li, S., Yu, Y., Liu, W., et al. (2022a). Genetic Diversity and Population Structure of the Swimming Crab (Portunus Trituberculatus) in China Seas Determined by Genotyping-By-Sequencing (GBS). Aquaculture 555, 738233. doi:10.1016/j.aquaculture.2022.738233
Duan, B., Liu, W., Li, S., Yu, Y., Guan, Y., Mu, S., et al. (2022b). Microsatellite Analysis of Genetic Diversity in Wild and Cultivated Portunus Trituberculatus in Bohai Bay. Mol. Biol. Rep. 49, 2543–2551. doi:10.1007/s11033-021-07054-w
Earl, D. A., and Vonholdt, B. M. (2012). STRUCTURE HARVESTER: A Website and Program for Visualizing Structure Output and Implementing the Evanno Method. Conserv. Genet. Resour. 4, 359–361. doi:10.1007/s12686-011-9548-7
Evanno, G., Regnaut, S., and Goudet, J. (2005). Detecting the Number of Clusters of Individuals Using the Software Structure: A Simulation Study. Mol. Ecol. 14, 2611–2620. doi:10.1111/j.1365-294X.2005.02553.x
Fan, X., Gao, B., Liu, P., and Li, J. (2009). Genetic Variation Analysis of Four Wild Populations of Portunus Trituberculatus by Isozyme. Prog. Fish. Sci. 30, 84–89. doi:10.3969/j.issn.1000-7075.2009.04.013
Fang, F., Yuan, Y., Jin, M., Shi, B., Zhu, T., Luo, J., et al. (2021a). Hepatopancreas Transcriptome Analysis Reveals the Molecular Responses to Different Dietary N-3 PUFA Lipid Sources in the Swimming Crab Portunus Trituberculatus. Aquaculture 543, 737016. doi:10.1016/j.aquaculture.2021.737016
Fang, J., Shen, Y., Zhang, L., Li, T., and Zhao, Y. (2020). Development and Verification of SSR Markers in Cyclina Sinensis. J. Phys. Oceanogr. 39, 214–220. doi:10.3969/J.ISSN.2095-4972.2020.02.008
Fang, Y., Chen, J., Ruan, H., Xu, N., Que, Z., and Liu, H. (2021b). Genetic Diversity and Population Structure of Metaphire Vulgaris Based on the Mitochondrial Coi Gene and Microsatellites. Front. Genet. 12, 686246. doi:10.3389/fgene.2021.686246
Franchini, P., Van der Merwe, M., and Roodt-Wilding, R. (2011). Transcriptome Characterization of the South African Abalone Haliotis Midae Using Sequencing-By-Synthesis. BMC Res. Notes 4, 59. doi:10.1186/1756-0500-4-59
Gong, S., Wang, Z., Xiao, S., Lin, A., and Xie, Y. (2016). Development and Verification of SSR Based on Transcriptome of Yellow Drum. Nibea Albiflora. J. Jimei Univ. Nat. Sci. 21, 241–246. doi:10.19715/j.jmuzr.2016.04.001
Götz, S., García-Gómez, J. M., Terol, J., Williams, T. D., Nagaraj, S. H., Nueda, M. J., et al. (2008). High-Throughput Functional Annotation and Data Mining with the Blast2Go Suite. Nucleic Acids Res. 36, 3420–3435. doi:10.1093/nar/gkn176
Gou, X., Shi, H., Yu, S., Wang, Z., Li, C., Liu, S., et al. (2020). SSRMMD: A Rapid and Accurate Algorithm for Mining SSR Feature Loci and Candidate Polymorphic SSRs Based on Assembled Sequences. Front. Genet. 11, 706. doi:10.3389/fgene.2020.00706
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length Transcriptome Assembly from RNA-Seq Data without a Reference Genome. Nat. Biotechnol. 29, 644–652. doi:10.1038/nbt.1883
Guo, E., Cui, Z., Wu, D., Hui, M., Liu, Y., and Wang, H. (2013). Genetic Structure and Diversity of Portunus Trituberculatus in Chinese Population Revealed by Microsatellite Markers. Biochem. Syst. Ecol. 50, 313–321. doi:10.1016/j.bse.2013.05.006
