
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 09 August 2022
Sec. Epigenomics and Epigenetics
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.928150
This article is part of the Research Topic Non-coding RNAs in Gastrointestinal and Gynecological Cancers: New Insights into the Mechanisms of Cancer Therapeutic Resistance, volume II View all 6 articles
 Lei Zhang1†
Lei Zhang1† Dapeng Li1†
Dapeng Li1† Lijing Gao1
Lijing Gao1 Jinming Fu1Simin Sun1Hao Huang1
Jinming Fu1Simin Sun1Hao Huang1 Ding Zhang1Chenyang Jia1Ting Zheng1Binbin Cui2*
Ding Zhang1Chenyang Jia1Ting Zheng1Binbin Cui2* Yanlong Liu2*
Yanlong Liu2* Yashuang Zhao1*
Yashuang Zhao1*Early and specific detection of cancer provides an opportunity for appropriate treatment. Although studies have suggested that QKI is a tumor suppressor gene, no studies have evaluated the diagnostic utility of QKI methylation in colorectal cancer (CRC). Here, we evaluated the methylation status of QKI by integrating the methylation data of tissues and cell lines of multiple cancer types. The diagnostic performance of QKI was analyzed in the discovery dataset from the TCGA CRC 450K array (n = 440) and tested in the test sets (n = 845) from the GEO. The methylation level of QKI was further validated in our independent dataset (n = 388) using targeted bisulfite sequencing. All detected CpG sites in the QKI promoter showed CRC-specific hypermethylation in 31 types of tumor tissues. In the discovery dataset, six consecutive CpG sites achieved high diagnostic performances, with AUCs ranging from 0.821 to 0.930. In the test set, a region (chr6: 163,834,452–163,834,924) including four consecutive CpG sites had robust diagnostic ability in distinguishing CRC and adenoma from normal samples. In the validation dataset, similar robust results were observed in both early- and advanced-stage CRC patients. In addition, QKI exhibited hypermethylation in the cfDNA of patients with CRC (n = 14). Collectively, the QKI promoter is a CRC-specific methylation biomarker and holds great promise for improving the diagnosis using minimally invasive biopsy.
With an estimated 1.9 million new cases occurring in 2020, colorectal cancer (CRC) is the second most common cause of cancer-related death worldwide (Sung et al., 2021). CRC occurs via a multistep process of transition from benign neoplasm to overt adenocarcinoma that takes 10–15 years, which provides a window for cancer intervention and prevention (Vogelstein et al., 1988; Arber and Levin, 2005). With the current treatment options, the 5-year survival rate of patients with stage I or II is more than 80%. However, less than half of CRC cases are detected at early stages, and the early detection of CRC remains a challenge (Miller et al., 2019).
DNA methylation is a heritable epigenetic modification that modulates gene expression without changing the underlying DNA sequence (Bird, 2002). Aberrant hypermethylation of tumor suppressor genes occurs early in tumorigenesis, making it a promising biomarker for cancers, including CRC (Irizarry et al., 2009; Baylin and Jones, 2011; 2016). Meanwhile, there are several potential advantages of assessing methylation over tumor somatic mutations, including geometrical increases in the number of detectable sites, and the methylation patterns often reflect the epigenetic origin of specific cancers to reveal the origin of the unknown primary cancers (Chen et al., 2016; Moran et al., 2016).
In a liquid biopsy, circulating cell-free DNA (cfDNA) has emerged as a key biomarker for early diagnoses (Hao et al., 2017; Jin et al., 2021). Genetic or epigenetic changes can distinguish circulating tumor DNA (ctDNA) released from circulating tumor cells (CTCs) or primary tumor tissue from cfDNA derived from leukocytes (Schwarzenbach et al., 2011; Moss et al., 2018). For instance, the blood-based test of SEPT9 methylation has been approved by the FDA for CRC screening (Lamb and Dhillon, 2017). However, this test showed varied performance in different studies and a high positive rate for other cancer types (Grützmann et al., 2008; Lee et al., 2013; Schrock et al., 2017; Jiao et al., 2019; Petit et al., 2019). Therefore, tests for cfDNA can be improved if they are combined with cancer-specific methylation patterns.
The RNA-binding protein quaking (QKI), which is a member of the signal transduction and activator of RNA metabolism (STAR) family (Vernet and Artzt, 1997), has been proven to affect pre-mRNA splicing, mRNA turnover, and translation (Saccomanno et al., 1999; Wu et al., 2002; Larocque et al., 2005). The function of QKI was first glimpsed in brain tumors, suggesting that it may have a role in tumor suppression (Li et al., 2002). Furthermore, the expression of QKI was greatly reduced or absent in the tissue of patients with CRC, and the change was accomplished at least partially by DNA hypermethylation (Yang et al., 2010; Iwata et al., 2017). A long non-coding RNA (lncRNA) named CAHM (chr6: 163,834,097–163,834,982), located adjacent to the QKI gene, has been demonstrated to be specifically hypermethylated in CRC and adenoma, with a low methylation level in the tissue of lung, prostate, and breast cancers (Pedersen et al., 2014). Given the fuzzy boundary of QKI and CAHM, the methylation of the QKI gene in CRC is worth exploring.
By integrating large-scale genome-wide methylation data of multiple cancer types from TCGA, we discovered that the QKI promoter was specifically hypermethylated in the CRC tissues but not in other cancer types or adjacent normal tissues. Although studies have found that QKI is hypermethylated in colorectal cancer tissues compared with normal tissues, whether hypermethylation of QKI is a promising molecular marker for early detection of CRC has not been investigated. Therefore, we further validated the potential diagnostic performance of QKI methylation for CRC in tissues, cell lines, and blood from public and in-house data.
Publicly available datasets were obtained from the TCGA and GEO. The Illumina 450K methylation array of 31 types of cancer, including CRC, was downloaded from UCSC Xena1. Meanwhile, RNA-Seq gene expression data of CRC, as in log2 (x + 1) transformed RSEM-normalized count was obtained from UCSC Xena. The 450K array of cell lines of 29 different tissues was obtained from GSE68379 (Iorio et al., 2016). Whole-blood DNA methylation profiles from healthy donors were generated in GSE40279 (Hannum et al., 2013). Moreover, two independent test sets, A and B, were established using the 450K array from GEO, including GSE42752 (Naumov et al., 2013), GSE48684 (Luo et al., 2014), GSE101764 (Barrow et al., 2017), GSE131013 (Díez-Villanueva et al., 2020), GSE77954 (Qu et al., 2016), GSE129364 (Fiedler et al., 2019), and GSE139404 (Fan et al., 2020). An additional methylation dataset array was obtained from GSE122126, which provided the methylation profile of three cfDNA samples from CRC and four samples from healthy controls with the Illumina EPIC (Moss et al., 2018). The methylation level of the CpG sites was represented as beta values, which is a ratio of intensities between the methylated (M) allele and the sum of methylated (M) and unmethylated (U) alleles. The beta values are continuous and range from 0 (unmethylated) to 1 (completely methylated). Details of the included subjects are presented in Table 1. The genome information of CpG sites of the QKI gene in the 450K array is shown in Supplementary Table S1, and the CpG sites with the missing value above 10% in the TCGA CRC dataset were excluded from further analysis.
