
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 15 July 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.927504
This article is part of the Research TopicGenetics of Familial Hypercholesterolemia: New Insight - Volume IIView all 17 articles
 Ilhame Diboun1,2
Ilhame Diboun1,2 Yasser Al-Sarraj3
Yasser Al-Sarraj3 Salman M. Toor1
Salman M. Toor1 Shaban Mohammed4Nadeem Qureshi5Moza S. H. Al Hail4
Shaban Mohammed4Nadeem Qureshi5Moza S. H. Al Hail4 Amin Jayyousi6
Amin Jayyousi6 Jassim Al Suwaidi7
Jassim Al Suwaidi7 Omar M. E. Albagha1,8*
Omar M. E. Albagha1,8*Familial hypercholesterolemia (FH) is an inherited disease characterized by reduced efficiency of low-density lipoprotein-cholesterol (LDL-C) removal from the blood and, consequently, an increased risk of life-threatening early cardiovascular complications. In Qatar, the prevalence of FH has not been determined and the disease, as in many countries, is largely underdiagnosed. In this study, we combined whole-genome sequencing data from the Qatar Genome Program with deep phenotype data from Qatar Biobank for 14,056 subjects to determine the genetic spectrum and estimate the prevalence of FH in Qatar. We used the Dutch Lipid Clinic Network (DLCN) as a diagnostic tool and scrutinized 11 FH-related genes for known pathogenic and possibly pathogenic mutations. Results revealed an estimated prevalence of 0.8% (1:125) for definite/probable cases of FH in the Qatari population. We detected 16 known pathogenic/likely pathogenic mutations in LDLR and one in PCSK9; all in a heterozygous state with high penetrance. The most common mutation was rs1064793799 (c.313+3A >C) followed by rs771019366 (p.Asp90Gly); both in LDLR. In addition, we identified 18 highly penetrant possibly pathogenic variants, of which 5 were Qatari-specific, in LDLR, APOB, PCSK9 and APOE, which are predicted to be among the top 1% most deleterious mutations in the human genome but further validations are required to confirm their pathogenicity. We did not detect any homozygous FH or autosomal recessive mutations in our study cohort. This pioneering study provides a reliable estimate of FH prevalence in Qatar based on a significantly large population-based cohort, whilst uncovering the spectrum of genetic variants associated with FH.
Familial hypercholesterolemia (FH) is a common autosomal disease characterized by elevated levels of low-density lipoprotein cholesterol (LDL-C), leading to an increased risk of atherosclerosis and premature coronary heart disease (CHD) (Bouhairie and Goldberg, 2015). The prevalence of FH in Caucasian populations has been typically considered around 1:500 (Austin et al., 2004), but more recent estimates show around 1:310 prevalence, with up to a 20-fold higher prevalence in those with premature CHD (Beheshti et al., 2020; Hu et al., 2020). Indeed, a recent study based on 225 Chinese subjects with premature myocardial infarction revealed up to 23.6% prevalence of FH (Cui et al., 2019). Notably, FH has a strong genetic basis compounded by environmental factors and its prevalence varies amongst different populations. Homozygous FH (HoFH) is rare; typically considered to affect 1 in 1,000,000 worldwide, but recent reports have estimated a higher prevalence of 1 in 160,000–300,000 (Cuchel et al., 2014; Singh and Bittner, 2015; Sjouke et al., 2015). HoFH is characterized by a drastic increase in LDL-C (>13 mmol/L before therapy) and the early development of atherosclerotic complications (Cuchel et al., 2014). Heterozygous FH (HeFH) is more common with an estimated prevalence of 1 in 250 in European populations (Vrablik et al., 2020). However, variations in FH prevalence have been reported in certain populations such as 1 in 137 in the Danish population (Benn et al., 2012), 1 in 270 in the United Kingdom population (Wald et al., 2016), and 1 in 311 in the Russian population (Meshkov et al., 2021). Notably, FH is considerably higher in certain populations such as Ashkenazi Jews (1 in 67) or Christian Lebanese (1 in 85) and South African Afrikaners (1 in 72), attributing to the founder effect which contributes to the significantly higher incidence (Austin et al., 2004; Henderson et al., 2016).
Mutations in LDLR, APOB and PCSK9 account for most FH cases and have been reported in both HoFH and HeFH. These three genes are members of the low-density lipoprotein receptor (LDL-R) pathway, which is responsible for maintaining healthy levels of plasma and intracellular cholesterol in the body. The vast majority of LDLR variants are deleterious (79%), affecting the splicing or regulatory aspects of early transcription or alternatively, leading to a defective receptor function (Henderson et al., 2016). Owing to the extensive spectrum of LDLR variants in FH, mutations in the LDLR gene can be population specific (Vrablik et al., 2020). Less stricter forms of FH have been associated with variants in APOB, a gene whose protein product helps LDL bind to its receptor LDL-R, but it is observed in about 5% of FH cases in European populations (Henderson et al., 2016; Vrablik et al., 2020). Mutations leading to a gain of function of PCSK9, an enzyme that promotes the degradation of LDL-R, could account for <1% of FH cases (Henderson et al., 2016). Less commonly observed variants of FH have been described in the LDLRAP1 gene causing an autosomal recessive form of the disease termed autosomal recessive hypercholesterolemia (ARH) (Garcia et al., 2001). Interestingly, ARH is characterized with LDL-C levels in between the HeFH and HoFH known levels, and unlike HoFH, the early-onset phenotype is comparatively rare (Henderson et al., 2016). Further to monogenic variants, polygenic effects have also been described to account for hypercholesterolemia (HC) cases (Saadatagah et al., 2021). Notably, more than 900 lipid-associated genomic loci have been identified in genome-wide studies (GWAS) based on investigating genetic variants in diverse ancestries (Graham et al., 2021).
The most widely accepted guidelines for the diagnosis of FH include the Simon-Broome Register criteria (Mortality, 1999), the Dutch Lipid Clinic Network (DLCN) criteria (Umans-Eckenhausen et al., 2001) and the US MedPed program (Williams et al., 1993). The DLCN criteria is a comprehensive tool, which considers multiple parameters in the assessment of FH including patients’ personal and family history related to the onset of early cardiovascular (CVD) or HC, untreated LDL-C concentration, disease clinical manifestations (including tendinous xanthomas and arcus cornealis) and functional mutation in a pathogenic FH-related gene (Defesche et al., 2004). However, despite the comprehensiveness of available diagnostic schemes, FH remains largely underdiagnosed.
The prevalence of FH in Qatar is not yet known. However, FH prevalence (probable and definite cases) in neighboring Gulf countries (Saudi Arabia, Oman, United Arab Emirates, Bahrain and Kuwait) was estimated at 1:232 (Al-Rasadi et al., 2020) and subsequently stratified to fulfill the DLCN criteria to reveal a markedly higher prevalence of 1:112, but genetic screening was not performed in this study (Alhabib et al., 2021). The high burden of lipid disorders in the region has also led to continued recommendations to improve the management of lipid disorders (Alsayed et al., 2022). In addition, accumulating studies have also explored the genetic spectrum of FH in the wider Middle Eastern region (Awan et al., 2019), with reports of mutations occurring predominantly in LDLR (Alhababi and Zayed, 2018). Notably, a recent study reported mutations in 6 LDLR variants in a subset of ∼6,000 individuals from Qatar, but no phenotypic analyses or evidence were sought for FH (Elfatih et al., 2021). Moreover, GWAS have also linked specific lipid risk variants to various populations in the region (Hebbar et al., 2019), specifically in Qatar (Thareja et al., 2021). Therefore, comprehensive investigations into FH etiology in Qatar are warranted.