Guo, E., Liu, Y., Cui, Z., Li, X., Cheng, Y., and Wu, X. (2012). Genetic Variation and Population Structure of Swimming Crab (Portunus Trituberculatus) Inferred from Mitochondrial Control Region. Mol. Biol. Rep. 39, 1453–1463. doi:10.1007/s11033-011-0882-3
Hou, R., Bao, Z., Wang, S., Su, H., Li, Y., Du, H., et al. (2011). Transcriptome Sequencing and De Novo Analysis for Yesso Scallop (Patinopecten Yessoensis) Using 454 GS FLX. PLoS One 6, e21560. doi:10.1371/journal.pone.0021560
Huang, C.-W., Chu, P.-Y., Wu, Y.-F., Chan, W.-R., and Wang, Y.-H. (2020). Identification of Functional SSR Markers in Freshwater Ornamental Shrimps Neocaridina Denticulata Using Transcriptome Sequencing. Mar. Biotechnol. 22, 772–785. doi:10.1007/s10126-020-09979-y
Huang, H., Du, H., Wuyun, T., and Liu, P. (2013). Development of SSR Molecular Markers Based on Transcriptome Sequencing of. Eucommia Ulmoides. Sci. Silvae Sin. 49, 176–181. doi:10.11707/j.1001-7488.20130523
Huang, X., Jiang, Y., Jiang, X., and Yang, T. (2021). Analysis of Microsatellite Markers in Harpadon Nehereus Based on Transcriptome Sequencing Illumina Hiseqtm 2500. J. Zhejiang Ocean. Univ. Nat. Sci.) 40, 189–197. doi:10.3969/j.issn.1008-830X.2021.03.001
Hui, M., Shi, G., Sha, Z., Liu, Y., and Cui, Z. (2018). Genetic Population Structure in the Swimming Crab, Portunus Trituberculatus and its Implications for Fishery Management. J. Mar. Biol. Ass. 99, 891–899. doi:10.1017/S0025315418000796
Jorge, P. H., Mastrochirico-Filho, V. A., Hata, M. E., Mendes, N. J., Ariede, R. B., Freitas, M. V. d., et al. (2018). Genetic Characterization of the Fish Piaractus Brachypomus by Microsatellites Derived from Transcriptome Sequencing. Front. Genet. 9, 46. doi:10.3389/fgene.2018.00046
Jung, H., Lyons, R. E., Dinh, H., Hurwood, D. A., McWilliam, S., Mather, P. B., et al. (2011). Transcriptomics of a Giant Freshwater Prawn (Macrobrachium Rosenbergii): De Novo Assembly, Annotation and Marker Discovery. PLoS One 6, e27938. doi:10.1371/journal.pone.0027938
Kong, X., Li, M., Chen, Z., Gong, Y., Zhang, J., and Zhang, P. (2019). Development and Evaluation of Di-/Tri-Nucleotide-repeated Microsatellites by RAD-Seq in. Decapterus Macrosoma. South China Fish. Sci. 15, 97–103. doi:10.12131/20180256
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 33, 1870–1874. doi:10.1093/molbev/msw054
Lee, H. J., Lee, D.-H., Yoon, S.-J., Kim, D. H., Kim, S.-G., Hyun, Y. S., et al. (2013). Characterization of 20 Microsatellite Loci by Multiplex PCR in Swimming Crab, Portunus Trituberculatus. Genes. Genom 35, 77–85. doi:10.1007/s13258-013-0062-z
Li, C., Ling, Q., Ge, C., Ye, Z., and Han, X. (2015a). Transcriptome Characterization and SSR Discovery in Large-Scale Loach Paramisgurnus dabryanus (Cobitidae, Cypriniformes). Gene 557, 201–208. doi:10.1016/j.gene.2014.12.034
Li, H., Liu, X., Du, X., Song, R., Zhang, G., Hu, J., et al. (2009). Development of Microsatellite Markers in Bay Scallop and Their Inheritance Patterns in an F1 Hybrid Family. Mar. Sci. 33, 4–8.