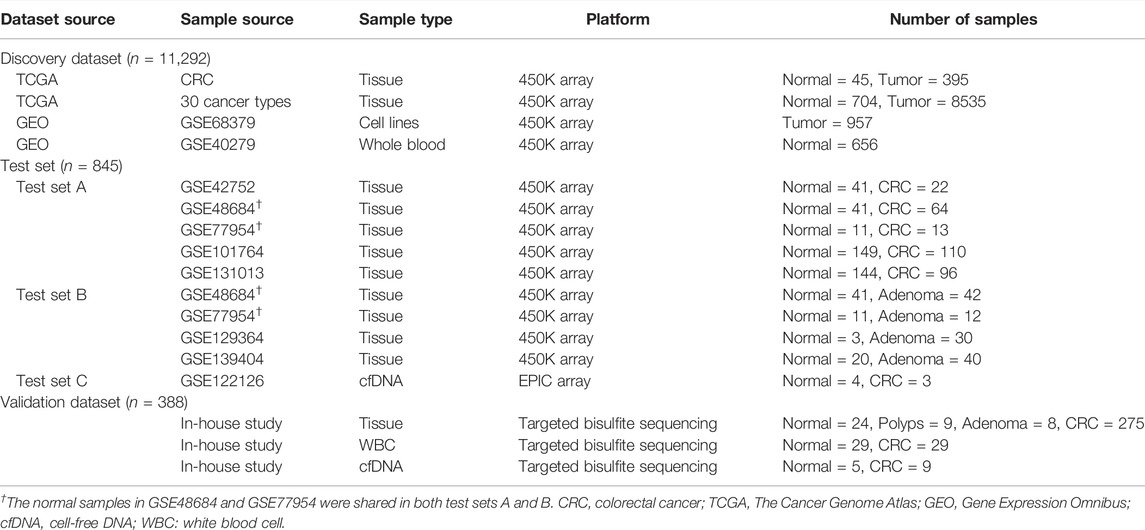
TABLE 1. The basic information of datasets used in the current study.
In total, 316 fresh frozen tissue samples were collected, including 275 CRC tissues, 24 adjacent normal tissues, eight adenoma tissues, and nine hyperplastic polyp tissues. We also collected 29 white blood cell (WBC) samples from CRC patients and 29 WBC samples from healthy controls. Blood specimens of nine patients with CRC were collected in ardent cell-free DNA BCT before surgical and neoadjuvant chemotherapy, as well as five healthy normal individuals. The plasma was separated from the blood (8–10 ml) via double centrifugation (3,000 rpm for 10 min at RT, and then, 16,000 g at 4°C for another 10 min) within 4 h after the blood was drawn and stored at −80°C until cfDNA isolation. These samples were collected at the Third Affiliated Hospital of Harbin Medical University. All patients with CRC or precancerous lesions were newly diagnosed by postoperative pathology or colonoscopy pathology and underwent surgery without neoadjuvant chemotherapy. Healthy controls were free from a history of malignancy, and without any benign or malignant diseases confirmed by colonoscopy. Demographic and clinical information including age, gender, tumor location, and tumor stage were obtained from medical records and pathological reports (Supplementary Table S2). All procedures performed in studies involving human participants complied with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Tissue and WBC genomic DNA (gDNA) were extracted using the phenol-chloroform method and QIAamp DNA Mini kit (Qiagen, Cat# 51306). cfDNA was isolated from 4–5 ml plasma using the QIAamp Circulating Nucleic Acid Kit (Qiagen, Cat# 55114) following the manufacturer’s protocol. The concentration of cfDNA was measured using the Qubit™ dsDNA HS Assay Kit (Thermo Fisher Scientific, Cat# Q32851). All samples were bisulfite-converted using the EZ DNA Methylation-Gold™ Kit (Zymo Research, Cat# D5006) according to the instruction manual. Targeted bisulfite sequencing is a next-generation sequencing-based technology for the quantization of the methylation of targeted regions at a base-specific level. We designed three regions to measure the methylation level of QKI in tissues and WBCs, and a short overlapping region in cfDNA (Supplementary Table S3). Multiplex PCR was performed first and amplified further using indexed primers, and target capture libraries were sequenced on Illumina’s MiSeq with a 2 × 150 bp paired-end mode (Genesky Biotechnologies Inc.). In data analysis, samples with bisulfite conversion rate <98%, or average coverage less than ×10 were filtered out.
The Student’s t-test was used to compare the differences in the methylation level of QKI between the two groups. The Spearman correlation method was used to assess the co-methylation of adjacent CpG sites and the correlation between methylation and expression (R package ‘corrplot’, version 0.84). The area under the curve (AUC) obtained from a receiver operating characteristic (ROC) curve analysis was used to test the diagnostic accuracy, and the optimal cut-off value for the CpG site of QKI was selected at the maximal Youden index. AUC values between subgroups were compared using the DeLong method (R package ‘pROC’, version 1.17.0.1). A trend test of the methylation level of QKI in different neoplastic progression states was performed with a linear regression model. We also used R packages such as ggplot2 (version 3.3.3), ggsignif (version 0.6.1), tidyverse (version 1.3.0), pheatmap (version 1.0.12), and survival (version 3.2-7). All statistical analyses were conducted using R software (version 4.0.4). A two-sided p < 0.05 was considered statistically significant. The human genome version GRCh37/hg19 was used as the reference for genomic coordinates.
The workflow is described in Figure 1. There were 22 available CpG sites of QKI after removing three CpG sites with missing values above 10% in the TCGA CRC dataset. The methylation level of 10 CpG sites in the QKI promoter was significantly higher in CRC than those in normal tissues, and the neighboring CpG sites were highly correlated with each other (termed as co-methylation, Figures 2A,B, Supplementary Table S4). In 391 CRC samples with both methylation and expression data, QKI promotor methylation was negatively correlated with the gene expression (average r = −0.331, p < 0.001; Supplementary Figure S1).
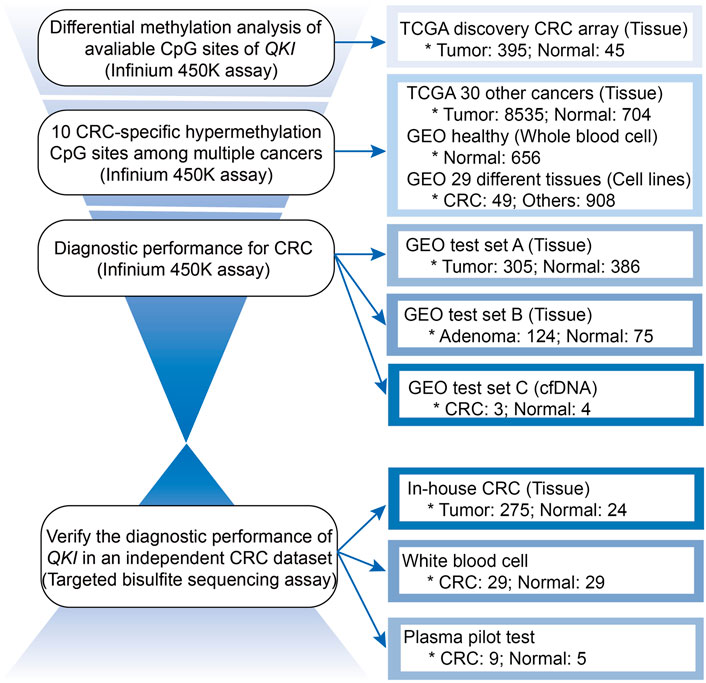
FIGURE 1. Workflow for analyzing the methylation level of the QKI gene in multiple datasets.