In this study, we estimated the prevalence of FH in Qatar, while uncovering the mutational spectrum and penetrance of population-specific FH-related monogenic variants using whole-genome sequencing of the population-based cohort of Qatar biobank (QBB; n = 13,808). We sought to decipher mutations in genes previously linked with FH pathogenicity and identify highly penetrant Qatari-specific variants. Our findings have the potential to empower the current diagnostic approaches by allowing targeted genetic screening of high-risk individuals and cascade screening of suspected FH cases.
This study was based on 14,056 Qatari subjects from the population-based Qatar Biobank (QBB) study (Al Kuwari et al., 2015) and was executed under ethical approvals from the institutional review boards of QBB, Doha, Qatar (Protocol no. E-2020-QF-QBB-RES-ACC-0154-0133) and Hamad Bin Khalifa University (Approval no. 2021-03-081). All participants provided written informed consent prior to sample donation. Participants also underwent a medical examination and filled out an approved and standardized questionnaire, which captured information on medical history. The study cohort is deeply phenotyped, featuring lipid-related measurements including total cholesterol, HDL-C, LDL-C, and triglycerides, and detailed information on diagnosis, comorbidities related to heart disease and administration of cholesterol-lowering medication was also accessible. However, while information on the family history of HC was recorded, lipid measurements from close relatives were not available.
Whole-genome sequencing of the cohort was available through the Qatar Genome Program (QGP) and was performed as previously described (Mbarek et al., 2022). Briefly, libraries were constructed using the Illumina TruSeq DNA nano kit and indexed using Illumina TruSeq Single Indexes (Illumina, San Diego, CA, United States). Quality-passed libraries were sequenced on an Illumina HiSeq X instrument. The quality metrics for generated Fastq files were assessed using FastQC (v. 0.11.2). The raw reads were trimmed and aligned to hs37d5 reference genome using bwa.kit (v0.7.12) (Li and Durbin, 2009) to generate mapped reads on BAM files. The coverage of each sample was evaluated using Picard (v1.117) (CollectWgsMetrics), while variant calling was performed using base quality score recalibration (BQSR) and intermediate genomic gVCF (gVCF) was generated by running HaplotypeCaller (Genome Analysis Toolkit; GATK). The genotype data were processed for quality control using Hail (Team, 2022) and Plink (Slifer, 2018). Genetic variants with <98% call rate, chi-square test p value for Hardy-Weinberg equilibrium <1 × 10−10, or those with a depth of coverage <10X were removed, while only variants with minor allele frequency <0.005 were included in the analysis, calculated based on estimating FH prevalence of ∼1 in 200. Subjects with excess heterozygosity, gender ambiguity, or with call rate <95% were also excluded. The final study cohort comprised 13,808 subjects with both whole-genome sequence data and complete phenotype data.
The DLCN scheme draws evidence from high LDL-C levels, the presence of functional mutations in LDLR, APOB or PCSK9 genes, xanthomas and corneal deposits of fat, and evidence of coronary and vascular disease in subjects and their close relatives for diagnosing FH (Austin et al., 2004). The assessment of the phenotypic or genetic parameters assigns numerical scores to classify subjects with unlikely FH (DLCN score: <3), possible FH (DLCN score: 3–5), probable FH (DLCN score: 6–8) or definite FH (DLCN score >8) (Austin et al., 2004). We followed these criteria to characterize FH cases, while performing correction of LDL-C levels for cholesterol-lowering medication as defined by Haralambos et al. (Haralambos et al., 2015). In instances where the medication dose was not available, we used the minimum dose for correction. Of note, the presence of xanthomas and corneal fat deposition was not recorded by QBB and these parameters were not taken into consideration in our study. Importantly, the remaining criteria were sufficient to classify patients into definite/probable/possible or unlikely FH cases based on DLCN scores.
We used the bcftools software (version 1.10.2) to extract records of variants located in selected FH genes from the variant calling format (VCF) genotype file. The selection of FH genes was based on a literature search and included LDLR, APOB, PCSK9, APOE, LDLRAP1, CYP7A1, STAP1, ITIH4, EPHX2, GHR and PPP1R17 (also known as GSBS) genes (Takada et al., 2003; Fujita et al., 2004; Sato et al., 2004; Paththinige et al., 2017; Wang et al., 2020). Variant annotation was performed using SnpEff/SnpSift (v4.3t) (Cingolani et al., 2012) utilizing dbSNP build version 151 (Sherry et al., 1999), ClinVar (Landrum et al., 2018), Human Gene Mutation Database (HGMD) variant categorization (Stenson et al., 2020) and Leiden Open Variation Database (version: 3.0; LOVD3) (Fokkema et al., 2021). Variants were considered pathogenic for FH if reported as pathogenic/likely pathogenic or disease-causing mutation (DM) or likely disease-causing mutation (DM?) by at least two of the databases described earlier. We assessed the penetrance of genetic variants using two approaches; the first was based on the DLCN score (DLCN-penetrance), which takes into consideration multiple phenotypic variables related to FH risk as described earlier. Using this approach, penetrance was defined as the proportion of mutation carriers with a DLCN score ≥3. The second approach was based on LDL-C and self-reported HC (HC-penetrance) by which penetrance was defined as the proportion of mutation carriers with LDL-C > 3.3 mmol/L, or taking cholesterol-lowering medication, or self-reported HC. For variants of uncertain significance (VUS), we considered them possibly pathogenic if their HC-penetrance value was >50% and their predictive combined annotation dependent depletion (CADD) (Kircher et al., 2014) score was greater than 20, which represents the top 1% most deleterious mutations in the human genome.
The Hardy-Weinberg equilibrium of genotypes was evaluated by χ2 tests. Unpaired Student’s t test was used for comparison between individuals with definite, probable, and possible FH relative to those with unlikely FH. The prevalence of each definition of FH was estimated as a percentage for all study subjects. Fisher’s exact test was used to compare the frequency of factors among the definitions of FH. A gene-based burden test was performed using SAIGE-Gene (Zhou et al., 2020) by collapsing all pathogenic/possibly pathogenic variants detected in each gene into a single burden variable, and in this analysis model, we adjusted for age, sex, genomic kinship and the first four population principal components.
This study was based on phenotypic and genotypic data from 13,808 individuals. However, the analysis cohort comprised 13,677 subjects since LDL-C profiling measurements were not available for 107 subjects, while additional 24 subjects with hypothyroidism were removed from the analysis (Figure 1). We first categorized our cohort based on self-reports of HC; 4,066 subjects declared HC, while 9,346 subjects reported no HC and the remaining 265 subjects did not provide any information. Moreover, among the self-reported HC subjects, 2,010 subjects declared taking cholesterol-lowering medication. Importantly, our initial classifications showed that 19 subjects were classified as definite cases of FH based on the fulfillment of the DLCN criteria (DLCN score >8) by phenotypic evidence only (Figure 1). Similarly, probable, possible and unlikely FH cases were also first classified based on phenotypic evidences only. The characteristic features of the overall study cohort classified into FH categories based on phenotypic evidence are listed in Table 1. Of note, the definite FH cases cohort comprised individuals with considerably higher percentages of self-reported HC and a history of paternal and maternal heart disease compared to those in the unlikely FH group (Table 1). Moreover, subjects in the definite FH group had a lower age of HC diagnosis (35.5 ± 11.6) compared to subjects in the unlikely FH category (40.6 ± 10.6), but this was of borderline significance (p = 0.06).