Li, N., Zheng, Y.-Q., Ding, H.-M., Li, H.-P., Peng, H.-Z., Jiang, B., et al. (2018). Development and Validation of SSR Markers Based on Transcriptome Sequencing of Casuarina equisetifolia. Trees 32, 41–49. doi:10.1007/s00468-017-1607-6
Li, P., Xu, K., and Zhou, H. (2015b). Genetic Diversity of Portunus Trituberculatus Among the Cultured and Wild Populations in the Offing of Zhejiang Using Microsatellite Markers. J. Zhejiang Ocean. Univ. Nat. Sci.) 34, 537–542. doi:10.3969/j.issn.1008-830X.2015.06.007
Li, X., Li, P., Li, J., and Gao, B. (2011). Genetic Diversity Among Five Wild Populations of Portunus Trituberculatus. J. Fish. Sci. China 18, 1327–1334. doi:10.3724/SP.J.1118.2011.01327
Liu, B., Zhang, X., Wang, Z., Li, W., Zhang, Q., Liu, Q., et al. (2021b). Genetic Pattern Fluctuations in Wild Swimming Crab Populations, Under the Influence of Continuous Mass Stock Enhancement. Fish. Res. 243, 106075. doi:10.1016/j.fishres.2021.106075
Liu, H., Ju, Y., Tamate, H., Wang, T., and Xing, X. (2021a). Phylogeography of Sika Deer (Cervus Nippon) Inferred from Mitochondrial Cytochrome-B Gene and Microsatellite DNA. Gene 772, 145375. doi:10.1016/j.gene.2020.145375
Liu, H., Liu, J., Yang, M., He, Y., and Wang, Y. (2020). SSR and SNP Polymorphic Feature Analysis Based on Cheilinus Undulatus Transcriptome. Genomics Appl. Biol. 39, 2451–2461. doi:10.13417/j.gab.039.002451
Liu, H., Zhang, Y., Wang, Z., Su, Y., and Wang, T. (2021c). Development and Application of EST-SSR Markers in Cephalotaxus Oliveri from Transcriptome Sequences. Front. Genet. 12, 759557. doi:10.3389/fgene.2021.759557
Liu, L.-X., Liu, Y.-G., and Xing, S.-C. (2014). An Analysis of Genetic Variability in Wild and Hatchery Populations of Swimming Crab (Portunus Trituberculatus) Using AFLP Markers. Fish. Aquac. J. 05, 1000104. doi:10.4172/2150-3508.1000104
Liu, L., Fu, Y., Zhu, F., Mu, C., Li, R., Song, W., et al. (2018a). Transcriptomic Analysis of Portunus Trituberculatus Reveals A Critical Role for WNT4 and WNT Signalling in Limb Regeneration. Gene 658, 113–122. doi:10.1016/j.gene.2018.03.015
Liu, M., Pan, J., Dong, Z., Cheng, Y., Gong, J., and Wu, X. (2019). Comparative Transcriptome Reveals the Potential Modulation Mechanisms of Estradiol Affecting Ovarian Development of Female Portunus Trituberculatus. PLoS One 14, e0226698. doi:10.1371/journal.pone.0226698
Liu, Q., Cui, F., Hu, P., Yi, G., Ge, Y., Liu, W., et al. (2018b). Using of Microsatellite DNA Profiling to Identify Hatchery-Reared Seed and Assess Potential Genetic Risks Associated with Large-Scale Release of Swimming Crab Portunus Trituberculatus in Panjin, Chinafiling to Identify Hatchery-Reared Seed and Assess Potential Genetic Risks Associated with Large-Scale Release of Swimming Crab Portunus Trituberculatus in Panjin, China. Fish. Res. 207, 187–196. doi:10.1016/j.fishres.2018.05.003
Liu, S., Sun, J., and Hurtado, L. A. (2013). Genetic Differentiation of Portunus Trituberculatus, the World's Largest Crab Fishery, Among its Three Main Fishing Areas. Fish. Res. 148, 38–46. doi:10.1016/j.fishres.2013.08.003
Liu, X., Xie, X., Liu, H., Nie, H., Ma, H., Li, D., et al. (2022). Population Genomic Evidence for Genetic Divergence in the Northwest Pacific Ark Shell (Scapharca Broughtonii). Aquac. Rep. 24, 101100. doi:10.1016/j.aqrep.2022.101100