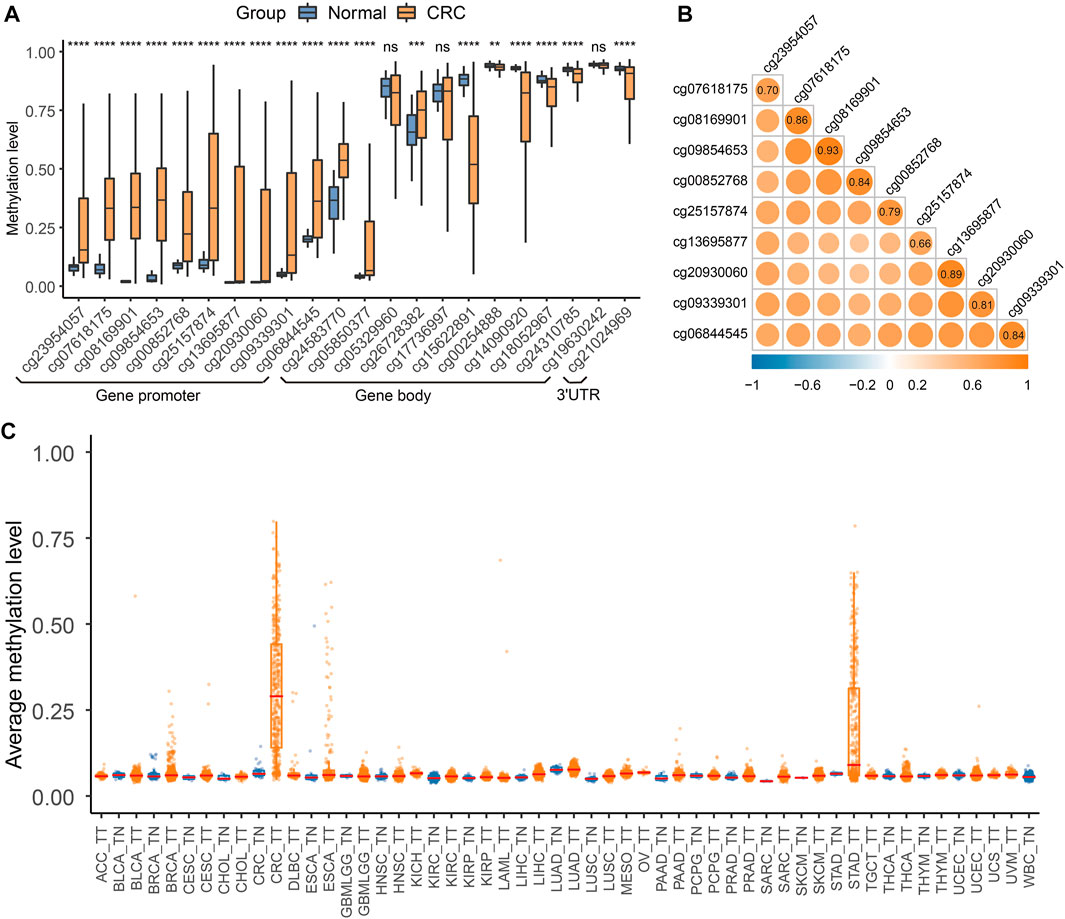
FIGURE 2. QKI methylation analysis in the discovery dataset. (A) Differential methylation analysis for 22 CpG sites of the QKI gene between colorectal cancer tissues and normal tissues in the TCGA CRC dataset. Gene promoter was defined as the region containing TSS200, TSS1500, 5′UTR, and the first exon. Symbols indicating statistical significance: NS, p > 0.05; *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001. (B) Co-methylation analysis of 10 CpG sites in the promoter region of QKI. (C) Boxplot of the average methylation level of the QKI promoter in multiple cancers.
We first assessed whether QKI methylation was a tissue-specific alteration of CRC by comparing the genome-wide methylation data of tumor tissues (n = 8930) and adjacent normal tissues (n = 749) of 31 cancer types from TCGA. We found that 10 CpG sites in the QKI promoter were specifically hypermethylated in CRC tissues but not in tumor tissues from 30 other cancer types or normal tissues (Figure 2C). Given that peripheral blood leukocytes are the main source of cfDNA, methylation levels of the QKI promotor were relatively low in whole-blood samples from healthy individuals (n = 656, Supplementary Figure S2). Moreover, the QKI promoter was specifically hypermethylated in cell lines of CRC (n = 49) compared with cell lines of other cancer cell lines (n = 908, Supplementary Figure S3). In summary, QKI promoter methylation is a tissue-specific marker of CRC that can distinguish CRC tissues from the tumor or normal tissues of other cancer types.
We evaluated the diagnostic performance of 10 CpG sites in distinguishing CRC or adenoma from normal tissues. The six consecutive CpG sites (cg23954057, cg07618175, cg08169901, cg09854653, cg00852768, and cg25157874) showed high discriminating ability, with AUC values ranging from 0.821 to 0.930 in the TCGA CRC dataset. Furthermore, an elevated methylation level of 10 CpG sites in the QKI promoter was found in the CRC of the test set A, and five of six CpG sites had a stable performance in distinguishing 305 CRC patients from 386 normal tissues (average AUC = 0.863). The average sensitivity and specificity of the five CpG sites were 80.3 and 75.2% (Figure 3), respectively. Next, we assessed the diagnostic performance of the 10 CpG sites to distinguish 124 adenomas from 75 normal tissues in test set B. The 10 CpG sites were significantly hypermethylated in adenomas. Four CpG sites (cg07618175, cg08169901, cg09854653, and cg00852768) achieved high diagnostic ability, with AUCs >0.800 (Figure 3). Collectively, the region including four consecutive CpG sites (chr6: 163,834,452–163,834,924) was identified as a candidate marker for diagnosing CRC and its precancerous lesions.