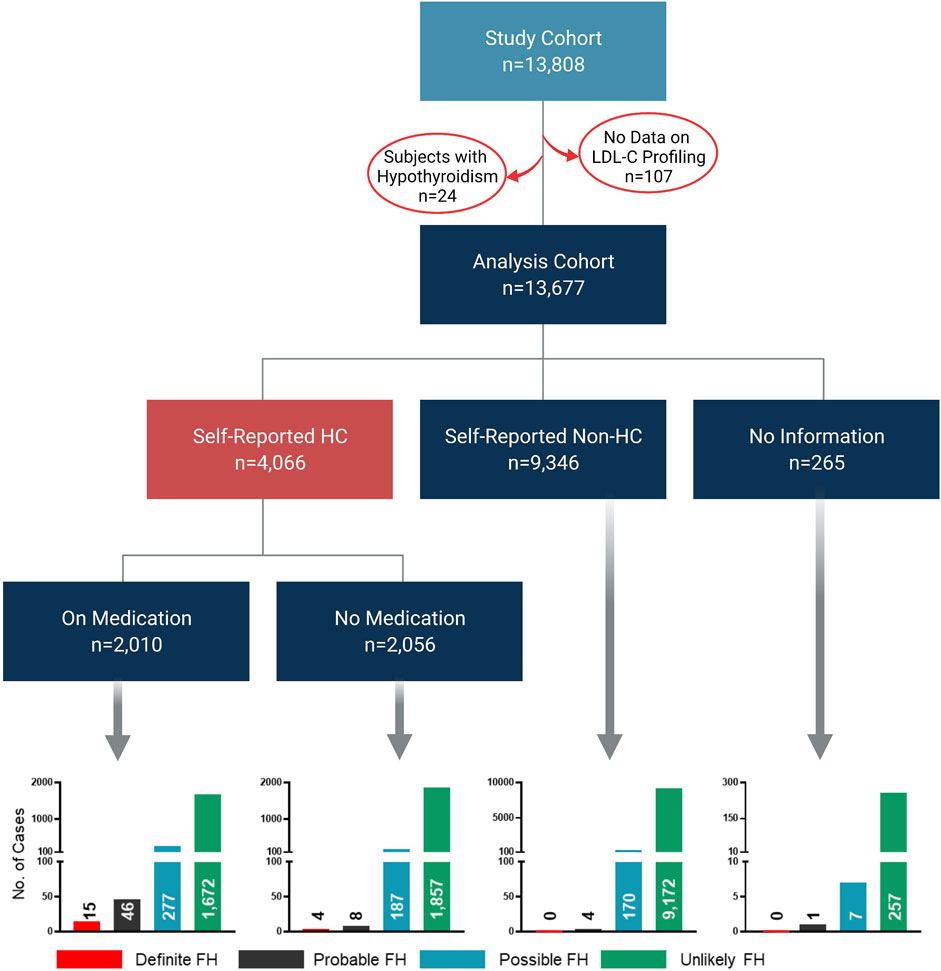
FIGURE 1. Classification of study cohort into FH subtypes based on phenotypic data. The flow chart depicts the workflow for the classification of study cohorts into FH subtypes based on phenotypic data only to fulfill DLCN criteria for FH classification. Bar charts represent numbers of individuals classified as definite, probable, possible and unlikely cases of FH in the analysis cohort.
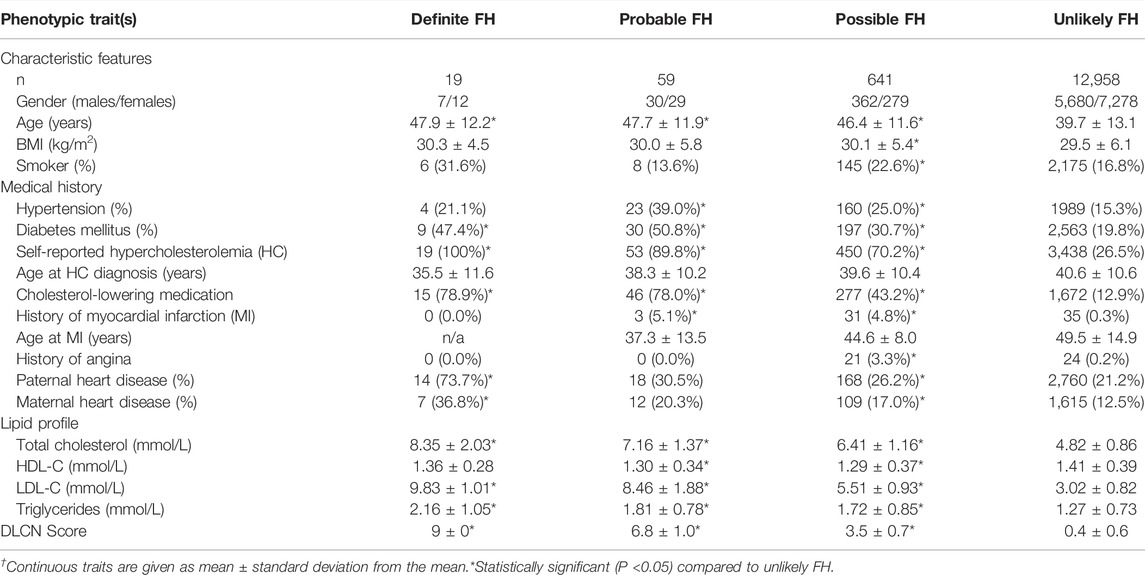
TABLE 1. Characteristics of Qatar Biobank study subjects according to the Dutch Lipid Clinic Network (DLCN) criteria based on phenotypic traits only.
Applying the DLCN criteria for FH diagnosis entails the identification of functional mutations in LDLR, APOB and PCSK9 genes. To this end, variants were scrutinized for their pathogenicity using ClinVar, LOVD3 and HGMD significance terms to identify previously reported pathogenic variants in the Qatari population. Our search for variants covered 11 FH-related genes (refer to methods). We detected 17 variants that were reported as pathogenic/likely pathogenic or DM/DM? by at least two of the ClinVar/LOVD3/HGMD databases (Table 2); all as heterozygous. Notably, 16 variants were located in LDLR (Figure 2) with rs1064793799 (c.313+3A>C) being the most frequent mutation (n = 13), followed by rs771019366 (p.Asp90Gly; n = 6) and rs747507019 (p.His327Tyr; n = 4), while one mutation was located in PCSK9 (rs891322948, n = 2). Moreover, the majority of these were missense mutations, which lead to specific protein changes while others were splice variants or associated with regulatory regions. Most mutations showed high DLCN-penetrance (>50%) and the majority had complete HC-penetrance (100%). We did not detect any previously reported pathogenic mutations in APOB, APOE, LDLRAP1, CYP7A1, STAP1, ITIH4, EPHX2, GHR, or PPP1R17.
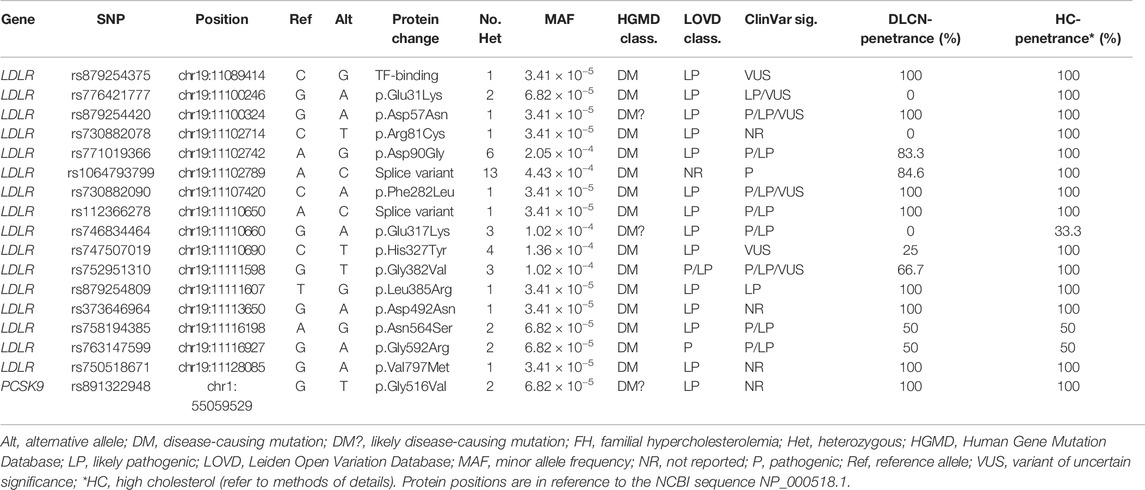
TABLE 2. Known pathogenic variants detected in the study subjects.