Liu, Y.-G., Guo, Y.-H., Hao, J., and Liu, L.-X. (2012). Genetic Diversity of Swimming Crab (Portunus Trituberculatus) Populations from Shandong Peninsula as Assessed by Microsatellite Markers. Biochem. Syst. Ecol. 41, 91–97. doi:10.1016/j.bse.2011.12.024
Liu, Y., Liu, R., Ye, L., Liang, J., Xuan, F., and Xu, Q. (2009). Genetic Differentiation between Populations of Swimming Crab Portunus Trituberculatus along the Coastal Waters of the East China Sea. Hydrobiologia 618, 125–137. doi:10.1007/s10750-008-9570-2
Lu, C., Sun, Z., Xu, P., Na, R., Lv, W., Cao, D., et al. (2022). Novel Microsatellites Reveal Wild Populations Genetic Variance in Pike-Perch (Sander lucioperca) in China. Aquac. Rep. 23, 101031. doi:10.1016/j.aqrep.2022.101031
Lv, J., Gao, B., Liu, P., Li, J., and Meng, X. (2017). Linkage Mapping Aided by De Novo Genome and Transcriptome Assembly in Portunus Trituberculatus: Applications in Growth-Related QTL and Gene Identification. Sci. Rep. 7, 7874. doi:10.1038/s41598-017-08256-8
Lv, J., Li, R., Su, Z., Gao, B., Ti, X., Yan, D., et al. (2021). A Chromosome-Level Genome of Portunus Trituberculatus Provides Insights into its Evolution, Salinity Adaptation and Sex Determination. Mol. Ecol. Resour. 22, 1606–1625. doi:10.1111/1755–0998.13564
Lv, J., Liu, P., Gao, B., Wang, Y., Wang, Z., Chen, P., et al. (2014). Transcriptome Analysis of the Portunus Trituberculatus: De Novo Assembly, Growth-Related Gene Identification and Marker Discovery. PLoS One 9, e94055. doi:10.1371/journal.pone.0094055
Lv, J., Wang, Y., Gao, B., Li, J., and Liu, P. (2013). Identification of Type Ⅰ Microsatellite Markers and Their Polymorphism in Portunus Trituberculatus. J. Fish. China 37, 816–822. doi:10.3724/SP.J.1231.2013.38343
Ma, H., Jiang, W., Liu, P., Feng, N., Ma, Q., Ma, C., et al. (2014). Identification of Transcriptome-Derived Microsatellite Markers and Their Association with the Growth Performance of the Mud Crab (Scylla Paramamosain). PLoS One 9, e89134. doi:10.1371/journal.pone.0089134
Ma, H., Li, L., Xiao, S., Zhang, Y., and Yu, Z. (2021). Microsatellite-Based Study of Population Genetics of Crassostrea Hongkongensis in Southern China. Aquac. Rep. 19, 100591. doi:10.1016/j.aqrep.2021.100591
Ma, K., Qiu, G., Feng, J., and Li, J. (2012). Transcriptome Analysis of the Oriental River Prawn, Macrobrachium Nipponense Using 454 Pyrosequencing for Discovery of Genes and Markers. PLoS One 7, e39727. doi:10.1371/journal.pone.0039727
Meng, W., Jiang, Y., Zhang, L., Yang, T., and Zhou, J. (2019). SSR Loci Information Analysis in the Transcriptome of Burbot (Lota Lota) Based on RNA-Seq. Freshw. Fish. 49, 10–14. doi:10.13721/j.cnki.dsyy.2019.06.002
Nagy, S., Poczai, P., Cernák, I., Gorji, A. M., Hegedűs, G., and Taller, J. (2012). PICcalc: An Online Program to Calculate Polymorphic Information Content for Molecular Genetic Studies. Biochem. Genet. 50, 670–672. doi:10.1007/s10528-012-9509-1
Nguyen, C., Nguyen, T. G., Nguyen, L. V., Pham, H. Q., Nguyen, T. H., Pham, H. T., et al. (2016). De Novo assembly and Transcriptome Characterization of Major Growth-Related Genes in Various Tissues of Penaeus monodon. Aquaculture 464, 545–553. doi:10.1016/j.aquaculture.2016.08.003