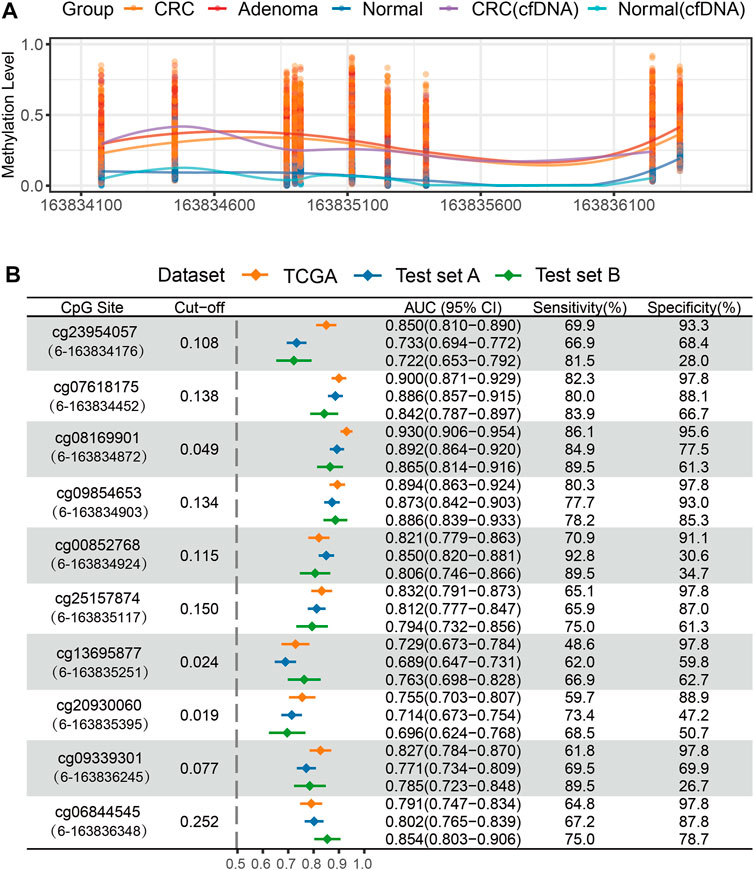
FIGURE 3. Methylation analysis of QKI promoter in the test set. (A) The methylation level of 10 CpG sites of the QKI promoter in the test set. (B) ROC curve analysis of 10 CpG sites of the QKI promoter for discriminating CRC or adenoma tissues from normal tissues in the discovery and test dataset. The content in brackets denotes the “chromosome”-“coordinate” of each CpG site.
In addition, we evaluated the methylation status of the QKI promoter in cfDNA samples from three CRC patients and four healthy controls. Hypermethylation of eight available CpG sites was observed in at least one cfDNA sample from CRC, but not in cfDNA samples from healthy individuals (Figure 3A, Supplementary Figure S4).
Based on the genomic coordinates of the QKI promoter in the discovery dataset, we designed three amplification regions (chr6: 163,834,632–163,835,160; Supplementary Tables S5–S7) for targeted bisulfite sequencing. A total of 76 CpG sites were detected in the three target regions, covering four CpG sites in the 450K array. Three samples with bisulfite conversion rate <98% and one sample with average coverage less than 10× were excluded. Differential methylation analysis showed that all 76 CpG sites in the target regions were hypermethylated in 271 CRC samples compared to 24 normal tissues (Figure 4). The cg08169901 (CpG39) was detected repeatedly in region 1 and region 2, and the AUC values were 0.896 and 0.911, respectively. We preferred the detection result of region 2 since it was located in the middle of target region 2, and its AUC was slightly lower than that of the discovery dataset (AUC = 0.930). The AUC value of cg09854653 (CpG41) was consistent with the discovery dataset (0.895 vs. 0.894). The cg00852768 (CpG44) appeared at the boundary of both target 2 and target 3, with AUC values (0.872 and 0.867, respectively) similar to those of the discovery dataset (AUC = 0.821). The AUC value of cg25157874 (CpG75) was lower than that of the TCGA dataset (0.782 vs. 0.830). The sensitivity of the four target CpG sites was 84.5, 82.3, 77.9, and 60.5%, with corresponding specificities of 95.8, 95.8, 91.7, and 100%, respectively (Figures 5A–D, Supplementary Tables S5–S7).
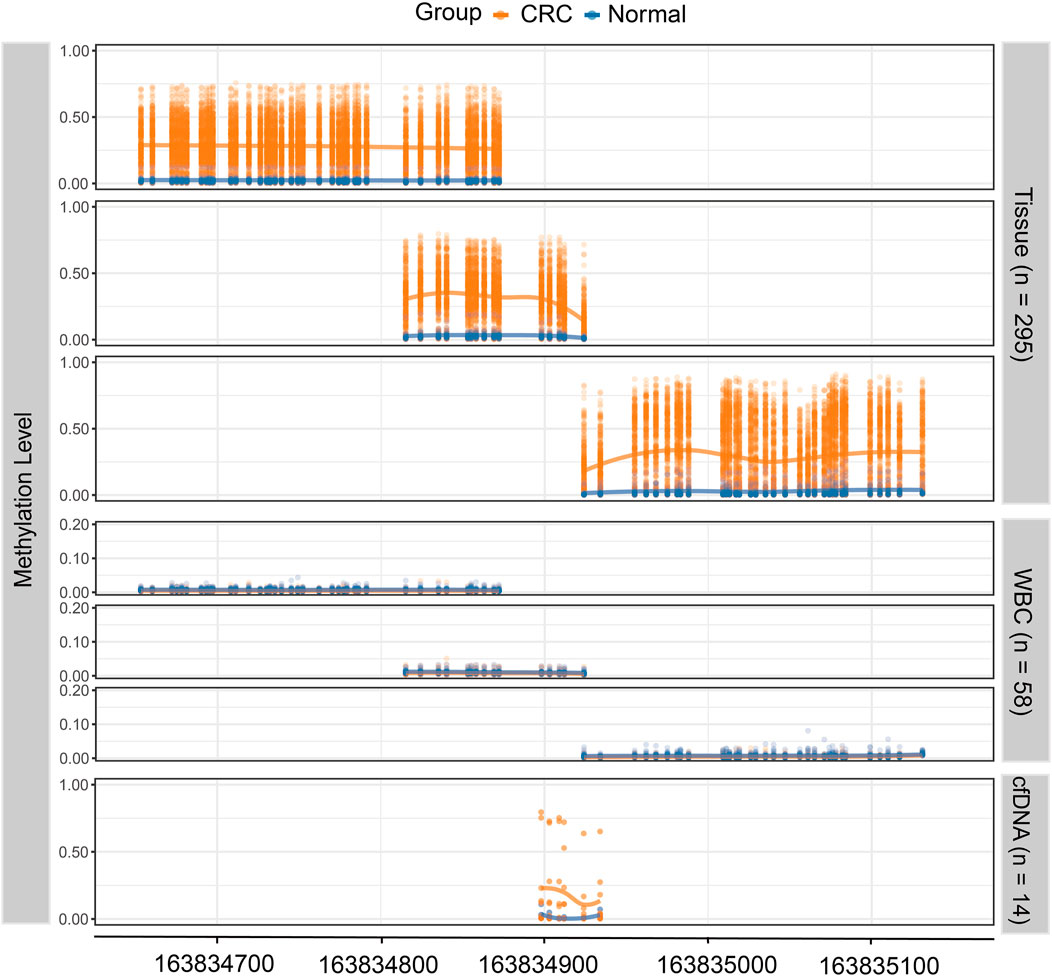
FIGURE 4. The methylation level of the QKI target region in the validation dataset. The top three panels show the methylation level of 271 CRC tissues and 24 normal tissues, where each dot represents one CpG site for each sample. The middle three panels show the methylation level of white blood cells from CRC patients (n = 29) and healthy controls (n = 29). The lowest panel shows the methylation level of cell-free DNA samples from CRC patients (n = 9) and healthy controls (n = 5).