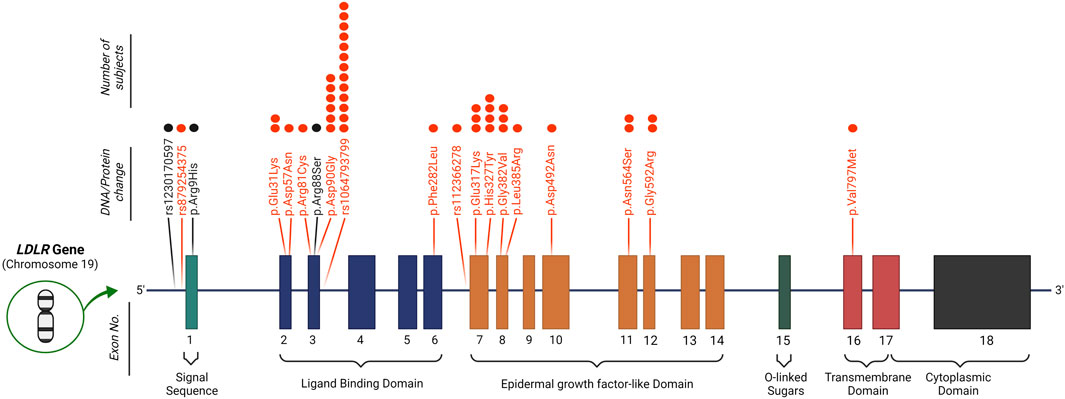
FIGURE 2. Identified mutations in the LDLR gene. 16 known pathogenic variants (marked in red) and 3 possibly pathogenic mutations (marked in black) were detected in LDLR in our study cohort. The number of subjects carrying each mutation is depicted by dots above each variant. Protein change is in reference to the NCBI sequence NP_000518.1.
We identified 18 possibly pathogenic variants as those which were predicted to be in the top 1% of most deleterious mutations in the human genome (CADD score >20) and their HC-penetrance is ≥50% (Table 3). These mutations were detected in FH-related genes including LDLR, APOB, PCSK9 and APOE, and featured a wide range of deleterious effects, including stop-gained and missense, in addition to impacting regulatory features. Five of the 18 detected variants were novel since they were not reported in genetic databases such as dbSNP and the Genome Aggregation Database (gnomAD), and were considered to be Qatari-specific. The most common possibly pathogenic variants were located in APOB (rs775231207; n = 7, rs13306190; n = 4, and rs12713559; n = 4). Overall, the majority of mutations observed were missense, while one stop-gain was detected in PCSK9. Notably, rs1230170597 is located 169 bp upstream of the coding region of LDLR and overlaps LDLR-AS1, an antisense non-coding RNA predicted to downregulate production of the LDL-R. However, these possibly pathogenic mutations were not used in DLCN classification because functional validation will be required to confirm their pathogenicity. Gene-based burden test LDL-R for the pathogenic and possibly pathogenic variants detected in our cohort showed significant associations with HC for LDLR (p <0.0001; Beta (β) = 3.2; standard error (SE) = 0.52), PCSK9 (p = 0.004; β = 2.6; SE = 1.08), and APOB (p = 0.004; β = 1.1; SE = 0.37) but not for APOE (p = 0.303). Genetic variations that fit the “possibly pathogenic” criteria were not detected in the other FH-related genes: LDLRAP1, CYP7A1, STAP1, ITIH4, EPHX2, GHR, or PPP1R17.
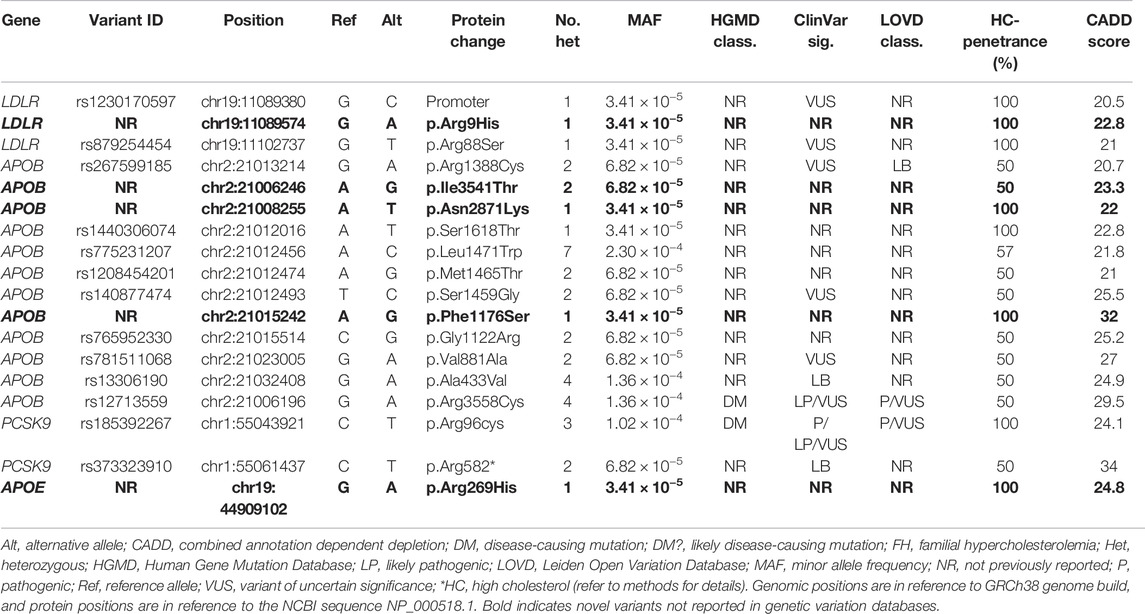
TABLE 3. Possibly pathogenic variants with high penetrance detected in the study subjects.
Assessment of QBB study participants using DLCN criteria after adding information from genotypes and known FH-pathogenic mutations revealed 52 subjects as definite cases of FH (Table 4). Moreover, the probable, possible and unlikely FH cases classified based on combining phenotypic and genotypic evidence also showed some differences in numbers as opposed to classification based on FH-associated phenotypes only (Tables 1, 4). A number of probable FH (n = 8), possible FH (n = 14) and unlikely FH (n = 10) subjects were re-classified as definite FH because they were found to carry one of the known pathogenic mutations listed in Table 2. Additionally, 6 subjects who were originally classified as unlikely FH were re-classified as probable FH after considering genetic data. The proportions of smokers, myocardial infractions and family history of heart disease was considerably higher in definite cases of FH, while age at diagnosis of HC was significantly lower than those with unlikely FH (Table 4). Next, we investigated differences in lipid profile measurements of FH definition categorized by the DLCN criteria based on phenotypic and genotypic data (Figure 3). We found that total cholesterol, LDL-C and triglyceride levels were significantly higher in definite, probable and possible FH cases compared to subjects with unlikely FH (Figure 3). Moreover, HDL-C levels were significantly lower in probable and possible FH groups compared to unlikely FH.