Ni, S., Yang, Y., Liu, S., and Zhuang, Z. (2018). Microsatellite Analysis of Patinopecten Yessoensis Using Next-Generation Sequencing Method. Prog. Fish. Sci. 39, 107–113. doi:10.11758/yykxjz.20161209001
Pavan Kumar, P., Janakiram, T., and Bhat, K. V. (2020). Microsatellite Based DNA Fingerprinting and Assessment of Genetic Diversity in Bougainvillea Cultivars. Gene 753, 144794. doi:10.1016/j.gene.2020.144794
Peakall, R., and Smouse, P. E. (2012). GenAlEx 6.5: Genetic Analysis in Excel. Population Genetic Software for Teaching and Research-Aan Update. Bioinformatics 28, 2537–2539. doi:10.1093/bioinformatics/bts460
Pritchard, J. K., Stephens, M., and Donnelly, P. (2000). Inference of Population Structure Using Multilocus Genotype Data. Genetics 155, 945–959. doi:10.1093/genetics/155.2.945
Qin, Y., Shi, G., and Sun, Y. (2013). Evaluation of Genetic Diversity in Pampus Argenteus Using SSR Markers. Genet. Mol. Res. 12, 5833–5841. doi:10.4238/2013.November.22.10
Rousset, F. (2008). genepop'007: a Complete Re-implementation of the Genepop Software for Windows and Linux. Mol. Ecol. Resour. 8, 103–106. doi:10.1111/j.1471-8286.2007.01931.x
Schlötterer, C., and Tautz, D. (1992). Slippage Synthesis of Simple Sequence DNA. Nucl. Acids Res. 20, 211–215. doi:10.1093/nar/20.2.211
Shan, B., Hui, M., Zhang, X., Liu, S., Cai, S., Song, N., et al. (2017). Genetic Effects of Released Swimming Crab (Portunus Trituberculatus) on Wild Populations Inferred from Mitochondrial Control Region Sequences. Mitochondrial DNA Part A 29, 856–861. doi:10.1080/24701394.2017.1373108
Singh, R. B., Mahenderakar, M. D., Jugran, A. K., Singh, R. K., and Srivastava, R. K. (2020). Assessing Genetic Diversity and Population Structure of Sugarcane Cultivars, Progenitor Species and Genera Using Microsatellite (SSR) Markers. Gene 753, 144800. doi:10.1016/j.gene.2020.144800
Song, L., Liu, P., Li, J., and Liu, Z. (2008). Analysis of Microsatellite Sequences in Genome of Crab, Portunus Trituberculatus. J. Fish. Sci. China 15, 738–744. doi:10.3321/j.issn:1005-8737.2008.05.004
Sun, H., Sun, C., Dong, J., Tian, Y., Hu, J., and Ye, X. (2019). Transcriptome Sequencing and Development and Application of Novel SSR Markers for Siniperca chuatsi. Genomics Appl. Biol. 38, 4413–4421. doi:10.13417/j.gab.038.004413
Tang, B., Zhang, D., Li, H., Jiang, S., Zhang, H., Xuan, F., et al. (2020). Chromosome-Level Genome Assembly Reveals the Unique Genome Evolution of the Swimming Crab (Portunus Trituberculatus). GigaScience 9, 1–10. doi:10.1093/gigascience/giz161
Tian, Z., Chen, A., Wu, Y., Chen, S., Zhang, Y., Cao, Y., et al. (2021). Bioinformatics Analysis of Microsatellite Sites in the RNA-Sequencing of <italic>Meretrix Meretrix</italic>. Mf 43, 160–167. doi:10.13233/j.cnki.mar.fish.20210422.001
Tulsani, N. J., Hamid, R., Jacob, F., Umretiya, N. G., Nandha, A. K., Tomar, R. S., et al. (2020). Transcriptome Landscaping for Gene Mining and SSR Marker Development in Coriander (Coriandrum Sativum L.). Genomics 112, 1545–1553. doi:10.1016/j.ygeno.2019.09.004