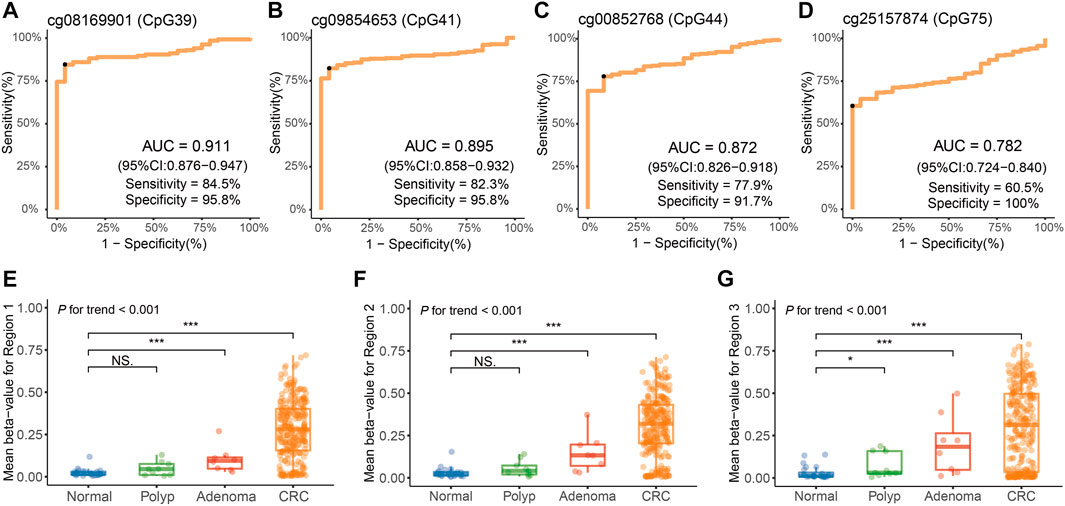
FIGURE 5. Methylation analysis of the QKI promoter in the validation dataset. (A–D) Diagnostic performance of four target CpG sites for distinguishing CRC from adjacent normal tissues. The black point indicates the optimal cut-off value for each CpG. (E–G) Average methylation levels of three regions of the QKI promoter at different disease stages during neoplastic progression of colorectal cancer tissues. Symbols indicating statistical significance: NS, p > 0.05; *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001.
Furthermore, we evaluated the diagnostic performance of the target CpG sites in different subgroups, including age, gender, tumor location, and tumor stage (Table 2). AUC values of the cg08169901 (CpG39), cg09854653 (CpG41), and cg00852768 (CpG44) were consistent among the different subgroups. However, cg25157874 (CpG75) had a higher AUC value when the tumor was located at the rectum (0.710 vs. 0.822, p = 0.043). Indeed, four CpG sites had consistent diagnostic performance in early-stage (stage I/II) and advanced-stage (stage III/IV) patients with CRC. In addition, the remaining CpG sites in region 1 (Supplementary Table S5) and region 2 (Supplementary Table S6) had similar AUC values ranging from 0.885 to 0.918, while the AUC values of region 3 (Supplementary Table S7) were slightly lower, having a range from 0.757 to 0.861.
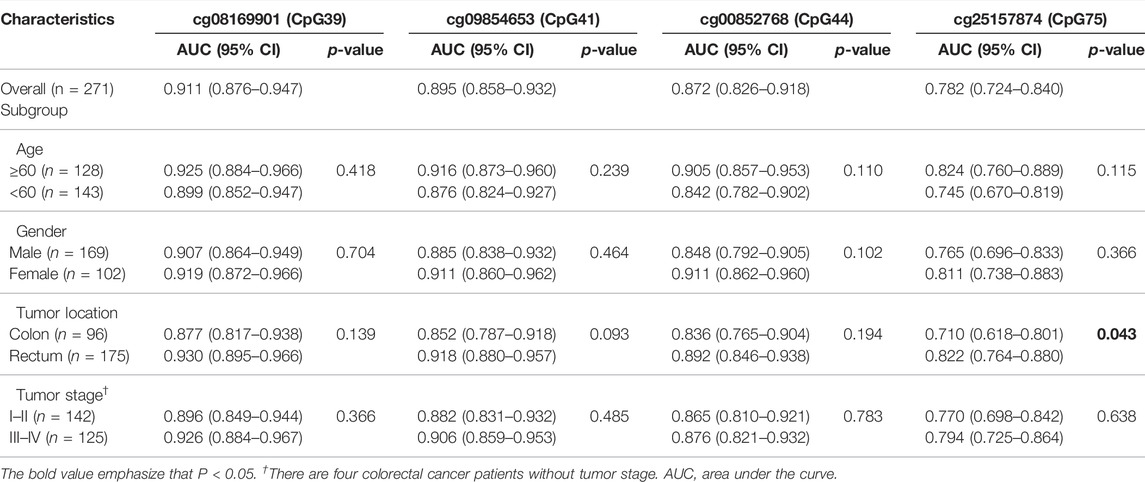
TABLE 2. The AUC values of four CpG sites in the subgroup of different clinicopathological factors in the validation dataset.
Furthermore, we detected the methylation level of the QKI promoter in eight adenoma tissues and nine hyperplastic polyp tissues. In three target regions, the average methylation of adenoma (median beta-value of 0.094 for target region 1, 0.133 for target region 2, and 0.184 for target region 3) was significantly higher than that in normal tissues (median beta-value of 0.016 for target region 1, 0.022 for target region 2, and 0.012 for target region 3). No statistically significant difference was observed between polyps and normal tissues in target region 1 and region 2. There was an obviously increasing trend from polyps to adenomas to CRC (P for trend <0.001, Figures 5E–G).
Methylation levels of three target regions were detected in WBC samples from 29 CRC patients and 29 healthy controls. The median methylation level of the three target regions was 0.005 for CRC (range, 0.003–0.012) and 0.008 for healthy controls (range, 0.004–0.014; Figure 4, Supplementary Tables S8–S10). The low level of methylation in WBCs from both CRC and healthy controls is unlikely to cause confusion in the detection of the cfDNA methylation level.
There are six CpG sites in the target region, covering cg09854653 (CpG41) and cg00852768 (CpG44, Supplementary Table S11). As shown in Figure 4, the methylation level of six CpG sites was significantly higher in nine cfDNA samples of CRC (range, 0.106–0.230) compared to five healthy controls (range, 0.003–0.036). Potentially, the methylation status of six CpG sites in this region was extracted to form methylation haplotypes, which accounted for a higher proportion of cfDNA from CRC patients than healthy controls (Supplementary Figure S5). In short, the methylation of the QKI promoter is promising as a specific marker for the non-invasive detection of CRC.