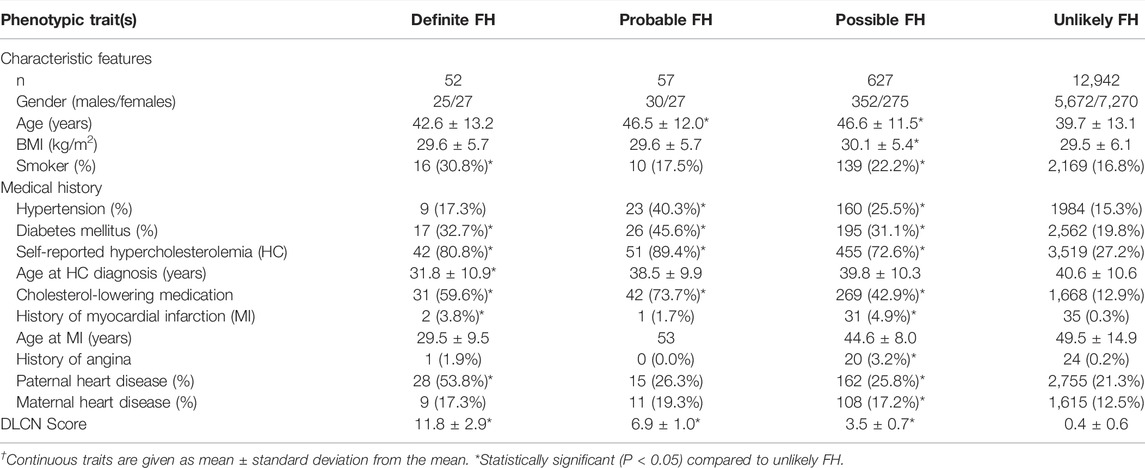
TABLE 4. Characteristics of study subjects classified according to the Dutch Lipid Clinic Network (DLCN) criteria based on phenotypic and genotypic data.
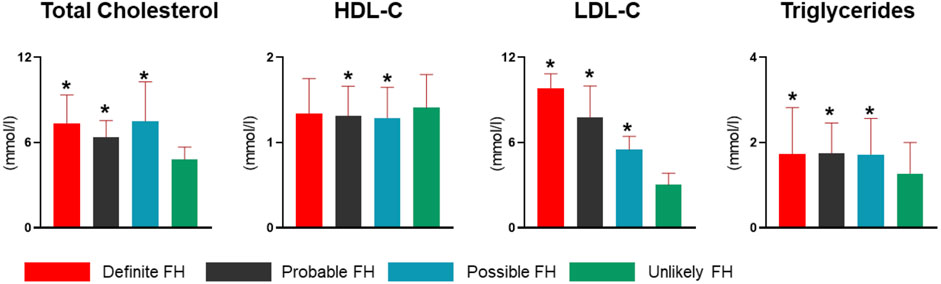
FIGURE 3. Lipid profiles of FH definition groups in the study cohort. Study subjects were categorized into definite, probable, possible and unlikely FH cases based on DLCN criteria, and using phenotype and genotype data. Bar charts represent levels of total cholesterol, HDL-C, LDL-C and triglycerides. LDL-C measurements were corrected for medication. Statistically significant (P < 0.05) differences among each FH definition compared to unlikely FH are denoted with an asterisk (*) in each plot.
Of the total 13,677 subjects used in this study, we identified 109 as definite or probable cases of FH based on the fulfillment of the DLCN criteria (Figure 3). Based on this, the prevalence of FH in Qatar was therefore estimated at 0.8% (1 in 125). Notably, 39 subjects were diagnosed as definite cases of FH (DLCN score >8) based on genotypic mutation and phenotypic evidence, while additional 13 subjects showed a DLCN score >8 based on phenotype alone and did not carry any of the known pathogenic FH variants. Combined these definite FH cases accounted for 52 subjects and yielded an overall prevalence of 0.38% (∼1 in 263) for definite FH (Figure 4). In contrast, the number of subjects with probable FH (DLCN score ranging between 6–8), who carried a known pathogenic mutation but showed no phenotypic evidence, was 6, while 51 subjects were classified as probable FH based on phenotype. These subjects accounted for approximately half of the total suspected FH cases in our cohort. Of note, the prevalence of possibly pathogenic variants was 1:351 and the overall prevalence of FH in Qatar would be considerably higher (∼1 in 92) when these mutations are included in the estimation of the prevalence. However, due to the lack of evidence on the pathogenicity of these variants, further investigations are required to incorporate them in the assessment of population-based FH prevalence. In addition, we did not detect any homozygous FH or autosomal recessive mutations in our study cohort, indicating their low prevalence in our study population.
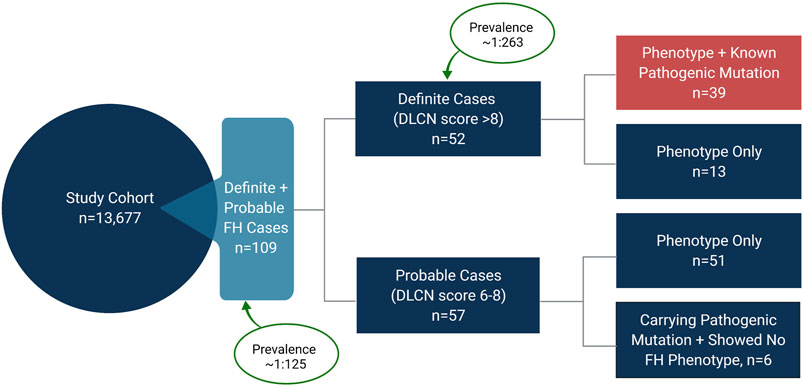
FIGURE 4. Prevalence of familial hypercholesterolemia (FH) in Qatar. The study cohort comprised 13,677 individuals. 109 subjects were identified as definite and probable cases of FH based on the Dutch Lipid Clinic Network (DLCN) criteria, yielding a prevalence of ∼1 in 125 in Qatar. 52 subjects were identified as definite cases of FH, indicating a prevalence of ∼1 in 263 for definite FH. Of the 52 definite cases, 39 subjects carried a known pathogenic mutation in LDLR or PCSK9 and showed FH-related phenotypes. 57 subjects were identified as probable cases of FH with 51 subjects showing FH-related phenotypes (DLCN score 6–8), while 6 subjects carried a known pathogenic mutation (DLCN score 8), but their LDL-C levels were below the threshold of 4.0 mmol/L defined by DLCN criteria.
This study provides the first reliable estimate of the prevalence of FH in the Qatari population and presents a comprehensive survey of population-specific monogenic FH variants in a considerably large population-based cohort. Combining FH-related phenotypes with whole-genome sequence data revealed a prevalence of definite and probable FH cases in Qatar of ∼1 in 125, which puts Qatari subjects at a higher risk of FH than the global average of 1:250, previously determined from a meta-analysis of 19 published studies (Akioyamen et al., 2017), but at a moderately lower risk than neighboring countries in the Arabian peninsula (1:112) (Alhabib et al., 2021). The higher prevalence than global estimates could be attributed to the relatively consanguineous nature of the populations in the region and in Qatar, but the prevalence is considerably lower than those reported for populations with founder effects such as the African Ashkenazi and Lebanese (Seftel et al., 1989; Abifadel et al., 2009). In addition, we also did not detect any cases of HoFH, indicating its rare prevalence in Qatar.
We identified 17 mutations, characterized in the literature as pathogenic, out of which 16 were located in LDLR. Notably, 6 of these variants in LDLR were previously reported in ∼6000 subjects from Qatar but without any phenotypic associations with FH (Elfatih et al., 2021). In contrast, some of these 16 variants we identified have been directly associated with FH phenotypes in other populations. The p.Asp90Gly mutation in LDLR was reported in phenotypic FH patients from Western Australia who were screened for LDLR, APOB and PCSK9 mutations (Hooper et al., 2012), while p.Glu31Lys has been reported in Asian Indian FH patients (Setia et al., 2020) and p.Phe282Leu was observed in Czech probands suspected to have FH (Tichý et al., 2012). Additionally, data from 6 studies investigating the influence of genotypes on response to PCSK9-targeting monoclonal antibody alirocumab also reported LDLR pGly382Val among the mutations identified in 898 HC patients (Defesche et al., 2004). While these mutations have been linked with FH, some of these variants are associated with severe disease complications related to FH. For instance, Cui et al. reported LDLR p.Arg81Cys mutation in patients with premature myocardial infarction (Cui et al., 2019).