Vidya, V., Prasath, D., Snigdha, M., Gobu, R., Sona, C., and Maiti, C. S. (2021). Development of EST-SSR Markers Based on Transcriptome and its Validation in Ginger (Zingiber Officinale Rosc.). PLoS ONE 16, e0259146. doi:10.1371/journal.pone.0259146
Waiho, K., Ikhwanuddin, M., Afiqah‐Aleng, N., Shu‐Chien, A. C., Wang, Y., Ma, H., et al. (2022). Transcriptomics in Advancing Portunid Aquaculture: A Systematic Review. Rev. Aquac. 14, 1–25. doi:10.1111/raq.12689
Wang, H., Cui, Z., Wu, D., Guo, E., Liu, Y., Wang, C., et al. (2012). Application of Microsatellite DNA Parentage Markers in the Swimming Crab Portunus Trituberculatus. Aquacult Int. 20, 649–656. doi:10.1007/s10499-011-9493-1
Wang, Q., Liu, Y., Yan, L., Chen, L., and Li, B. (2021). Genome-Wide SNP Discovery and Population Genetic Analysis of Mesocentrotus Nudus in China Seas. Front. Genet. 12, 717764. doi:10.3389/fgene.2021.717764
Wang, S.-H., Zhang, C., Shang, M., Wu, X.-G., and Cheng, Y.-X. (2019b). Genetic Diversity and Population Structure of Native Mitten Crab (Eriocheir Sensu Stricto) by Microsatellite Markers and Mitochondrial COI Gene Sequence. Gene 693, 101–113. doi:10.1016/j.gene.2018.12.083
Wang, Z., Li, J., Hao, R., Adzigbli, L., and Deng, Y. (2019a). Characterization and Development of SSR Markers of Pinctada Maxima by RNA-Seq Approach. Aquac. Rep. 15, 100230. doi:10.1016/j.aqrep.2019.100230
Wang, Z., Li, X., Jiang, Y., Zuo, B., Si, Y., and Zheng, Y. (2018a). Population Genetic Analysis on Eriocheir Sinensis with the Red Shell Using Microsatellite Markers. Chin. Fish. Qual. Stand. 8, 34–41. doi:10.3969/j.issn.2095-1833.2018.03.005
Wang, Z., Sun, L., Guan, W., Zhou, C., Tang, B., Cheng, Y., et al. (2018b). De Novo Transcriptome Sequencing and Analysis of Male and Female Swimming Crab (Portunus Trituberculatus) Reproductive Systems During Mating Embrace (Stage Ii). BMC Genet. 19, 3. doi:10.1186/s12863-017-0592-5
Wong, L. L., Razali, S. A., Deris, Z. M., Danish-Daniel, M., Tan, M. P., Nor, S. A. M., et al. (2022). Application of Second-Generation Sequencing (SGS) and Third Generation Sequencing (TGS) in Aquaculture Breeding Program. Aquaculture 548, 737633. doi:10.1016/j.aquaculture.2021.737633
Xia, W., Xiao, Y., Liu, Z., Luo, Y., Mason, A. S., Fan, H., et al. (2014). Development of Gene-Based Simple Sequence Repeat Markers for Association Analysis in Cocos nucifera. Mol. Breed. 34, 525–535. doi:10.1007/s11032-014-0055-x
Xu, Q., and Liu, R. (2011). Development and Characterization of Microsatellite Markers for Genetic Analysis of the Swimming Crab, Portunus Trituberculatus. Biochem. Genet. 49, 202–212. doi:10.1007/s10528-010-9399-z
Xu, R., Wang, Z., Su, Y., and Wang, T. (2020). Characterization and Development of Microsatellite Markers in Pseudotaxus Chienii (Taxaceae) Based on Transcriptome Sequencing. Front. Genet. 11, 574304. doi:10.3389/fgene.2020.574304
Yang, W., Zheng, J., Jia, B., Wei, H., Wang, G., and Yang, F. (2018). Isolation of Novel Microsatellite Markers and Their Application for Genetic Diversity and Parentage Analyses in Sika Deer. Gene 643, 68–73. doi:10.1016/j.gene.2017.12.007
Yeh, F. C., Yang, R., and Boyle, T. (1999). POPGENE Microsoft Window-Base Software for Population Genetic Analysis Version1.32. Alberta, Canada: University of Alberta Center for International Forestry Research. A Quick User's Guide.