In this study, we demonstrated that the QKI promoter is a tissue-specific methylation marker for CRC by integrating large-scale public methylation datasets. A region including four consecutive CpG sites (chr6: 163,834,452–163,834,924) has excellent diagnostic performance in distinguishing CRC and adenoma tissues from normal tissues, which was further validated by a PCR-based method. As expected, methylation of the QKI promoter remains robust in diagnosing CRC, even in the early stages of cancer. A gradually increasing methylation level was observed during the tumorigenesis of CRC. The pilot cfDNA test suggested that methylation of the QKI promoter has the potential to be a non-invasive diagnostic marker for CRC.
An ideal biomarker of CRC should identify the tissue of origin of the tumor in clinical practice, and have considerable ability to diagnose colorectal adenoma. Previous studies have demonstrated that the combination of cancer-specific DNA methylation patterns and ctDNA can achieve highly specific detection in more than 38 cancer types, and detect asymptomatic individuals who were diagnosed later (Moran et al., 2016; Chen et al., 2020; Liu et al., 2020). Integrating large-scale datasets of multiple cancer types, our genome-wide methylation analysis identified that QKI was specifically hypermethylated in CRC tissues but not in other cancer types. We found that QKI methylation can distinguish both CRC and colorectal adenoma tissues from adjacent normal tissues. Collectively, methylation of the QKI promoter is a tissue-specific methylation alteration of CRC and it occurs early in the process of CRC formation.
Although previous genome-wide methylation studies have achieved high accuracy in diagnosing multiple types of cancers, the single-gene approach is more suitable for screening CRC in large-scale populations. Therefore, we validated hypermethylation of the QKI promotor in CRC tissues using a cost-effective PCR-based technology, with results consistent with the genome-wide methylation data. In addition, our analysis suggested that QKI methylation in cfDNA of CRC patients was not derived from DNA released from WBCs. Moreover, our plasma pilot study suggested that the QKI gene was hypermethylated in cfDNA samples from CRC patients. Both the high-throughput detection method and single-gene approach suggested that methylation of the QKI promoter was a candidate early diagnostic marker of CRC.
We extended the analysis of QKI methylation in cfDNA to the analysis of the methylated haplotypes. Early sequencing studies have indicated that adjacent CpG sites share similar methylation patterns (Eckhardt et al., 2006). Locally disordered methylation has also been observed, which indicates that methylation heterogeneity is primarily from variability within DNA fragments (Landau et al., 2014). Therefore, some studies have extended the theoretical framework of linkage disequilibrium (Slatkin, 2008) to CpG co-methylation analysis to annotate these methylation blocks as methylation haplotypes with a unique set of genomic features (Shoemaker et al., 2010). Studies have shown that methylated haplotypes show high accuracy in predicting the tissue of origin and the cancer status in cfDNA, whether it is built on 450K sparse genome coverage or the whole-genome sequencing analysis (Lehmann-Werman et al., 2016; Guo et al., 2017). Therefore, methylation haplotype detection may be more suitable for tumor-derived ctDNA without a clear source. As shown in our results, the methylated haplotype of cfDNA provides a reference for the primer design of PCR-based methods, and we hypothesize that identifying different haplotypes of patients at different stages can achieve an accurate diagnosis.
Although Yang et al. (2010) and Novikov et al. (2011) have provided evidence that QKI acts as a tumor suppressor gene and the down-regulation of QKI expression may be involved in CRC onset and progression, no study has assessed the diagnostic utility of QKI methylation in CRC. Particularly, a PCR-based region (chr6: 163,834,393–163,834,455) of CAHM demonstrated frequent methylation, with a positive rate of 81% (17/21) in adenomas and 71% (56/79) in CRC tissues. Indeed, increased methylation of CAHM was also found in the plasma DNA of 55% (40/73) of CRC patients, but not in 73 adenomas (4%) and 74 subjects without neoplasia (7%) (Pedersen et al., 2014). Complementing this, our study demonstrates that a region of the QKI gene (chr6: 163,834,452–163,834,924) has similar robustness in distinguishing CRC from normal tissues and is a CRC-specific methylation marker relative to 30 other types of cancer. Iwata et al. (2017) demonstrated that gene expression of QKI was significantly down-regulated in CRC tissues (p = 0.049), and patients with low tumor QKI expression were associated with worse prognoses (n = 153). In contrast, our survival analysis based on the TCGA CRC dataset (n = 361) and our in-house validation dataset (n = 246) did not suggest a significant association between the methylation level of the QKI promoter and the overall survival of CRC (Supplementary Figure S6).
This study has several limitations. First, all samples in the validation dataset were collected from a single center. Second, although our study demonstrated that the QKI gene was hypermethylated in cfDNA samples from CRC patients, its clinical utility should be further evaluated in cohorts with more cfDNA samples.
By integrating genome-wide methylation data from tissues and cell lines of multiple cancer types, this study revealed that methylation of the QKI promoter was a CRC-specific marker. Its diagnostic performance is stable in patients with CRC and its precursor lesions from the GEO database and our in-house dataset. Hypermethylation in cfDNA of CRC patients indicates that QKI methylation is a tissue-specific and non-invasive marker for the detection of CRC, and is worthy of further verification in a large-scale cohort.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://doi.org/10.6084/m9.figshare.20055134.
The studies involving human participants were reviewed and approved by Ethical Committee of Harbin Medical University. The patients/participants provided their written informed consent to participate in this study.
YZ and YL contributed to the study design. LZ, DL, and LG contributed to data collection. LZ, DL, JF, and SS performed data analysis and interpretation. LZ, HH, DZ, CJ, and TZ performed experiments. LZ and DL drafted the manuscript. YZ, YL, and BC revised the manuscript. YZ and BC provided funding and supervision. All authors read and approved the final manuscript.
This research was funded by National Natural Science Foundation of China, grant number 81473055, and Nn10 Program of Harbin Medical University Cancer Hospital (Nn102017-02).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank all the patients who participated in the studies from TCGA and GEO datasets and our study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.928150/full#supplementary-material
1https://xena.ucsc.edu/public/.