Subjects carrying any of the known pathogenic mutations and with the presentation of FH-related phenotypes were identified as definite FH according to the DLCN. Notably, the definite FH identified in our study comprised a significantly higher proportion of subjects with self-reported HC and more importantly, a higher proportion of myocardial infarction and those diagnosed with HC at a younger age compared to subjects classified as unlikely FH. Moreover, a family history of heart disease was also considerably higher in definite FH cases. These clinical manifestations and prerequisites for FH classifications supported their classification into definite cases of FH. Considering the genotypic aberrations, the most frequently occurring variant was rs1064793799 found in 13 Qatari FH subjects, suspected to cause abnormal gene splicing. Single base changes occurring near or within introns can lead to intron retention, exon skipping, or activation of cryptic splice sites (Bourbon et al., 2009). Notably, an extreme reduction in LDLR expression, recorded in two families of Arab descent, was attributed to the activation of a cryptic splice acceptor site in LDLR due to a single substitution mutation (Shawar et al., 2012), however, this mutation was not detected in our study. In contrast, the majority of the mutations we detected in LDLR corresponded to missense mutations, which may be detected via PCR-based genotyping protocols for FH detection. For instance, Pandey et al. (2016) validated the detection of mutations in APOB and LDLR, by PCR, including p.Asp492Asn, also detected in our cohort. We also detected a pathogenic variant in PCSK9 (p.Gly516Val) in two subjects with definite FH. This variant was detected in South African FH patients and was found to be pathogenic by functional studies (Huijgen et al., 2021).
Our study highlighted the importance of genetic testing to confirm FH diagnosis since only 19 subjects were classified as definite FH based on phenotype data while additional 33 subjects were considered definite FH when genetic testing was taken into consideration.
We also detected several possibly pathogenic variants in LDLR, APOB, PCSK9 and APOE in our study subjects which were not previously confirmed in the literature as disease-causing mutations. These variants, however, were predicted to be among the top 1% of most deleterious mutations in the human genome and showed high penetrance for HC in our study. These variants could be presented as novel variants related to FH, however, further functional investigations are warranted for their validation. Notably, all variants detected in APOB were point mutations that could lead to defects in lipid hemostasis. Point mutations in APOB have been reported to cause FH by affecting its affinity for the LDL-R, which causes disruptions in LDL clearance via LDL-R-mediated internalization (Sharifi et al., 2017). These mutations differ from truncation mutations in APOB associated with hypobetalipoproteinemia. However, further functional investigations are warranted to confirm their pathogenicity in FH. Notably, five of the possibly pathogenic variants were novel and appear to be predominant in the Qatari population. Novel Qatari-predominant loci have been identified for many clinically relevant traits in a recent GWAS study of the QBB study participants (Thareja et al., 2021). A gene-based burden test for the pathogenic and possibly pathogenic variants in LDLR, PCSK9 and APOB confirmed the association of these variants with HC. However, the gene-based burden test was not significant for APOE possibly because of the small number of pathogenic variants detected in our cohort. Overall, our findings reiterate that mutations in LDLR are the most common cause of FH in the Qatari population.
In this study, a substantially large cohort of 13,677 was used to achieve a reliable estimate for the prevalence of definite and probable cases of FH in the Qatari population, which was approximated to 1 in 125. The prevalence of subjects with possibly pathogenic variants was 1:351 and if future functional studies confirm the pathogenicity of these variants, the overall prevalence of FH will be 1:92 which is higher than our original estimate of 1:125 based on known pathogenic variants. We did not detect any cases of HoFH in our study cohort which indicates that HoFH is extremely rare in Qatar, as estimated in many European populations, but a larger sample size would be required to accurately estimate the prevalence of HoFH. However, one key limitation of our data collection was the lack of phenotypic data related to xanthomas and arcus cornealis for subjects or their first-degree relatives in our cohort. In addition, we did not investigate structural variants in FH-related genes, however, FH is predominantly caused by point mutations and a small fraction of FH cases are attributed to structural variants in FH-related genes. Overall, we detected 17 known pathogenic mutations in FH subjects and identified further 18 possibly pathogenic variants as novel candidates for FH. However, further functional investigations are warranted to investigate their pathogenicity. Overall, the clinical translation of FH-related variants reported herein may be explored further to design FH diagnostic tools.
The data analyzed in this study is subject to the following licenses/restrictions: The raw whole-genome sequence data from Qatar Biobank are protected and are not available due to data privacy laws. Access to QBB/QGP phenotype and whole-genome sequence data can be obtained through an ISO-certified protocol, which involves submitting a project request at https://www.qatarbiobank.org.qa/research/how-apply, subject to approval by the Institutional Review Board of the QBB. Requests to access these datasets should be directed to https://www.qatarbiobank.org.qa/research/how-apply.
The studies involving human participants were reviewed and approved by Qatar Biobank, Hamad Bin Khalifa University. The patients/participants provided their written informed consent to participate in this study.
ID: data curation, formal analysis, investigation, and writing—original draft. YA-S: investigation and methodology. ST: writing—review and editing and visualization. NQ: writing—review and editing. SM, MA, AJ, and JA: resources. OMEA: conceptualization, funding acquisition, formal analysis, supervision, investigation, and writing—review and editing.
This study was supported by a grant from the Qatar National Research Fund (PPM 03-0324-190038) awarded to OMEA.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We are grateful to the Qatar Biobank (QBB) and the Qatar Genome Program Research (QGPR) Consortium for providing us the genomic and phenotypic data. We are also thankful to all the participants of this study.