Yu, L., Zhu, X., Liang, J., Fan, J., and Chen, C. (2019). Analysis of Genetic Structure of Wild and Cultured Giant Freshwater Prawn (Macrobrachium Rosenbergii) Using Newly Developed Microsatellite. Front. Mar. Sci. 6, 323. doi:10.3389/fmars.2019.00323
Yue, H., Zhai, Q., Song, M., Ye, H., Yang, X., and Li, C. (2016). Development of Microsatellite Markers in Cyprinus carpio Var. Singuonensis Using Next-Generation Sequencing. Freshw. Fish. 46, 24–28. doi:10.13721/j.cnki.dsyy.2016.01.004
Yue, L., Wang, Y., Xian, W., and Zhang, H. (2022). Genetic Diversity and Population Structure of Portunustrituberculatus in Released and Wild Populations Based on Microsatellite DNA Markers from the Yangtze Estuary. Diversity 14, 374. doi:10.3390/d14050374
Zhai, Y., Wu, R., Niu, S., Shen, R., and Zhang, H. (2020). Development of Dinucleotide Microsatellite Loci for Lateolabrax Maculates Using Next Generation Sequencing Technology. Genomics Appl. Biol. 39, 507–513. doi:10.13417/j.gab.039.000507
Zhan, A., Hu, J., Hu, X., Lu, W., Wang, M., Peng, W., et al. (2008). Fast Identification of Scallop Adductor Muscles Using Species-specific Microsatellite Markers. Eur. Food Res. Technol. 227, 353–359. doi:10.1007/s00217-007-0728-3
Zhang, W., Lv, J., Lan, W., Gao, B., and Liu, P. (2022). Discovery of Sex-Determining Genes in Portunus Trituberculatus: A Comparison of Male and Female Transcriptomes During Early Developmental Stages. Front. Mar. Sci. 8, 811052. doi:10.3389/fmars.2021.811052
Zhao, Y., Zhu, X., Li, Z., Xu, W., Dong, J., Wei, H., et al. (2019). Genetic Diversity and Structure of Chinese Grass Shrimp, Palaemonetes Sinensis, Inferred from Transcriptome-Derived Microsatellite Markers. BMC Genet. 20, 75. doi:10.1186/s12863-019-0779-z
Zheng, X., Kuang, Y., Lü, W., Cao, D., and Sun, X. (2014). Transcriptome-derived EST-SSR Markers and Their Correlations with Growth Traits in Crucian Carp Carassius auratus. Fish. Sci. 80, 977–984. doi:10.1007/s12562-014-0782-2
Zhou, Q.-C., Shi, B., Jiao, L.-F., Jin, M., Sun, P., Ding, L.-Y., et al. (2019a). Hepatopancreas and Ovarian Transcriptome Response to Different Dietary Soybean Lecithin Levels in Portunus Trituberculatus. Comp. Biochem. Physiology Part D Genomics Proteomics 31, 100600. doi:10.1016/j.cbd.2019.100600
Zhou, Q., Mu, K., Ni, Z., Liu, X., Li, Y., and Xu, L.-a. (2020b). Analysis of Genetic Diversity of Ancient Ginkgo Populations Using SSR Markers. Industrial Crops Prod. 145, 111942. doi:10.1016/j.indcrop.2019.111942
Keywords: Portunus trituberculatus, transcriptome, SSR, genetic diversity, Bohai Sea
Citation: Duan B, Mu S, Guan Y, Liu W, Kang T, Cheng Y, Li Z, Tian Y and Kang X (2022) Development of Microsatellite Markers Based on Transcriptome Sequencing and Evaluation of Genetic Diversity in Swimming Crab (Portunus trituberculatus). Front. Genet. 13:932173. doi: 10.3389/fgene.2022.932173
Received: 29 April 2022; Accepted: 16 June 2022;
Published: 18 July 2022.
Edited by:
Siti Nor, University of Malaysia Terengganu, MalaysiaReviewed by:
Xiaoming Pang, Beijing Forestry University, ChinaCopyright © 2022 Duan, Mu, Guan, Liu, Kang, Cheng, Li, Tian and Kang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xianjiang Kang, eGprYW5nQGhidS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.