Arber, N., and Levin, B. (2005). Chemoprevention of Colorectal Cancer: Ready for Routine Use? Ctmc 5 (5), 517–525. doi:10.2174/1568026054201659
Barrow, T. M., Klett, H., Toth, R., Böhm, J., Gigic, B., Habermann, N., et al. (2017). Smoking Is Associated with Hypermethylation of the APC 1A Promoter in Colorectal Cancer: the ColoCare Study. J. Pathol. 243 (3), 366–375. doi:10.1002/path.4955
Baylin, S. B., and Jones, P. A. (2011). A Decade of Exploring the Cancer Epigenome - Biological and Translational Implications. Nat. Rev. Cancer 11 (10), 726–734. doi:10.1038/nrc3130
Baylin, S. B., and Jones, P. A. (2016). Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 8 (9), a019505. doi:10.1101/cshperspect.a019505
Bird, A. (2002). DNA Methylation Patterns and Epigenetic Memory. Genes Dev. 16 (1), 6–21. doi:10.1101/gad.947102
Chen, X., Gole, J., Gore, A., He, Q., Lu, M., Min, J., et al. (2020). Non-invasive Early Detection of Cancer Four Years before Conventional Diagnosis Using a Blood Test. Nat. Commun. 11 (1), 3475. doi:10.1038/s41467-020-17316-z
Chen, Y., Breeze, C. E., Zhen, S., Beck, S., and Teschendorff, A. E. (2016). Tissue-independent and Tissue-specific Patterns of DNA Methylation Alteration in Cancer. Epigenetics Chromatin 9, 10. doi:10.1186/s13072-016-0058-4
Díez-Villanueva, A., Sanz-Pamplona, R., Carreras-Torres, R., Moratalla-Navarro, F., Alonso, M. H., Paré-Brunet, L., et al. (2020). DNA Methylation Events in Transcription Factors and Gene Expression Changes in Colon Cancer. Epigenomics 12 (18), 1593–1610. doi:10.2217/epi-2020-0029
Eckhardt, F., Lewin, J., Cortese, R., Rakyan, V. K., Attwood, J., Burger, M., et al. (2006). DNA Methylation Profiling of Human Chromosomes 6, 20 and 22. Nat. Genet. 38 (12), 1378–1385. doi:10.1038/ng1909
Fan, J., Li, J., Guo, S., Tao, C., Zhang, H., Wang, W., et al. (2020). Genome-wide DNA Methylation Profiles of Low- and High-Grade Adenoma Reveals Potential Biomarkers for Early Detection of Colorectal Carcinoma. Clin. Epigenet 12 (1), 56. doi:10.1186/s13148-020-00851-3
Fiedler, D., Hirsch, D., El Hajj, N., Yang, H. H., Hu, Y., Sticht, C., et al. (2019). Genome‐wide DNA Methylation Analysis of Colorectal Adenomas with and without Recurrence Reveals an Association between Cytosine‐phosphate‐guanine Methylation and Histological Subtypes. Genes Chromosom. Cancer 58 (11), 783–797. doi:10.1002/gcc.22787
Grützmann, R., Molnar, B., Pilarsky, C., Habermann, J. K., Schlag, P. M., Saeger, H. D., et al. (2008). Sensitive Detection of Colorectal Cancer in Peripheral Blood by Septin 9 DNA Methylation Assay. PLoS One 3 (11), e3759. doi:10.1371/journal.pone.0003759
Guo, S., Diep, D., Plongthongkum, N., Fung, H. L., Zhang, K., and Zhang, K. (2017). Identification of Methylation Haplotype Blocks Aids in Deconvolution of Heterogeneous Tissue Samples and Tumor Tissue-Of-Origin Mapping from Plasma DNA. Nat. Genet. 49 (4), 635–642. doi:10.1038/ng.3805
Hannum, G., Guinney, J., Zhao, L., Zhang, L., Hughes, G., Sadda, S., et al. (2013). Genome-wide Methylation Profiles Reveal Quantitative Views of Human Aging Rates. Mol. Cell 49 (2), 359–367. doi:10.1016/j.molcel.2012.10.016
Hao, X., Luo, H., Krawczyk, M., Wei, W., Wang, W., Wang, J., et al. (2017). DNA Methylation Markers for Diagnosis and Prognosis of Common Cancers. Proc. Natl. Acad. Sci. U.S.A. 114 (28), 7414–7419. doi:10.1073/pnas.1703577114
Iorio, F., Knijnenburg, T. A., Vis, D. J., Bignell, G. R., Menden, M. P., Schubert, M., et al. (2016). A Landscape of Pharmacogenomic Interactions in Cancer. Cell 166 (3), 740–754. doi:10.1016/j.cell.2016.06.017
Irizarry, R. A., Ladd-Acosta, C., Wen, B., Wu, Z., Montano, C., Onyango, P., et al. (2009). The Human Colon Cancer Methylome Shows Similar Hypo- and Hypermethylation at Conserved Tissue-specific CpG Island Shores. Nat. Genet. 41 (2), 178–186. doi:10.1038/ng.298
Iwata, N., Ishikawa, T., Okazaki, S., Mogushi, K., Baba, H., Ishiguro, M., et al. (2017). Clinical Significance of Methylation and Reduced Expression of the Quaking Gene in Colorectal Cancer. Ar 37 (2), 489–498. doi:10.21873/anticanres.11341
Jiao, X., Zhang, S., Jiao, J., Zhang, T., Qu, W., Muloye, G. M., et al. (2019). Promoter Methylation of SEPT9 as a Potential Biomarker for Early Detection of Cervical Cancer and its Overexpression Predicts Radioresistance. Clin. Epigenet 11 (1), 120. doi:10.1186/s13148-019-0719-9
Jin, S., Zhu, D., Shao, F., Chen, S., Guo, Y., Li, K., et al. (2021). Efficient Detection and Post-surgical Monitoring of Colon Cancer with a Multi-Marker DNA Methylation Liquid Biopsy. Proc. Natl. Acad. Sci. U.S.A. 118 (5). doi:10.1073/pnas.2017421118
Lamb, Y. N., and Dhillon, S. (2017). Epi proColon(®) 2.0 CE: A Blood-Based Screening Test for Colorectal Cancer. Mol. Diagn Ther. 21 (2), 225–232. doi:10.1007/s40291-017-0259-y
Landau, D. A., Clement, K., Ziller, M. J., Boyle, P., Fan, J., Gu, H., et al. (2014). Locally Disordered Methylation Forms the Basis of Intratumor Methylome Variation in Chronic Lymphocytic Leukemia. Cancer Cell 26 (6), 813–825. doi:10.1016/j.ccell.2014.10.012
Larocque, D., Galarneau, A., Liu, H. N., Scott, M., Almazan, G., and Richard, S. (2005). Protection of p27(Kip1) mRNA by Quaking RNA Binding Proteins Promotes Oligodendrocyte Differentiation. Nat. Neurosci. 8 (1), 27–33. doi:10.1038/nn1359
Lee, H. S., Hwang, S. M., Kim, T. S., Kim, D. W., Park, D. J., Kang, S. B., et al. (2013). Circulating Methylated Septin 9 Nucleic Acid in the Plasma of Patients with Gastrointestinal Cancer in the Stomach and Colon. Transl. Oncol. 6 (3), 290–IN4. doi:10.1593/tlo.13118
Lehmann-Werman, R., Neiman, D., Zemmour, H., Moss, J., Magenheim, J., Vaknin-Dembinsky, A., et al. (2016). Identification of Tissue-specific Cell Death Using Methylation Patterns of Circulating DNA. Proc. Natl. Acad. Sci. U.S.A. 113 (13), E1826–E1834. doi:10.1073/pnas.1519286113