Abifadel, M., Rabès, J.-P., Jambart, S. l., Halaby, G., Gannagé-Yared, M.-H. l. n., Sarkis, A., et al. (2009). The Molecular Basis of Familial Hypercholesterolemia in Lebanon: Spectrum ofLDLRmutations and Role ofPCSK9as a Modifier Gene. Hum. Mutat. 30, E682–E691. doi:10.1002/humu.21002
Akioyamen, L. E., Genest, J., Shan, S. D., Reel, R. L., Albaum, J. M., Chu, A., et al. (2017). Estimating the Prevalence of Heterozygous Familial Hypercholesterolaemia: a Systematic Review and Meta-Analysis. BMJ Open 7 (9), e016461. doi:10.1136/bmjopen-2017-016461
Al Kuwari, H., Al Thani, A., Al Marri, A., Al Kaabi, A., Abderrahim, H., Afifi, N., et al. (2015). The Qatar Biobank: Background and Methods. BMC Public Health 15, 1208. doi:10.1186/s12889-015-2522-7
Al-Rasadi, K., Alhabib, K. F., Al-Allaf, F., Al-Waili, K., Al-Zakwani, I., AlSarraf, A., et al. (2020). The Gulf Familial Hypercholesterolemia Registry (Gulf FH): Design, Rationale and Preliminary Results. Curr. Vasc. Pharmacol. 18 (1), 57–64. doi:10.2174/1570161116666181005125459
Alhababi, D., and Zayed, H. (2018). Spectrum of Mutations of Familial Hypercholesterolemia in the 22 Arab Countries. Atherosclerosis 279, 62–72. doi:10.1016/j.atherosclerosis.2018.10.022
Alhabib, K. F., Al-Rasadi, K., Almigbal, T. H., Batais, M. A., Al-Zakwani, I., Al-Allaf, F. A., et al. (2021). Familial Hypercholesterolemia in the Arabian Gulf Region: Clinical Results of the Gulf FH Registry. PLoS One 16 (6), e0251560. doi:10.1371/journal.pone.0251560
Alsayed, N., Almahmeed, W., Alnouri, F., Al-Waili, K., Sabbour, H., Sulaiman, K., et al. (2022). Consensus Clinical Recommendations for the Management of Plasma Lipid Disorders in the Middle East: 2021 Update. Atherosclerosis 343, 28–50. doi:10.1016/j.atherosclerosis.2021.11.022
Austin, M. A., Hutter, C. M., Zimmern, R. L., and Humphries, S. E. (2004). Genetic Causes of Monogenic Heterozygous Familial Hypercholesterolemia: a HuGE Prevalence Review. Am. J. Epidemiol. 160 (5), 407–420. doi:10.1093/aje/kwh236
Awan, Z. A., Bondagji, N. S., and Bamimore, M. A. (2019). Recently Reported Familial Hypercholesterolemia-Related Mutations from Cases in the Middle East and North Africa Region. Curr. Opin. Lipidol. 30 (2), 88–93. doi:10.1097/mol.0000000000000586
Beheshti, S. O., Madsen, C. M., Varbo, A., and Nordestgaard, B. G. (2020). Worldwide Prevalence of Familial Hypercholesterolemia. J. Am. Coll. Cardiol. 75 (20), 2553–2566. doi:10.1016/j.jacc.2020.03.057
Benn, M., Watts, G. F., Tybjaerg-Hansen, A., and Nordestgaard, B. G. (2012). Familial Hypercholesterolemia in the Danish General Population: Prevalence, Coronary Artery Disease, and Cholesterol-Lowering Medication. J. Clin. Endocrinol. Metab. 97 (11), 3956–3964. doi:10.1210/jc.2012-1563
Bouhairie, V. E., and Goldberg, A. C. (2015). Familial Hypercholesterolemia. Cardiol. Clin. 33 (2), 169–179. doi:10.1016/j.ccl.2015.01.001
Bourbon, M., Duarte, M. A., Alves, A. C., Medeiros, A. M., Marques, L., and Soutar, A. K. (2009). Genetic Diagnosis of Familial Hypercholesterolaemia: the Importance of Functional Analysis of Potential Splice-Site Mutations. J. Med. Genet. 46 (5), 352–357. doi:10.1136/jmg.2007.057000
Cingolani, P., Platts, A., Wang, L. L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff. Fly 6 (2), 80–92. doi:10.4161/fly.19695
Cuchel, M., Bruckert, E., Ginsberg, H. N., Raal, F. J., Santos, R. D., Hegele, R. A., et al. (2014). Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur. Heart J. 35 (32), 2146–2157. doi:10.1093/eurheartj/ehu274
Cui, Y., Li, S., Zhang, F., Song, J., Lee, C., Wu, M., et al. (2019). Prevalence of familial hypercholesterolemia in patients with premature myocardial infarction. Clin. Cardiol. 42 (3), 385–390. doi:10.1002/clc.23154
Defesche, J. C., Lansberg, P. J., Umans-Eckenhausen, M. A., and Kastelein, J. J. (2004). Advanced method for the identification of patients with inherited hypercholesterolemia. Semin. Vasc. Med. 4 (1), 59–65. doi:10.1055/s-2004-822987
Elfatih, A., Mifsud, B., Syed, N., Badii, R., Mbarek, H., Abbaszadeh, F., et al. (2021). Actionable genomic variants in 6045 participants from the Qatar Genome Program. Hum. Mutat. 42, 1584–1601. doi:10.1002/humu.24278
Fokkema, I. F. A. C., Kroon, M., López Hernández, J. A., Asscheman, D., Lugtenburg, I., Hoogenboom, J., et al. (2021). The LOVD3 platform: efficient genome-wide sharing of genetic variants. Eur. J. Hum. Genet. 29 (12), 1796–1803. doi:10.1038/s41431-021-00959-x
Fujita, Y., Ezura, Y., Emi, M., Sato, K., Takada, D., Iino, Y., et al. (2004). Hypercholesterolemia associated with splice-junction variation of inter-α-trypsin inhibitor heavy chain 4 (ITIH4) gene. J. Hum. Genet. 49 (1), 24–28. doi:10.1007/s10038-003-0101-8
Garcia, C. K., Wilund, K., Arca, M., Zuliani, G., Fellin, R., Maioli, M., et al. (2001). Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein. Science 292 (5520), 1394–1398. doi:10.1126/science.1060458
Graham, S. E., Clarke, S. L., Wu, K. H., Kanoni, S., Zajac, G. J. M., Ramdas, S., et al. (2021). The power of genetic diversity in genome-wide association studies of lipids. Nature 600 (7890), 675–679. doi:10.1038/s41586-021-04064-3
Haralambos, K., Whatley, S. D., Edwards, R., Gingell, R., Townsend, D., Ashfield-Watt, P., et al. (2015). Clinical experience of scoring criteria for Familial Hypercholesterolaemia (FH) genetic testing in Wales. Atherosclerosis 240 (1), 190–196. doi:10.1016/j.atherosclerosis.2015.03.003
Hebbar, P., Abubaker, J. A., Abu-Farha, M., Tuomilehto, J., Al-Mulla, F., and Thanaraj, T. A. (2019). A Perception on Genome-Wide Genetic Analysis of Metabolic Traits in Arab Populations. Front. Endocrinol. 10, 8. doi:10.3389/fendo.2019.00008
Henderson, R., O’Kane, M., McGilligan, V., and Watterson, S. (2016). The genetics and screening of familial hypercholesterolaemia. J. Biomed. Sci. 23, 39. doi:10.1186/s12929-016-0256-1
Hooper, A. J., Nguyen, L. T., Burnett, J. R., Bates, T. R., Bell, D. A., Redgrave, T. G., et al. (2012). Genetic analysis of familial hypercholesterolaemia in Western Australia. Atherosclerosis 224 (2), 430–434. doi:10.1016/j.atherosclerosis.2012.07.030
Hu, P., Dharmayat, K. I., Stevens, C. A. T., Sharabiani, M. T. A., Jones, R. S., Watts, G. F., et al. (2020). Prevalence of Familial Hypercholesterolemia Among the General Population and Patients With Atherosclerotic Cardiovascular Disease. Circulation 141 (22), 1742–1759. doi:10.1161/circulationaha.119.044795
Huijgen, R., Blom, D. J., Hartgers, M. L., Chemello, K., Benito-Vicente, A., Uribe, K. B., et al. (2021). Novel PCSK9 (Proprotein Convertase Subtilisin Kexin Type 9) Variants in Patients With Familial Hypercholesterolemia From Cape Town. Atvb 41 (2), 934–943. doi:10.1161/atvbaha.120.314482
Kircher, M., Witten, D. M., Jain, P., O'Roak, B. J., Cooper, G. M., and Shendure, J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46 (3), 310–315. doi:10.1038/ng.2892
Landrum, M. J., Lee, J. M., Benson, M., Brown, G. R., Chao, C., Chitipiralla, S., et al. (2018). ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46 (D1), D1062–D1067. doi:10.1093/nar/gkx1153
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 (14), 1754–1760. doi:10.1093/bioinformatics/btp324
Mbarek, H., Devadoss Gandhi, G., Selvaraj, S., Al-Muftah, W., Badji, R., Al-Sarraj, Y., et al. (2022). Qatar genome: Insights on genomics from the Middle East. Hum. Mutat. 43, 499–510. doi:10.1002/humu.24336
Meshkov, A., Ershova, A., Kiseleva, A., Zotova, E., Sotnikova, E., Petukhova, A., et al. (2021). The LDLR, APOB, and PCSK9 Variants of Index Patients with Familial Hypercholesterolemia in Russia. Genes (Basel). 12 (1). doi:10.3390/genes12010066
Mortality (1999). Mortality in treated heterozygous familial hypercholesterolaemia: implications for clinical management. Scientific Steering Committee on behalf of the Simon Broome Register Group. Atherosclerosis 142 (1), 105–112. doi:10.1016/S0021-9150(98)00200-7
Pandey, S., Leider, M., Khan, M., and Grammatopoulos, D. K. (2016). Cascade Screening for Familial Hypercholesterolemia: PCR Methods with Melting-Curve Genotyping for the Targeted Molecular Detection of Apolipoprotein B and LDL Receptor Gene Mutations to Identify Affected Relatives. J. Appl. Lab. Med. 1 (2), 109–118. doi:10.1373/jalm.2016.020610
Paththinige, C., Sirisena, N., and Dissanayake, V. (2017). Genetic determinants of inherited susceptibility to hypercholesterolemia - a comprehensive literature review. Lipids Health Dis. 16 (1), 103. doi:10.1186/s12944-017-0488-4
Saadatagah, S., Jose, M., Dikilitas, O., Alhalabi, L., Miller, A. A., Fan, X., et al. (2021). Genetic basis of hypercholesterolemia in adults. npj Genom. Med. 6 (1), 28. doi:10.1038/s41525-021-00190-z
Sato, K., Emi, M., Ezura, Y., Fujita, Y., Takada, D., Ishigami, T., et al. (2004). Soluble epoxide hydrolase variant (Glu287Arg) modifies plasma total cholesterol and triglyceride phenotype in familial hypercholesterolemia: intrafamilial association study in an eight-generation hyperlipidemic kindred. J. Hum. Genet. 49 (1), 29–34. doi:10.1007/s10038-003-0103-6
Seftel, H. C., Baker, S. G., Jenkins, T., and Mendelsohn, D. (1989). Prevalence of familial hypercholesterolemia in Johannesburg Jews. Am. J. Med. Genet. 34, 545–547. doi:10.1002/ajmg.1320340418
Setia, N., Movva, S., Balakrishnan, P., Biji, I. K., Sawhney, J. P. S., Puri, R., et al. (2020). Genetic analysis of familial hypercholesterolemia in Asian Indians: A single-center study. J. Clin. Lipidol. 14 (1), 35–45. doi:10.1016/j.jacl.2019.12.010
Sharifi, M., Futema, M., Nair, D., and Humphries, S. E. (2017). Genetic Architecture of Familial Hypercholesterolaemia. Curr. Cardiol. Rep. 19 (5), 44. doi:10.1007/s11886-017-0848-8
Shawar, S. M., Al-Drees, M. A., Ramadan, A. R., Ali, N. H., and Alfadhli, S. M. (2012). The Arabic allele: a single base pair substitution activates a 10-base downstream cryptic splice acceptor site in exon 12 of LDLR and severely decreases LDLR expression in two unrelated Arab families with familial hypercholesterolemia. Atherosclerosis 220 (2), 429–436. doi:10.1016/j.atherosclerosis.2011.10.045
Sherry, S. T., Ward, M., and Sirotkin, K. (1999). dbSNP-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res. 9 (8), 677–679. doi:10.1101/gr.9.8.677
Singh, S., and Bittner, V. (2015). Familial hypercholesterolemia--epidemiology, diagnosis, and screening. Curr. Atheroscler. Rep. 17 (2), 482. doi:10.1007/s11883-014-0482-5
Sjouke, B., Kusters, D. M., Kindt, I., Besseling, J., Defesche, J. C., Sijbrands, E. J. G., et al. (2015). Homozygous autosomal dominant hypercholesterolaemia in the Netherlands: prevalence, genotype-phenotype relationship, and clinical outcome. Eur. Heart J. 36 (9), 560–565. doi:10.1093/eurheartj/ehu058
Slifer, S. H. (2018). PLINK: Key Functions for Data Analysis. Curr. Protoc. Hum. Genet. 97 (1), e59. doi:10.1002/cphg.59
Stenson, P. D., Mort, M., Ball, E. V., Chapman, M., Evans, K., Azevedo, L., et al. (2020). The Human Gene Mutation Database (HGMD): optimizing its use in a clinical diagnostic or research setting. Hum. Genet. 139 (10), 1197–1207. doi:10.1007/s00439-020-02199-3
Takada, D., Ezura, Y., Ono, S., Iino, Y., Katayama, Y., Xin, Y., et al. (2003). Growth hormone receptor variant (L526I) modifies plasma HDL cholesterol phenotype in familial hypercholesterolemia: intra-familial association study in an eight-generation hyperlipidemic kindred. Am. J. Med. Genet. 121A (2), 136–140. doi:10.1002/ajmg.a.20172
Team, H. (2022). Hail 0.2. Available at: https://github.com/hail-is/hail.
Thareja, G., Al-Sarraj, Y., Al-Sarraj, Y., Belkadi, A., Qatar Genome Program Research, C., Almotawa, M., et al. (2021). Whole genome sequencing in the Middle Eastern Qatari population identifies genetic associations with 45 clinically relevant traits. Nat. Commun. 12 (1), 1250. doi:10.1038/s41467-021-21381-3
Tichý, L., Freiberger, T., Zapletalová, P., Soška, V., Ravčuková, B., and Fajkusová, L. (2012). The molecular basis of familial hypercholesterolemia in the Czech Republic: spectrum of LDLR mutations and genotype-phenotype correlations. Atherosclerosis 223 (2), 401–408. doi:10.1016/j.atherosclerosis.2012.05.014
Umans-Eckenhausen, M. A., Defesche, J. C., Sijbrands, E. J., Scheerder, R. L., and Kastelein, J. J. (2001). Review of first 5 years of screening for familial hypercholesterolaemia in the Netherlands. Lancet 357 (9251), 165–168. doi:10.1016/s0140-6736(00)03587-x
Vrablik, M., Tichý, L., Freiberger, T., Blaha, V., Satny, M., and Hubacek, J. A. (2020). Genetics of Familial Hypercholesterolemia: New Insights. Front. Genet. 11, 574474. doi:10.3389/fgene.2020.574474
Wald, D. S., Bestwick, J. P., Morris, J. K., Whyte, K., Jenkins, L., and Wald, N. J. (2016). Child-Parent Familial Hypercholesterolemia Screening in Primary Care. N. Engl. J. Med. 375 (17), 1628–1637. doi:10.1056/nejmoa1602777
Wang, H., Yang, H., Liu, Z., Cui, K., Zhang, Y., Zhang, Y., et al. (2020). Targeted Genetic Analysis in a Chinese Cohort of 208 Patients Related to Familial Hypercholesterolemia. Jat 27 (12), 1288–1298. doi:10.5551/jat.54593
Williams, R. R., Hunt, S. C., Schumacher, M. C., Hegele, R. A., Leppert, M. F., Ludwig, E. H., et al. (1993). Diagnosing heterozygous familial hypercholesterolemia using new practical criteria validated by molecular genetics. Am. J. Cardiol. 72 (2), 171–176. doi:10.1016/0002-9149(93)90155-6
Keywords: dyslipidemias, hypercholesterolemia, familial hypercholesterolemia, monogenic, FH, LDL-C, LDLR
Citation: Diboun I, Al-Sarraj Y, Toor SM, Mohammed S, Qureshi N, Al Hail MSH, Jayyousi A, Al Suwaidi J and Albagha OME (2022) The Prevalence and Genetic Spectrum of Familial Hypercholesterolemia in Qatar Based on Whole Genome Sequencing of 14,000 Subjects. Front. Genet. 13:927504. doi: 10.3389/fgene.2022.927504
Received: 24 April 2022; Accepted: 15 June 2022;
Published: 15 July 2022.
Edited by:
Uma Ramaswami, Royal Free London NHS Foundation Trust, United KingdomCopyright © 2022 Diboun, Al-Sarraj, Toor, Mohammed, Qureshi, Al Hail, Jayyousi, Al Suwaidi and Albagha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Omar M. E. Albagha, b2FsYmFnaGFAaGJrdS5lZHUucWE=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.