Li, Z. Z., Kondo, T., Murata, T., Ebersole, T. A., Nishi, T., Tada, K., et al. (2002). Expression ofHqkEncoding a KH RNA Binding Protein Is Altered in Human Glioma. Jpn. J. Cancer Res. 93 (2), 167–177. doi:10.1111/j.1349-7006.2002.tb01255.x
Liu, M. C., Oxnard, G. R., Klein, E. A., Swanton, C., and Seiden, M. V. (2020). Sensitive and Specific Multi-Cancer Detection and Localization Using Methylation Signatures in Cell-free DNA. Ann. Oncol. 31 (6), 745–759. doi:10.1016/j.annonc.2020.02.011
Luo, Y., Wong, C. J., Kaz, A. M., Dzieciatkowski, S., Carter, K. T., Morris, S. M., et al. (2014). Differences in DNA Methylation Signatures Reveal Multiple Pathways of Progression from Adenoma to Colorectal Cancer. Gastroenterology 147 (2), 418–429. e418. doi:10.1053/j.gastro.2014.04.039
Miller, K. D., Nogueira, L., Mariotto, A. B., Rowland, J. H., Yabroff, K. R., Alfano, C. M., et al. (2019). Cancer Treatment and Survivorship Statistics, 2019. CA A Cancer J. Clin. 69 (5), 363–385. doi:10.3322/caac.21565
Moran, S., Martínez-Cardús, A., Sayols, S., Musulén, E., Balañá, C., Estival-Gonzalez, A., et al. (2016). Epigenetic Profiling to Classify Cancer of Unknown Primary: a Multicentre, Retrospective Analysis. Lancet Oncol. 17 (10), 1386–1395. doi:10.1016/s1470-2045(16)30297-2
Moss, J., Magenheim, J., Neiman, D., Zemmour, H., Loyfer, N., Korach, A., et al. (2018). Comprehensive Human Cell-type Methylation Atlas Reveals Origins of Circulating Cell-free DNA in Health and Disease. Nat. Commun. 9 (1), 5068. doi:10.1038/s41467-018-07466-6
Naumov, V. A., Generozov, E. V., Zaharjevskaya, N. B., Matushkina, D. S., Larin, A. K., Chernyshov, S. V., et al. (2013). Genome-scale Analysis of DNA Methylation in Colorectal Cancer Using Infinium HumanMethylation450 BeadChips. Epigenetics 8 (9), 921–934. doi:10.4161/epi.25577
Novikov, L., Park, J. W., Chen, H., Klerman, H., Jalloh, A. S., and Gamble, M. J. (2011). QKI-Mediated Alternative Splicing of the Histone Variant MacroH2A1 Regulates Cancer Cell Proliferation. Mol. Cell Biol. 31 (20), 4244–4255. doi:10.1128/mcb.05244-11
Pedersen, S. K., Mitchell, S. M., Graham, L. D., McEvoy, A., Thomas, M. L., Baker, R. T., et al. (2014). CAHM, a Long Non-coding RNA Gene Hypermethylated in Colorectal Neoplasia. Epigenetics 9 (8), 1071–1082. doi:10.4161/epi.29046
Petit, J., Carroll, G., Gould, T., Pockney, P., Dun, M., and Scott, R. J. (2019). Cell-Free DNA as a Diagnostic Blood-Based Biomarker for Colorectal Cancer: A Systematic Review. J. Surg. Res. 236, 184–197. doi:10.1016/j.jss.2018.11.029
Qu, X., Sandmann, T., Frierson, H., Fu, L., Fuentes, E., Walter, K., et al. (2016). Integrated Genomic Analysis of Colorectal Cancer Progression Reveals Activation of EGFR through Demethylation of the EREG Promoter. Oncogene 35 (50), 6403–6415. doi:10.1038/onc.2016.170
Saccomanno, L., Loushin, C., Jan, E., Punkay, E., Artzt, K., and Goodwin, E. B. (1999). The STAR Protein QKI-6 Is a Translational Repressor. Proc. Natl. Acad. Sci. U.S.A. 96 (22), 12605–12610. doi:10.1073/pnas.96.22.12605
Schröck, A., Leisse, A., de Vos, L., Gevensleben, H., Dröge, F., Franzen, A., et al. (2017). Free-Circulating Methylated DNA in Blood for Diagnosis, Staging, Prognosis, and Monitoring of Head and Neck Squamous Cell Carcinoma Patients: An Observational Prospective Cohort Study. Clin. Chem. 63 (7), 1288–1296. doi:10.1373/clinchem.2016.270207
Schwarzenbach, H., Hoon, D. S., and Pantel, K. (2011). Cell-free Nucleic Acids as Biomarkers in Cancer Patients. Nat. Rev. Cancer 11 (6), 426–437. doi:10.1038/nrc3066
Shoemaker, R., Deng, J., Wang, W., and Zhang, K. (2010). Allele-specific Methylation Is Prevalent and Is Contributed by CpG-SNPs in the Human Genome. Genome Res. 20 (7), 883–889. doi:10.1101/gr.104695.109
Slatkin, M. (2008). Linkage Disequilibrium - Understanding the Evolutionary Past and Mapping the Medical Future. Nat. Rev. Genet. 9 (6), 477–485. doi:10.1038/nrg2361
Sung, H., Ferlay, J., Siegel, R. L., Laversanne, M., Soerjomataram, I., Jemal, A., et al. (2021). Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 71 (3), 209–249. doi:10.3322/caac.21660
Vernet, C., and Artzt, K. (1997). STAR, a Gene Family Involved in Signal Transduction and Activation of RNA. Trends Genet. 13 (12), 479–484. doi:10.1016/s0168-9525(97)01269-9
Vogelstein, B., Fearon, E. R., Hamilton, S. R., Kern, S. E., Preisinger, A. C., Leppert, M., et al. (1988). Genetic Alterations during Colorectal-Tumor Development. N. Engl. J. Med. 319 (9), 525–532. doi:10.1056/nejm198809013190901
Wu, J. I., Reed, R. B., Grabowski, P. J., and Artzt, K. (2002). Function of Quaking in Myelination: Regulation of Alternative Splicing. Proc. Natl. Acad. Sci. U.S.A. 99 (7), 4233–4238. doi:10.1073/pnas.072090399
Keywords: colorectal cancer, diagnostic, QKI, DNA methylation, biomarker
Citation: Zhang L, Li D, Gao L, Fu J, Sun S, Huang H, Zhang D, Jia C, Zheng T, Cui B, Liu Y and Zhao Y (2022) Promoter Methylation of QKI as a Potential Specific Biomarker for Early Detection of Colorectal Cancer. Front. Genet. 13:928150. doi: 10.3389/fgene.2022.928150
Received: 25 April 2022; Accepted: 24 June 2022;
Published: 09 August 2022.
Edited by:
Nejat Dalay, Istanbul University, TurkeyReviewed by:
Mohsen Nikbakht, Tehran University of Medical Sciences, IranCopyright © 2022 Zhang, Li, Gao, Fu, Sun, Huang, Zhang, Jia, Zheng, Cui, Liu and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yashuang Zhao, emhhb195YXNodWFuZ0AyNjMubmV0; Yanlong Liu, bGl1eWFubG9uZzE5NzlAMTYzLmNvbQ==; Binbin Cui, Y3VpYmluYmluQGhyYm11LmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.